

Introduction
Pelizaeus-Merzbacher disease (PMD; #312080) is a leukodystrophy associated with dysmyelination in the central nervous system (CNS) that usually segregates as an X-linked recessive trait. Clinical features include nystagmus, stridor, hypotonia, spasticity, ataxia, tremor, and deterioration of speech and mental function (1). The clinical severity, age of onset, and rate of progression in PMD vary widely, primarily depending on the nature of the causative mutation.
PMD can result from mutations in the highly conserved PLP1 gene that encodes an extremely hydrophobic, tetraspan, integral membrane protein (2). About 80% of PMD patients carry PLP1 gene duplications or missense mutations whereas deletions are extremely rare (3, 4). The PLP1 duplications range in size between 100 kb to 4.6 Mb with heterogeneous distal and proximal breakpoints; no correlation could be found between the size and position of the duplications and the severity of the disease (5, 6). In general, duplications convey a milder clinical phenotype than point mutations in the PLP1 gene, whereas patients with complete loss of PLP1 appear to be less severely affected, but have also peripheral neuropathy (7, 8).
Patients with the PMD phenotype but without mutations of the PLP1 gene are considered to have Pelizaeus-Merzbacher-like disease (PMLD; #608804). PMLD is a genetically heterogeneous and usually autosomal recessive trait. In a minority of PMLD cases, it is caused by mutations in gap junction protein α12 gene (GJA12/GJC2; #608803) coding for Connexin 47 (Cx47) (9). To date, 25 GJA12/GJC2 mutations have been reported in PMLD patients, including a point mutation in the promoter region that is characterized as a transcription factor SOX10 binding site in the mouse Gjc2 promoter (10-15).
The presence of phenotypic similarities in spite of genetic heterogeneity suggests Cx47 together with other connexins and PLP1 are critical parts of the myelination program that requires correct expression and targeting to the myelin sheath (16).
We investigated the genetic heterogeneity of the disease in a cohort of 19 families that were clinically diagnosed with PMD. The patients were screened for mutations in the PLP1 and GJA12/GJC2 genes by conventional PCR and Sanger sequencing methods and the chromosomal rearrangements in the PLP1 locus were investigated further by array CGH.
Patients and methods
Patients
Nineteen unrelated Turkish families with PMD phenotype, including seven consanguineous pedigrees, were analyzed. Among 22 patients studied 17 were males and 5 were females. In two families, there were at least two affected individuals. Informed consent was procured from all participating subjects.
The cases were followed during 1997-2010 in the Department of Child Neurology, Istanbul Medical Faculty. Pyramidal signs were classified as mild (only brisk reflexes and clonus, with/or without the increase in extensor tonus of the lower extremities), moderate (prominent spasticity), and severe (tendon contracture). Cerebellar signs were also evaluated as severe, moderate, and mild according to the Klockgether ataxia score (17). Screening tests for inherited metabolic diseases, Aryl-sulphatase-A activity, VEP, NCV, MRI, and MRS were studied. The involvement of white matter, U fiber, corpus callosum, brain stem, cerebellum, and basal ganglia were assessed by MRI. MRS studies were obtained by a 1.5 T clinical scanner using PROBE/SV software (GE, Milwaukee; WI).
Interphase FISH
FISH was performed using standard protocols (18). A cosmid clone, cU125A1, containing the PLP1 gene and a control cosmid, cU144A10, mapping approximately 850 kb distal to the PLP1 gene were used as probes.
Quantitative real-time PCR analysis
DNA samples were amplified using the primer pairs for PLP1 exons 3 and 6 and Periaxin exon 6 as an autosomal reference with Light Cycler (Roche). A normal male and female were included as controls. PCR was performed according to the manufacturer’s protocol.
aCGH
A tiling-path custom oligonucleotide microarray was designed for a 15.5 Mb genomic segment encompassing PLP1 as described previously (19).
