

Abstract
To ensure immune tolerance, regulatory T cell (Treg) numbers must be maintained by cell division. This process has been thought to be strictly dependent on the Treg TCR interacting with MHC class II (MHCII). Here, we report that Treg division does not absolutely require cell-autonomous TCR signaling in vivo, depending on the degree of IL-2-mediated stimulation provided. At steady state IL-2 levels, Tregs require cell-autonomous TCR signaling to divide. However, when given exogenous IL-2 or when STAT5 is selectively activated in Tregs, Treg division can occur independently of MHCII and TCR signaling. Thus, depending on the amount of IL-2 receptor stimulation, a wide range of TCR signals support Treg division, which may contribute to preservation of a diverse repertoire of Treg TCR specificities. These findings also have therapeutic implications, as TCR signaling by Tregs may not be required when using IL-2 to increase Treg numbers for treatment of inflammatory disorders.
Keywords: Regulatory T Cells, Cell Proliferation, T cell receptors, IL-2
Introduction
Suppression of immune responses against both self and foreign antigens by CD4+Foxp3+ regulatory T cells (Treg)s is an essential mechanism of self-tolerance (1). To maintain immune tolerance, a sufficient number of Tregs must be maintained in the periphery of the host, in part by their continuous cell division in the steady state (2). IL-2 is critical for the proliferation and maintenance of Tregs, as the acute neutralization of IL-2 in adult mice disrupts Treg homeostasis (3, 4), whereas IL-2 receptor (IL-2R) agonists augment Treg division and proliferation (5). Tregs do not produce IL-2 themselves but constitutively express CD25, the high affinity α subunit of the IL-2R, which allows them to respond to low levels of IL-2 produced by conventional CD4+ T cells (Tconvs) (3, 4).
In addition to IL-2, current dogma asserts that Treg division is absolutely dependent on TCR signaling by Tregs, since adoptively transferred Tregs fail to divide in MHC class II (MHCII) KO hosts and the deletion of TCR signaling proteins in T cells results in decreased Treg division and survival (6–9). However, in each of the experimental approaches utilized to date, the loss of TCR signaling is not confined to Tregs but also occurs in Tconvs, which are the major source of IL-2 that supports Treg division. Thus, the failure of Tregs to divide in previous studies may result from the lack of IL-2 production by Tconvs rather than a cell-autonomous requirement for TCR stimulation in Tregs. We propose this notion based on in vitro studies demonstrating that Tregs do not need MHCII to divide if exogenous IL-2 and contact with MHCII KO DCs are provided (10, 11).
Here, we examined the necessity of cell-autonomous TCR signaling in Tregs for their division in vivo. Through the complementary approaches of restricting the peptide repertoire presented on MHCII and disrupting TCR signal transduction specifically in Tregs, we find that Treg division is partially dependent on a diverse peptide repertoire and is completely dependent on TCR signaling at steady state IL-2 levels. However, exogenous IL-2R stimulation induced by IL-2 ICs or selective activation of the IL-2-induced STAT5 pathway in Tregs partially restored Treg division in the absence of MHCII or TCR signaling, indicating that TCR signals are not absolutely necessary in every setting. These data demonstrate for the first time that while steady state Treg division requires TCR signaling, targeted IL-2R stimulation can partially overcome this requirement to promote Treg division in vivo. We propose that the combination of TCR and IL-2R/STAT5 signaling together determine the extent of cell division individual Tregs undergo and ultimately control the homeostasis of this population.
Materials and Methods
Mice
SLP-76flox/− conditional knockout (cKO), SLP-76+/− heterozygous (Het), and SLP-76flox/+ conditional heterozygous (cHet) mice were generated as described (12). STAT5b-CA transgenic mice were generated as described (13) and were bred with SLP-76 cKO and SLP-76 cHet mice. H-2DMα KO mice were a generous gift from Dr. Terri Laufer. All other mice were purchased from The Jackson Laboratory or from Taconic Farms. Mice were housed in pathogen-free conditions and treated in strict compliance with Institutional Animal Care and Use Committee regulations of the University of Pennsylvania.