Screening of PLP1 and GJA12/GJC2 for point mutations
PLP1 and GJA12/GJC2 were screened for mutations by SSCP and subsequent bidirectional sequencing. Direct sequencing has been performed for GJA12/GJC2 promoter of the patients F7.3, F5.3, F11.3, F13.3, and F14.3. The primer sequences for PLP1 and GJA12/GJC2 promoter were as described previously (20, 21) and for GJA12/GJC2 are listed in Supplementary 1. Segregation of the identified sequence alterations among family members and absence in at least 100 control chromosomes were investigated by appropriate restriction enzyme analysis, SSCP analysis (p.G236R) or 8% denaturing polyacrylamide gels (p.D64fs214X).
Results
Clinical evaluation
Clinical and MRI findings were compatible with a PMD diagnosis in 22 subjects (Table 1, Supplementary 2). The age range of the patients was between six months-13 years at the time of admission (6.7±5.9). The follow-up period was between two-14 years (7.4±2.8), and at the end of follow-up, the ages were between 7-29 years (14±6.6). Patient F17.4 died because of a lung infection during the follow-up. The most common clinical manifestations consisted of rotatory or pendular nystagmus within the first 2 years of life in all and ataxic gait within 5 years. All were socially interactive and appeared to have better receptive ability than expressive ability because of the cerebellar dysarthria and cognitive delay, but no polyneuropathy. Inherited metabolic diseases were ruled out by screening tests. All patients had cerebellar signs that contributed to their motor impairment as they aged. None of them walked independently at the end of follow-up except F9.3 (29 years old) with PLP1 duplication and F23.3 (7 years old) with a 20 bp GJA12 duplication. Five patients (F15.3, F16.3, F17.3, F17.4, and F21.3) never achieved a stable sitting position because of the severe hypotonia. The course of the disease was slowly progressive during a prolonged time interval.
Table 1.
Clinical characteristics of patients analyzed in this study.
| Mutation | Patient number |
Consanguinity | Sex | Current age |
Onset age of nystagmus |
Course of nystagmus |
Onset age of stridor |
Dysphagia | Pyramidal signs |
Cerebellar signs |
|---|---|---|---|---|---|---|---|---|---|---|
| PLP1 dup | F4.3 | − | M | 17 | Congenital | Same | Absent | Absent | Moderate | Severe |
| PLP1 dup | F9.3 | − | M | 29 | Absent | Absent | Absent | Absent | Moderate | Mild |
| PLP1 dup | F19.3 | − | M | 7 | 2,5 months | Same | Absent | Absent | Mild | Moderate/severe |
| PLP1 dup | F20.3 | − | M | 8 | Congenital | Same | Absent | Absent | Moderate | Moderate/severe |
| PLP1 mut | F15.3 | − | M | 7 | Congenital | Decreasing | 2 years | Present | Severe | Severe |
| PLP1 mut | F16.3 | − | M | 8 | 2 months | Decreasing | Congenital | Present | Severe | Severe |
| CGH del | F6.3 | + | M | 17 | 3 months | Same | Absent | Absent | Severe | Severe |
| CGH dup, CNV? | F18.3 | + | M | 17 | Congenital | Decreasing | Absent | Absent | Moderate | Severe |
| CGH dup, CNV? | F22.3 | − | M | 29 | Absent | Absent | Absent | Absent | Mild | Moderate/Severe |
| GJA12/GJC2 mut | F1.3* | + | M | 14 | Congenital | Same | Absent | Absent | Severe | Severe |
| GJA12/GJC2 mut | F2.3* | + | M | 18 | 1,5 years | Same | Absent | Absent | Severe | Severe |
| GJA12/GJC2 mut | F3.3* | + | F | 14 | 2 months | Same | Absent | Present | Severe | Severe |
| GJA12/GJC2 mut | F7.3 | − | M | 22 | 1 month | Decreasing | Absent | Absent | Severe | Severe |
| GJA12/GJC2 mut | F12.3 | Same village | F | 18 | 3 months | Same | Absent | Present | Moderate | Severe |
| GJA12/GJC2 mut | F17.3 | + | F | 21 | 8 months | Decreasing | Absent | Absent | Moderate | Moderate/Severe |
| GJA12/GJC2 mut | F17.4 | + | F | died at 14 | 9 months | Decreasing | Absent | Absent | Moderate | Severe |
| GJA12/GJC2 mut | F21.3 | Same village | F | 9 | Congenital | Same | Absent | Absent | Moderate | Moderate/Severe |
| GJA12/GJC2 mut | F23.3 | + | M | 7 | Congenital | Decreasing | Absent | Absent | Mild | Mild |
| − | F5.3 | − | M | 5 | 1 month | Decreasing | Absent | Absent | Moderate | Moderate |
| − | F11.3 | − | M | 14 | 1,5 years | Decreasing | Absent | Absent | Moderate | Severe |
| − | F13.3 | + | M | 28 | Not known | Decreasing | Absent | Absent | Mild | Mild |
| − | F14.3 | + | M | 18 | Congenital | Decreasing | Absent | Present | Moderate | Moderate/Severe |
Patients are from different branches of the same consanguineous family.
MRI revealed bilateral diffuse superficial and deep white matter hyperintensity in T2 and PD weighted images with U fiber involvement in all and the brain had the appearance of a newborn in T1 (Fig. 1). N-acetyl-aspartate/creatinine (NAA/Cr) ratios were not significantly different from the normal variance found in control subjects (p>0.05) in MRS.
Fig. 1.
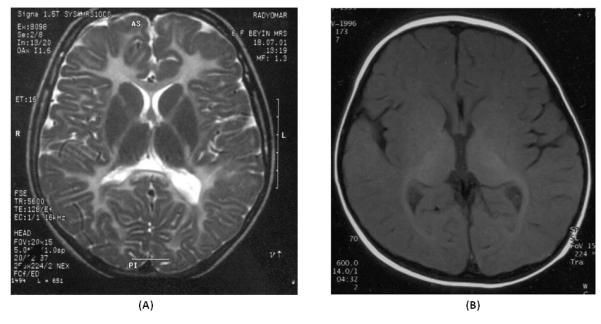
Brain MRIs of two patients analyzed in this study. a Axial T2-weighted image from patient F3.3 (with p.V182fs257X mutation in GJA12/GJC2) shows diffuse white matter hyperintensities with U fiber involvement. b Axial T1-weighted image from patient F4.3 (with PLP1 gene duplication) shows high signal intensity in the internal capsule, optic radiations like in a newborn indicating hypomyelination.
PLP1 gene rearrangements
PLP1 gene duplications were identified in patients F4.3, F9.3, F19.3, and F20.3 using FISH (data not shown) and quantitative real-time assays (Supplementary 3). The aCGH data provided further evidence for the presence of PLP1 duplications, determined the duplication breakpoints, and revealed three further rearrangements in patients F6.3, F18.3, and F22.3 (Fig. 2). Because of unavailability of DNA samples from mothers the origin of these rearrangements could not be investigated. Four patients (F5.3, F11.3, F13.3, and F14.3) did not show any alterations within the resolution limits of our array.
Fig. 2.
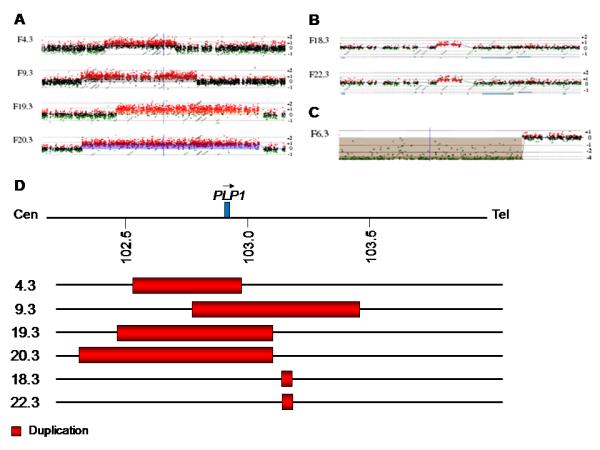
Results of aCGH analysis of the PLP1 gene rearrangements. The X-axis indicates the genomic positions of the probes. The positive and negative numbers on the Y-axis indicate gain and loss of genomic copy numbers, respectively. The vertical blue line indicates the position of the PLP1 gene. a The four PLP1 duplications identified in this study, b the two small duplications telomeric to PLP1, and c the deletion centromeric to PLP1 are shown. d Breakpoints of the duplications identified in this study and the position of the PLP1 gene is indicated.