Flow cytometry, cell sorting, and data analysis
Antibodies for flow cytometry were purchased from BD Pharmingen (San Diego, CA), eBioscience (San Diego, CA), or Molecular Probes, Invitrogen (Carlsbad, CA). Flow cytometry and FACS was performed with an LSR II, FACSCanto, or a FACSAria cell sorter (BD Biosciences). Data were analyzed with FlowJo software (TreeStar) and Prism (GraphPad).
Mixed BM chimeras and Tamoxifen administration
T cell-depleted bone marrow (BM) from CD90.1+CD45.2+ wildtype (WT) donor mice were mixed at a 1:1 ratio with CD90.2+CD45.2+ SLP-76 Het, SLP-76 cHet, or SLP-76 cKO BM and a total of 3–4 × 106 BM cells were injected i.v. into lethally irradiated (9.5 Gy) CD45.1+ recipient mice. Mice were used after eight to ten weeks. For deletion of the floxed SLP-76 allele, mice were given 200 μg/g body weight of Tamoxifen in corn oil every day for 5 days and rested for 5 days before use.
In vivo BrdU incorporation and staining
Mice were administered BrdU with an initial bolus of BrdU (2 mg/200 μl) i.p. and given drinking water containing BrdU (1 mg/mL) until the time of sacrifice. To detect BrdU incorporation in SLP-76 cHet or cKO Tregs, MACS-purified T cells were FACS-sorted into YFP+ and YFP− fractions prior to surface staining and subsequent intracellular staining for Foxp3 and BrdU.
Adoptive transfers
MACS-sorted CFSE-labeled T cells from Thy1.1+ WT mice and CD45.1+ H-2DMα KO mice were mixed and injected i.v. into CD45.2+ H-2DMα KO mice. 4 weeks later, the donor-derived T cells from the spleen were analyzed by flow cytometry. FACS-sorted CFSE-labeled T cells from CD45.1+ WT mice were adoptively transferred into CD45.2+ WT or MHCII KO (complete MHCII KO or I-Abβ KO mice). 1 day later, PBS or IL-2 ICs were injected for three consecutive days and mice were sacrificed for analysis of splenic donor-derived T by flow cytometry on the sixth day after the first injection. IL-2 ICs were prepared by incubating 5 μg of JES6-1 anti-mouse IL-2 antibody (BioXCell, West Lebanon, NH) with 1 μg of recombinant mouse IL-2 (eBioscience) for 30 minutes on ice. FACS-sorted CFSE-labeled T cells from CD90.1+CD45.2+ WT and CD90.2+CD45.2+ STAT5b-CA were mixed at a 1:1 ratio and adoptively transferred into CD90.2+CD45.2+ WT or MHCII KO recipient mice. Two weeks later, splenic and LN donor-derived T cells were analyzed by flow cytometry. FACS-sorted CFSE-labeled T cells from SLP-76 cKO, SLP-76 cHet, SLP-76 cKO/STAT5b-CA, and SLP-76 cHet/STAT5b-CA BM chimeras were adoptively transferred into CD45.1+ WT mice and given BrdU. 7 days later, splenic and LN donor-derived T cells were analyzed for BrdU incorporation by flow cytometry.
Results and Discussion
TCR signaling is required for Treg division in the steady state in vivo
To test whether a diverse peptide repertoire presented on MHCII was needed for Treg division, we adoptively transferred CFSE-labeled WT and H-2DMα KO CD4+ T cells into H-2DMα KO recipients. H-2DMα KO mice are defective in their ability to exchange peptides from maturing MHCII molecules (14), virtually making all surface MHCII molecules loaded with a single peptide, CLIP. We reasoned that WT Tregs that have been selected on a diverse array of peptides would fail to encounter their cognate antigen when adoptively transferred into H-2DMα KO hosts. In contrast, Tregs that have developed in H-2DMα KO mice would have TCR specificities that react most suitably with CLIP. One month after adoptive transfer, we found that the division (Fig. 1A, B) and absolute number (Supplemental Fig. 1A) of WT Tregs were significantly diminished compared to H-2DMα KO Tregs. Similarly, WT Tconvs divided significantly less compared to H-2DMα KO Tconvs (Supplemental Fig. 1B). These data suggest that cell-autonomous TCR interactions with their selecting antigen/MHCII complexes are important for optimal Treg division. However, some Tregs can still divide without these TCR signals.