Mutation analysis in PLP1 and GJA12/GJC2
The PLP1 AhaII polymorphism was observed in patients F7.3, F10.3, F14.3, F17.3, and F17.4. Patient F15.3 had the previously reported p.P215S mutation inherited from the mother (22). Patient F16.3 was hemizygous for the novel p.F232S mutation that was not observed in his mother (Fig. 3a and Supplementary 4).
Fig. 3.
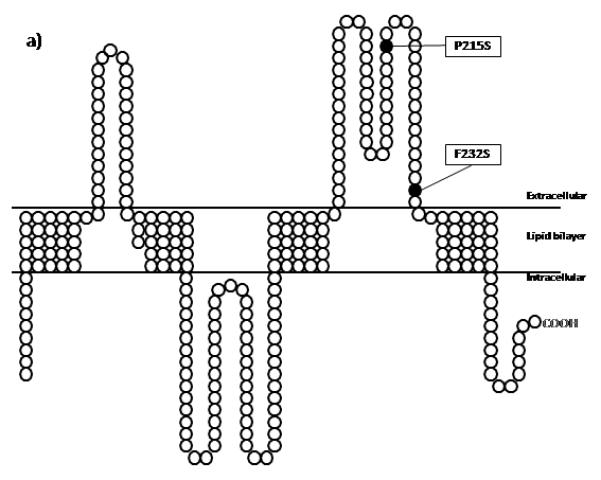
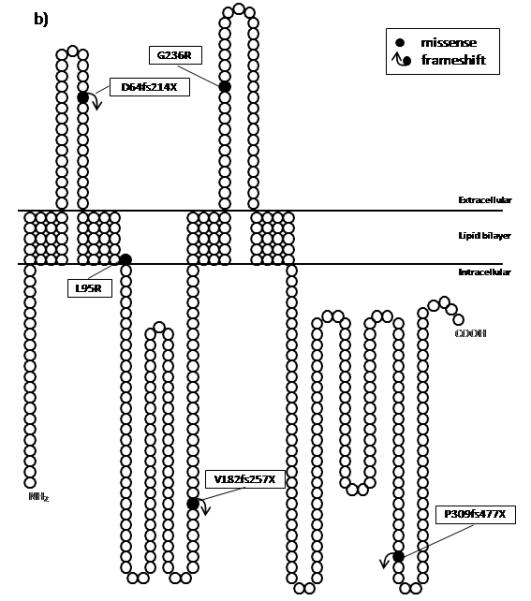
Schematic representations of both a PLP1 and b GJA12/GJC2 proteins with their known domains and the locations of the mutations identified in this study. Black circles represent the amino acid changes along the proteins. The positions of the domains in PLP1 is based on the website (http://www.uniprot.org/uniprot/P60201) and for GJA12/GJC2 on the work of Yeager M and Nicholson BJ, 1996 (31).
Five different sequence alterations were identified in the GJA12/GJC2 gene in six other families (Supplementary 4). A deletion of 17 basepairs (c.546-542del) was identified in GJA12/GJC2 in patients F1.3, F2.3, and F3.3 from the same large consanguineous family. HhaI restriction analysis in the family revealed that the parents and the healthy siblings were heterozygous whereas all three affected individuals were homozygous for the mutation. Patient F7.3 and his father were heterozygous for the previously reported c.706G>C (p.G236R) mutation and the mother was homozygous for the normal allele (13). The residue affected was highly conserved among different species (Pan troglodytes, Mus musculus, Rattus norvegicus, Gallus gallus). A novel homozygous c.T284G (p.L95R) variation was identified in patient F12.3. The parents were from the same village and heterozygous for the mutation. The mutant allele was absent in the two unaffected siblings. Patients F17.3, F17.4, and F21.3 had a 14 bp tandem duplication of nucleotides between 177 and 190 (c.177-190dup/p.D64fs214X). The parents in both families were heterozygous for the mutation. The last novel GJA12/GJC2 variation identified in family F23 was a 20 bp tandem duplication (c.924-925ins20bp) that is expected to encode 37 more triplets in the 3′UTR of the wild type sequence. The segragation of the mutation could not be tested due to unavailability of DNA samples from parents (Table 2, Fig. 3b).