Figure 1. TCR signaling is required for optimal Treg division in the steady state.
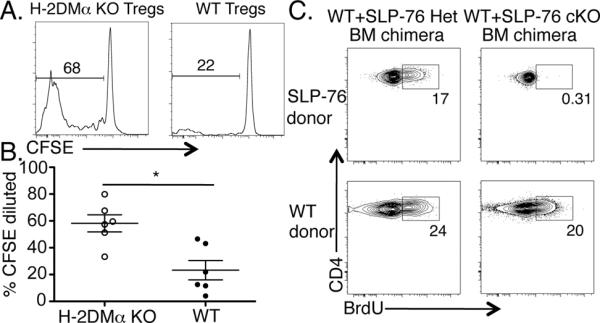
CFSE and congenically labeled CD4+ T cells from WT and H-2DMα KO mice were mixed and injected i.v. into H-2DMα KO mice. One month later, CFSE dilution of splenic donor Tregs was analyzed by flow cytometry. (A) One representative CFSE dilution plot of donor WT (right plot) or H-2DMα KO (left plot) CD4+Foxp3+ Tregs is shown. (B) Data from two independent experiments are represented as mean ± SEM of n = 6 mice per group. (C) WT/SLP-76 cHet and WT/SLP-76 cKO mixed BM chimeras were administered Tamoxifen and treated with BrdU for 13–14 days. BrdU incorporation by splenic Tregs was analyzed by flow cytometry. One representative contour plot (n = 7–8 mice/group; 2 independent experiments) gated on YFP+ Tregs from SLP-76 cHet (top left plot) or cKO (top right plot) and WT donor Tregs (bottom plots) is shown.
To investigate further the requirement of TCR signaling in Treg division, we devised a strategy to acutely and inducibly abrogate TCR signaling specifically in Tregs. This approach was needed to dissociate the TCR signaling capacity of Tregs from that of Tconvs, since Tconvs produce IL-2 in a TCR/MHCII-dependent manner to support Treg division. We utilized mice in which the TCR signaling molecule Src homology 2 domain-containing leukocyte protein of 76 kD (SLP-76) could be inducibly deleted by a Tamoxifen-inducible cre recombinase (15). A YFP reporter was used to mark cells with a history of cre-mediated recombination and thus deletion of the floxed SLP-76 allele. To preserve sufficient numbers of Tconvs to provide IL-2 but still delete SLP-76 from a fraction of Tregs, we employed a mixed BM chimera approach in which WT donor BM was mixed with BM from a SLP-76flox/− (cKO), SLP-76+/− (Het), or SLP-76flox/+ (cHet) donor and transplanted into irradiated WT recipients. After 8–10 weeks, Tamoxifen was administered to all BM chimeras to delete SLP-76 from the SLP-76flox BM-derived T cells and administered BrdU to assess the extent of Treg division. In each of the BM chimeras, 10–25% of the WT donor Tregs incorporated BrdU. While a fraction of the YFP+ SLP-76 Het/cHet Tregs also incorporated BrdU, the YFP+ SLP-76 cKO Tregs were nearly completely defective in BrdU incorporation (Fig. 1C). Thus, these data suggest that cell-autonomous TCR signaling is required to sustain Treg division at steady state levels of IL-2.
Exogenous IL-2 can partially restore Treg division in the absence of MHCII or SLP-76-mediated TCR signaling
To test the role of MHCII in Treg division, CFSE-labeled T cells were adoptively transferred into either WT or MHCII KO recipients. Some of the recipient mice were also given IL-2 ICs (5), since CD4+ T cells cannot produce the IL-2 necessary for Treg division in MHCII KO mice. As expected, adoptively transferred Tregs divided in WT but not in MHCII KO recipients (Fig. 2A, B). However, with IL-2 IC administration, donor Tregs divided in MHCII KO hosts, indicating that IL-2 IC-induced Treg division can occur independently of MHCII-mediated TCR signaling (Fig. 2 A, B). Moreover, the frequency and absolute number of Tregs were increased in IL-2 IC-treated MHCII KO recipients, although this increase was reduced compared to IL-2 IC-treated WT mice (Supplemental Fig. 2A, B). Treg proliferation was comparable in IL-2 IC-treated MHCII KO recipients at 1 and 3 weeks after adoptive transfer (Supplemental Fig. 2C), suggesting that IL-2 signaling in the absence of MHCII can sustain Treg division for extended periods of time. Furthermore, the proliferating Tregs maintained a diverse TCR Vβ repertoire in IL-2 IC-treated MHCII KO recipients (Supplemental Fig. 2D), suggesting that the proliferation of the Tregs was unlikely to represent an oligoclonal outgrowth of Tregs with selective TCRs.
Figure 2. Exogenous IL-2 signals induce Treg proliferation in the absence of MHCII in vivo.
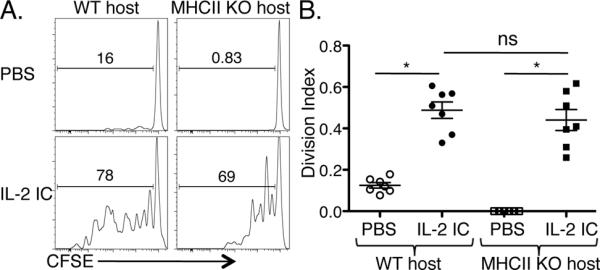
CFSE and congenically labeled WT T cells were adoptively transferred into WT or MHCII KO mice. (A) Histograms show representative CFSE profiles of WT donor Tregs in the indicated recipients treated with PBS (top plots) or IL-2 ICs (bottom plots). (B) The division index of Tregs from WT and MHCII KO recipient mice are plotted as mean ± SEM of n = 7–8 mice/group from two independent experiments. * p<0.001 and ns = not significant, by one-way ANOVA with Tukey's post test.
We next examined whether IL-2 ICs would allow Tregs to proliferate in the absence of SLP-76-mediated TCR signaling. Tamoxifen-treated WT/SLP-76 cHet and WT/SLP-76 cKO mixed BM chimeras were injected with PBS or IL-2 ICs at the start of a BrdU pulse. While PBS-treated YFP+ SLP-76 cKO Tregs did not incorporate BrdU, IL-2 IC-treated YFP+ SLP-76 cKO Tregs incorporated BrdU at a rate similar to PBS-treated YFP+ SLP-76 cHet Tregs (Fig. 3A, B). However, BrdU incorporation by IL-2 IC-treated YFP+ SLP-76 cKO Tregs was significantly less than that observed in both WT donor Tregs within the same mice and YFP+ SLP-76 cHet Tregs from IL-2 IC-treated mice (Fig. 3A, B). Together, these data suggest that IL-2 IC treatment partially restores the ability of Tregs to divide in the absence of MHCII- or SLP-76-mediated TCR signaling.
Figure 3. IL-2 ICs partially restore Treg division in the absence of SLP-76-mediated TCR signaling.

WT/SLP-76 cHet and WT/SLP-76 cKO mixed BM chimeras were administered Tamoxifen and treated with PBS or IL-2 IC and BrdU. (A) A representative contour plot shows BrdU incorporation by congenically disparate SLP-76 cHet or cKO Tregs (top row) and WT donor Tregs (bottom row). (B) BrdU incorporation by either SLP-76 cHet or cKO Tregs was normalized to that of WT donor Tregs within the same mouse and data from two independent experiments are represented as mean ± SEM of n = 8 mice/group (right graph). * p<0.001 by one-way ANOVA with Tukey's post test.
Isolated STAT5 activation is sufficient to partially restore Treg division in the absence of MHCII or TCR signaling in vivo
The IL-2R transmits its signals intracellularly through a JAK3-STAT5 pathway and two Shc dependent pathways that activate either PI3K/Akt or Ras/MAPK signaling (4). To provide a mechanism for the ability of IL-2 ICs to promote TCR-independent Treg division, we tested the involvement of the STAT5 pathway, since transgenic expression of a constitutively active form of STAT5b (STAT5b-CA) is sufficient to induce thymic Treg differentiation and maintain Treg homeostasis in the absence of IL-2Rβ (13). We adoptively transferred CFSE-labeled WT and STAT5b-CA T cells into either WT or MHCII KO hosts to examine whether transgenic expression of STAT5b-CA was sufficient for Tregs to proliferate in the absence of MHCII. This approach also allowed us to test whether IL-2 signals were acting directly or indirectly on Tregs for their proliferation, as IL-2 IC treatment could be affecting Tregs indirectly through other IL-2-responsive cell types. Two weeks later, we found that although WT Tregs did not divide at all in MHCII KO hosts, proliferation of Tregs was partially restored in MHCII KO hosts when the adoptively transferred Tregs expressed STAT5b-CA (Fig. 4A, B).