Table 2.
Sequence variations identified in this study.
| Patient number | Gene | Mutation | Putative effect on protein |
Affected domain |
|---|---|---|---|---|
| F1.3*, F2.3*, F3.3* | GJA12/GJC2 | c.546-562del | p.V182fs257X | IC-1 |
| F4.3 | PLP1 | PLP1 gene duplication (438 kb) | Overexpression | - |
| F6.3 | PLP1 | 325 kb deletion (~4.7 Mb centromeric to PLP1) |
Unknown | - |
| F7.3** | GJA12/GJC2 | c.706G→C | p.G236R | EC-2 |
| F9.3 | PLP1 | PLP1 gene duplication (700 kb) | Overexpression | - |
| F12.3 | GJA12/GJC2 | c.284T→G | p.L95R | TM-2 |
| F15.3 | PLP1 | c.643C→T | p.P215S | EC-2 |
| F16.3 | PLP1 | c.695T→C | p.F232S | EC-2 |
| F17.3, F17.4, F21.3 | GJA12/GJC2 | c.177-190dup | p.D64fs214X | EC-1 |
| F18.3 | PLP1 | 45 kb duplication (~212 kb telomeric to PLP1) |
Unknown | - |
| F19.3 | PLP1 | PLP1 gene duplication (640 kb) | Overexpression | - |
| F20.3 | PLP1 | PLP1 gene duplication (795 kb) | Overexpression | - |
| F22.3 | PLP1 | 46 kb duplication (~212 kb telomeric to PLP1) |
Unknown | - |
| F23.3 | GJA12/GJC2 | c.905-924dup | p.P309fs477X | Cytoplasmic (carboxyl end) |
Patients are from different branches of the same consanguineous family.
Patient was heterozygous for the c.706G→C mutation.
Discussion
We investigated 19 PMD families both clinically and genetically. PLP1 gene duplications and missense mutations were observed in six families (32%); four families had PLP1 gene duplications (21%) though with different clinical severity and two patients with PLP1 missense mutations (11%) presented with the most severe phenotype among all patients analyzed. GJA12/GJC2 gene mutations were identified in six other families (32%) three of which had consanguinity. aCGH analysis revealed chromosomal aberrations that did not include the PLP1 gene in three additional families with an unknown effect on the phenotype.
The low frequency of PLP1 duplications in our cohort compared to previous reports can be attributed to the high frequency of autosomal recessive cases, different ethnic backgrounds, and limitations in the detection systems or difficulty of distinguishing different myelin disorders. The severity of the disease did not show any correlation with the duplication size as found for other populations (5, 6). Distal breakpoints were either mapping within or spanning the low-copy repeat (LCR) positions providing further evidence that PLP1 duplications may be stimulated by LCR-PMDs (23).
The small noncoding duplications downstream of PLP1 might represent polymorphisms since copy number variations (CNVs) encompassing that region were reported, though the database resolution does not allow a correlation with the control samples. Another duplication downstream of PLP1, encompassing the region duplicated in our two patients, has been reported previously as the only genetic abnormality identified in one patient with spastic paraplegia type 2 (SPG2; #312920) which is an allelic disorder of PMD (24). Non-recurrent duplications upstream and exclusive of the peripheral myelin protein 22 gene (PMP22) have also been reported to be associated with Charcot-Marie-Tooth type 1A (CMT1A; #118220) peripheral neuropathy suggesting that CNV of regulatory regions may lead to pathogenicity via a position effect (25).