Figure 4. Selective activation of STAT5 is sufficient to sustain Treg division in the absence of MHCII and TCR signaling.
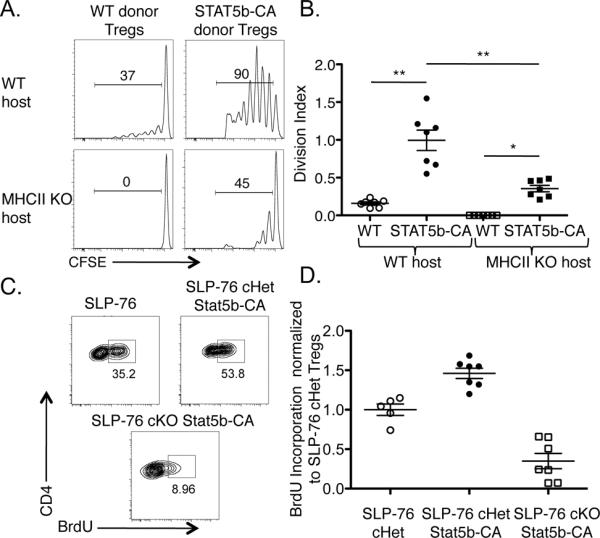
(A) CFSE and congenically labeled WT and STAT5b-CA total T cells were adoptively transferred into WT or MHCII KO recipient mice. Two weeks later, CFSE dilution of CFSE+ donor Tregs in spleen and LNs was analyzed by flow cytometry and shown as representative histograms or (B) as the division index of Tregs represented as mean ± SEM of n = 7 mice per group from two independent experiments. (C) T cells from Tamoxifen-treated WT/SLP-76 cHet, WT/SLP-76 cKO, WT/SLP-76 cHet STAT5b-CA, and WT/SLP-76 cKO STAT5b-CA mixed BM chimeras were FACS-sorted and injected i.v. into WT mice. The mice were treated with BrdU for 7 days and the splenic T cells were analyzed by flow cytometry. BrdU incorporation by SLP-76 cHet, SLP-76 cHet STAT5b-CA, or SLP-76 cKO STAT5b-CA Tregs is shown in representative contour plots or (D) normalized to the average BrdU incorporation by SLP-76 cHet Tregs and is represented as mean ± SEM of n = 5–7 mice per group. * p<0.01 and ** p<0.001 by unpaired, two-tailed Student's t test or one-way ANOVA with Tukey's post test.
We next examined whether STAT5b-CA expression would allow Tregs to divide in the absence of SLP-76-mediated TCR signaling. To this end, we intercrossed STAT5b-CA transgenic mice with SLP-76 cHet and SLP-76 cKO mice, treated them with Tamoxifen, adoptively transferred their T cells into WT hosts, and administered BrdU. Although SLP-76 cHet Tregs incorporated BrdU after a 7-day BrdU labeling period, almost no YFP+ SLP-76 cKO Tregs could be detected in recipient mice, precluding BrdU analysis on these cells (Fig. 4C). However, when the SLP-76 cKO Tregs expressed STAT5b-CA, YFP+ Tregs that incorporated BrdU were detected (Fig. 4C). Similar to the Tregs transferred into MHCII KO mice, the rescue of Treg survival and proliferation by expression of STAT5b-CA was partial, given that SLP-76 cHet Tregs incorporated significantly more BrdU than SLP-76 cKO Tregs expressing STAT5b-CA (Fig. 4D). Together, these data suggest that IL-2 promotes Treg proliferation by acting in a cell-autonomous manner and that IL-2-induced STAT5 signaling is sufficient to partially restore Treg division in the absence of TCR signaling. Thus, although TCR signals are important for Treg proliferation, they are not absolutely required for the division of Tregs.
Collectively, the experiments presented here demonstrate for the first time that depending on the nature of the IL-2R stimulation that is provided, TCR/MHCII contacts are partially dispensable for Treg division. We propose that progressively stronger IL-2R stimulation is required to offset the loss of cell-autonomous TCR signals in Tregs in order to sustain the division and homeostasis of this population. In limiting amounts of IL-2, Tregs that encounter their cognate antigens likely divide preferentially over Tregs bearing TCR specificities for rare antigens. However, in settings where IL-2 concentrations are high, such as during inflammation, even Tregs experiencing weak or non-specific TCR interactions may be able to divide. This model offers a mechanism for maintenance of Tregs bearing TCRs specific for rare or sequestered self-antigens. The preservation of these Treg populations may allow for the retention of a diverse Treg TCR repertoire that is important for self-tolerance (16).
The TCR-independent Treg division induced by exogenous IL-2R stimulation has important implications for treatment of autoimmunity, systemic inflammatory diseases, and the maintenance of transplant tolerance. In these clinical conditions, an imbalance exists between the activity of effector Tconvs and that of Tregs (1). Although IL-2R stimulation may enhance the function of effector Tconvs, mouse models of autoimmunity and human studies suggest that IL-2 therapy may preferentially expand Treg populations and alter this imbalance in favor of immune tolerance (17, 18). IL-2 treatment increases Tregs and provides benefit in mouse models of graft-versus-host disease (GVHD) and in humans with chronic GVHD (19, 20). Based on our data, we speculate that IL-2 may expand Tregs even when inhibitors of TCR signaling are concurrently provided. Although further studies are needed, our preliminary data suggest that IL-2 in combination with the TCR signaling inhibitor Cyclosporine A inhibits antigen-specific Tconv proliferation while expanding Tregs (A. Satake, unpublished observations). As Tregs clearly have remarkable therapeutic potential to modulate human disease, further understanding of the mechanisms that govern their division and homeostasis will be crucial for properly harnessing the immunosuppressive functions of Tregs.
Supplementary Material
Acknowledgements
We thank G. Koretzky, M. Jordan, R. Joshi, and S. Lieberman for critical reading of the manuscript and members of the Kambayashi, Maltzman, and Koretzky labs for discussions.
This work was supported by the Perelman School of Medicine at the University of Pennsylvania's new investigator start-up funds (T.K.), the National Blood Foundation (T.K.), American Society of Hematology (T.K.), NIH grants R01AI085160 (J.S.M.), R01HL107589 (T.K.), R01HL111501 (T.K.), K08HL086503 (T.K.), the American Societ y o f Nephrology/American Society of Transplantation (J.S.M.), and the Cancer Research Institute Predoctoral Emphasis Pathway in Tumor Immunology training grant (T.Z.).
Abbreviations used in this paper
- BM
bone marrow
- cHet
conditional heterozygous
- cKO
conditional KO
- Het
heterozygous
- IC
immune complex
- LN
lymph node
- MHCII
MHC class II
- SLP-76
Src homology 2 domain-containing leukocyte protein of 76 kD
- STAT5b-CA
constitutively active STAT5b
- Tconv
conventional CD4+ T cell
- Treg
regulatory T cell
- WT
wildtype
- YFP
yellow fluorescent protein
References
- 1.Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat. Immunol. 2010;11:7–13. doi: 10.1038/ni.1818. [DOI] [PubMed] [Google Scholar]