Clinical features of GJA12/GJC2-related PMD cases were variable, but milder in general than those observed with PLP1 mutations. We could not perform brainstem auditory evoked potential (BAEP) studies although it was reported to allow separation of PMD from PMLD (26). VEP and MRI/MRS results were very similar in all patients. However, MRI contributes to recognition and evaluation of hypomyelination and may aid in differentiating PMD from other leukodystrophies or other neurodegenerative disorders.
The truncation GJA12/GJC2 variations (p.V182fs257X/p.D64fs214X) identified in three families, hypothetically cause either proteins lacking the third and forth transmembrane domains or loss of downstream domains after the first transmembrane domain and may lead to formation of non-functional proteins by disrupting their membrane insertion or stability. Alternatively, nonsense mediated decay may result in mRNA instabilitry with no mutant protein being produced. Among those families with premature termination codon mutations, F17 and F21 showed the most severe clinical presentation almost comparable to those of patients with PLP1 missense mutations. The p.P309fs477X mutation, that may cause production of a longer protein than the wild type, was associated with the mildest phenotype in the cohort. This can be expected since the mutation affects only the intracellular carboxyl domain of the protein, possibly leading to a partially functional protein. The effect of truncation mutations were reported for another gap junction protein gene, GJB1/Cx32, that is known to be responsible for CMTX (#302800). Some of these mutations were shown to have decreased ability to form functional hemichannels and complete channels whereas for the ones that can form channels normally, channel activity is affected depending on length (27). Further studies are required to unravel the contribution of different domains of gap junction proteins to disease pathogenesis.
In one of our severely affected cases, only one mutant GJA12/GJC2 allele could be identified. An autosomal recessive mode of inheritance was the most likely pattern since the patient’s father was an unaffected carrier and the mutation was previously found in homozygous condition in another patient from Turkey (13). The second mutant allele may reside in either regulatory or intronic regions or in another connexin gene that forms heteromeric channels with Cx47.
Observation of equal frequencies for PLP1 and GJA12/GJC2 mutations in our cohort suggests that autosomal recessive GJA12/GJC2 gene mutations may be more frequently found in association with PMD than expected in populations where consanguineous marriages are common. Absence of mutations in four of our families and previously reported cases presenting prototypical features of PMD suggests the presence of further genetic heterogeneity for PMLD. These patients can be screened for mutations in HSP60, AIMP1/p43 and MCT8 that were reported to be associated with PMLD presentation (28-30).
Supplementary Material
Acknowledgements
The cosmid probes cU125A1 and cU144A10 used in FISH analyses were kindly provided by Karen Woodward. This project was funded by the Bogazici University Research Fund with project code 04B106D. This work was supported in part from the National Institute of Neurological Disorders and Stroke of the United States National Institutes of Health grant RO1 NS058529 to JRL.
Footnotes
Conflict of interest Dr. Lupski owns JRL, which is a paid consultant for Athena Diagnostics, Inc., has stock ownership in 23andMe and Ion Torrent Systems, Inc., and is a co-Inventor on multiple United States and European DNA diagnostic patents.
The Medical Genetics Laboratories (MGL) of the Dept of Molecular and Human Genetics at Baylor College of Medicine derives revenue from molecular diagnostic testing including genomewide assays by Chromosomal MicroArray Analysis (CMA). MGL, http://www.bcm.edu/geneticlabs/
The other authors declare no conflict of interest.
References
- 1.Boulloche J, Aicardi J. Pelizaeus-Merzbacher disease: clinical and nosological study. J Child Neurol. 1986;1:233–239. doi: 10.1177/088307388600100310. [DOI] [PubMed] [Google Scholar]
- 2.Campagnoni AT, Macklin WB. Cellular and molecular aspects of myelin protein gene expression. Mol Neurobiol. 1988;2:41–89. doi: 10.1007/BF02935632. [DOI] [PubMed] [Google Scholar]
- 3.Garbern J, Hobson G. Prenatal diagnosis of Pelizaeus-Merzbacher disease. Prenat Diagn. 2002;22:1033–1035. doi: 10.1002/pd.465. [DOI] [PubMed] [Google Scholar]
- 4.Inoue K, Osaka H, Thurston VC, et al. Genomic rearrangements resulting in PLP1 deletion occur by nonhomologous end joining and cause different dysmyelinating phenotypes in males and females. Am J Hum Genet. 2002;71:838–853. doi: 10.1086/342728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woodward K, Kendall E, Vetrie D, Malcolm S. Pelizaeus-Merzbacher disease: identification of Xq22 proteolipid-protein duplications and characterization of breakpoints by interphase FISH. Am J Hum Genet. 1998;63:207–217. doi: 10.1086/301933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Inoue K, Osaka H, Imaizumi K, et al. Proteolipid protein gene duplications causing Pelizaeus-Merzbacher disease: molecular mechanism and phenotypic manifestations. Ann Neurol. 1999;45:624–632. [PubMed] [Google Scholar]
- 7.Cailloux F, Gauthier-Barichard F, Mimault C, et al. Genotype-phenotype correlation in inherited brain myelination defects due to proteolipid protein gene mutations. Clinical European Network on Brain Dysmyelinating Disease. Eur J Hum Genet. 2000;8:837–845. doi: 10.1038/sj.ejhg.5200537. [DOI] [PubMed] [Google Scholar]
- 8.Garbern JY, Cambi F, Tang XM, et al. Proteolipid protein is necessary in peripheral as well as central myelin. Neuron. 1997;19:205–218. doi: 10.1016/s0896-6273(00)80360-8. [DOI] [PubMed] [Google Scholar]
- 9.Uhlenberg B, Schuelke M, Rüschendorf F, et al. Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. Am J Hum Genet. 2004;75:251–260. doi: 10.1086/422763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bugiani M, Al Shahwan S, Lamantea E, et al. GJA12 mutations in children with recessive hypomyelinating leukoencephalopathy. Neurology. 2006;67:273–279. doi: 10.1212/01.wnl.0000223832.66286.e4. [DOI] [PubMed] [Google Scholar]
- 11.Salviati L, Trevisson E, Baldoin MC, et al. A novel deletion in the GJA12 gene causes Pelizaeus-Merzbacher-like disease. Neurogenetics. 2007;8:57–60. doi: 10.1007/s10048-006-0065-x. [DOI] [PubMed] [Google Scholar]
- 12.Wolf NI, Cundall M, Rutland P, et al. Frameshift mutation in GJA12 leading to nystagmus, spastic ataxia and CNS dys-/demyelination. Neurogenetics. 2007;8:39–44. doi: 10.1007/s10048-006-0062-0. [DOI] [PubMed] [Google Scholar]
- 13.Henneke M, Combes P, Diekmann S, et al. GJA12 mutations are a rare cause of Pelizaeus-Merzbacher-like disease. Neurology. 2008;70:748–754. doi: 10.1212/01.wnl.0000284828.84464.35. [DOI] [PubMed] [Google Scholar]
- 14.Wang J, Wang H, Wang Y, Chen T, Wu X, Jiang Y. Two novel gap junction protein alpha 12 gene mutations in two Chinese patients with Pelizaeus-Merzbacher-like disease. Brain Dev. 2010;32:236–243. doi: 10.1016/j.braindev.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 15.Osaka H, Hamanoue H, Yamamoto R, et al. Disrupted SOX10 regulation of GJC2 transcription causes Pelizaeus-Merzbacher-like disease. Ann Neurol. 2010;68:250–254. doi: 10.1002/ana.22022. [DOI] [PubMed] [Google Scholar]
- 16.Woodward KJ. The molecular and cellular defects underlying Pelizaeus-Merzbacher disease. Expert Rev Mol Med. 2008;10:e14. doi: 10.1017/S1462399408000677. [DOI] [PubMed] [Google Scholar]
- 17.Klockgether T, Schroth G, Diener HC, Ichgans J. Idiopathic cerebellar ataxia of late onset: natural history and MRI morphology. J Neurol Neurosurg Psychiatr. 1990;53:297–305. doi: 10.1136/jnnp.53.4.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woodward K, Cundall M, Palmer R, Surtees R, Winter RM, Malcolm S. Complex chromosomal rearrangement and associated counseling issues in a family with Pelizaeus-Merzbacher disease. Am J Med Genet. 2003;A118:15–24. doi: 10.1002/ajmg.a.10103. [DOI] [PubMed] [Google Scholar]
- 19.Carvalho C, Bartnik M, Pehlivan D, Fang P, Shen J, Lupski J. Evidence for disease penetrance relating to CNV size: Pelizaeus-Merzbacher disease and manifesting carriers with a familial 11 Mb duplication at Xq22. Clin Genet. 2011 doi: 10.1111/j.1399-0004.2011.01716.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Osaka H, Kawanishi C, Inoue K, et al. Novel nonsense proteolipid protein gene mutation as a cause of X-linked spastic paraplegia in twin males. Biochem Biophys Res Commun. 1995;215:835–841. doi: 10.1006/bbrc.1995.2539. [DOI] [PubMed] [Google Scholar]
- 21.Ruf N, Uhlenberg B. Analysis of human alternative first exons and copy number variation of the GJA12 gene in patients with Pelizaeus-Merzbacher-like disease. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:226–232. doi: 10.1002/ajmg.b.30792. [DOI] [PubMed] [Google Scholar]
- 22.Gencic S, Abuelo D, Ambler M, Hudson LD. Pelizaeus-Merzbacher disease: an X-linked neurologic disorder of myelin metabolism with a novel mutation in the gene encoding proteolipid protein. Am J Hum Genet. 1989;45:435–442. [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JA, Inoue K, Cheung SW, Shaw CA, Stankiewicz P, Lupski JR. Role of genomic architecture in PLP1 duplication causing Pelizaeus-Merzbacher disease. Hum Mol Genet. 2006;15:2250–2265. doi: 10.1093/hmg/ddl150. [DOI] [PubMed] [Google Scholar]
- 24.Lee JA, Madrid RE, Sperle K, et al. Spastic paraplegia type 2 associated with axonal neuropathy and apparent PLP1 position effect. Ann Neurol. 2006;59:398–403. doi: 10.1002/ana.20732. [DOI] [PubMed] [Google Scholar]
- 25.Weterman MA, van Ruissen F, deWissel M, et al. Copy number variation upstream of PMP22 in Charcot-Marie-Tooth disease. Eur J Hum Genet. 2010;18:421–428. doi: 10.1038/ejhg.2009.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henneke M, Gegner S, Hahn A, et al. Clinical neurophysiology in GJA12-related hypomyelination vs Pelizaeus-Merzbacher disease. Neurology. 2010;74:1785–1789. doi: 10.1212/WNL.0b013e3181e0f820. [DOI] [PubMed] [Google Scholar]
- 27.Castro C, Gomez-Hernandez JM, Silander K, Barrio LC. Altered formation of hemichannels and gap junction channels caused by C-terminal connexin-32 mutations. J Neurosci. 1999;19:3752–3760. doi: 10.1523/JNEUROSCI.19-10-03752.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Magen D, Georgopoulos C, Bross P, et al. Mitochondrial hsp60 chaperonopathy cause an autosomal-recessive neurodegenereative disorder linked to brain hypomyelination and leukodystrophy. Am J Hum Genet. 2008;83:30–48. doi: 10.1016/j.ajhg.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feinstein M, Markus B, Noyman I, et al. Pelizaeus-Merzbacher-like disease caused by AIMP/p43 homozygous mutation. Am J Hum Genet. 2010;87:820–828. doi: 10.1016/j.ajhg.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vaurs-Barriére C, Deville M, Sarret C, et al. Pelizaeus-Merzbacher-Like disease presentation of MCT8 mutated male subjects. Ann Neurol. 2009;65:114–118. doi: 10.1002/ana.21579. [DOI] [PubMed] [Google Scholar]
- 31.Yeager M, Nicholson BJ. Structure of gap junction intercellular channels. Curr Opin Struct Biol. 1996;6:183–192. doi: 10.1016/s0959-440x(96)80073-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.