- 2.Fisson S, Darrasse-Jèze G, Litvinova E, Septier F, Klatzmann D, Liblau R, Salomon BL. Continuous activation of autoreactive CD4+ CD25+ regulatory T cells in the steady state. J. Exp. Med. 2003;198:737–746. doi: 10.1084/jem.20030686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J. Exp. Med. 2005;201:723–735. doi: 10.1084/jem.20041982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng G, Yu A, Malek TR. T-cell tolerance and the multi-functional role of IL-2R signaling in T-regulatory cells. Immunol. Rev. 2011;241:63–76. doi: 10.1111/j.1600-065X.2011.01004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Webster KE, Walters S, Kohler RE, Mrkvan T, Boyman O, Surh CD, Grey ST, Sprent J. In vivo expansion of T reg cells with IL-2-mAb complexes: induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J. Exp. Med. 2009;206:751–760. doi: 10.1084/jem.20082824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhandoola A, Tai X, Eckhaus M, Auchincloss H, Mason K, Rubin SA, Carbone KM, Grossman Z, Rosenberg AS, Singer A. Peripheral expression of self-MHC-II influences the reactivity and self-tolerance of mature CD4(+) T cells: evidence from a lymphopenic T cell model. Immunity. 2002;17:425–436. doi: 10.1016/s1074-7613(02)00417-x. [DOI] [PubMed] [Google Scholar]
- 7.Gavin MA, Clarke SR, Negrou E, Gallegos A, Rudensky A. Homeostasis and anergy of CD4(+)CD25(+) suppressor T cells in vivo. Nat. Immunol. 2002;3:33–41. doi: 10.1038/ni743. [DOI] [PubMed] [Google Scholar]
- 8.Kim JK, Klinger M, Benjamin J, Xiao Y, Erle DJ, Littman DR, Killeen N. Impact of the TCR signal on regulatory T cell homeostasis, function, and trafficking. PLoS ONE. 2009;4:e6580. doi: 10.1371/journal.pone.0006580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shen S, Chuck MI, Zhu M, Fuller DM, Yang C-WO, Zhang W. The Importance of LAT in the Activation, Homeostasis, and Regulatory Function of T Cells. J. Biol. Chem. 2010;285:35393–35405. doi: 10.1074/jbc.M110.145052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swee L, Bosco N, Malissen B, Ceredig R, Rolink A. Expansion of peripheral nTreg by FLT3L treatment. Blood. 2009;113:6277–87. doi: 10.1182/blood-2008-06-161026. [DOI] [PubMed] [Google Scholar]
- 11.Zou T, Caton AJ, Koretzky GA, Kambayashi T. Dendritic cells induce regulatory T cell proliferation through antigen-dependent and -independent interactions. J. Immunol. 2010;185:2790–2799. doi: 10.4049/jimmunol.0903740. [DOI] [PubMed] [Google Scholar]
- 12.Wu GF, Corbo E, Schmidt M, Smith-Garvin JE, Riese MJ, Jordan MS, Laufer TM, Brown EJ, Maltzman JS. Conditional deletion of SLP-76 in mature T cells abrogates peripheral immune responses. Eur. J. Immunol. 2011;41:2064–2073. doi: 10.1002/eji.201040809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J. Immunol. 2007;178:280–290. doi: 10.4049/jimmunol.178.1.280. [DOI] [PubMed] [Google Scholar]
- 14.Martin WD, Hicks GG, Mendiratta SK, Leva HI, Ruley HE, Van Kaer L. H2-M mutant mice are defective in the peptide loading of class II molecules, antigen presentation, and T cell repertoire selection. Cell. 1996;84:543–550. doi: 10.1016/s0092-8674(00)81030-2. [DOI] [PubMed] [Google Scholar]
- 15.Maltzman JS, Kovoor L, Clements JL, Koretzky GA. Conditional deletion reveals a cell-autonomous requirement of SLP-76 for thymocyte selection. J. Exp. Med. 2005;202:893–900. doi: 10.1084/jem.20051128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Föhse L, Suffner J, Suhre K, Wahl B, Lindner C, Lee C-W, Schmitz S, Haas JD, Lamprecht S, Koenecke C, Bleich A, Hämmerling GJ, Malissen B, Suerbaum S, Förster R, Prinz I. High TCR diversity ensures optimal function and homeostasis of Foxp3+ regulatory Tcells. Eur. J. Immunol. 2011;41:3101–3113. doi: 10.1002/eji.201141986. [DOI] [PubMed] [Google Scholar]
- 17.Zhang H, Chua KS, Guimond M, Kapoor V, Brown MV, Fleisher TA, Long LM, Bernstein D, Hill BJ, Douek DC, Berzofsky JA, Carter CS, Read EJ, Helman LJ, Mackall CL. Lymphopenia and interleukin-2 therapy alter homeostasis of CD4+CD25+ regulatory T cells. Nat. Med. 2005;11:1238–1243. doi: 10.1038/nm1312. [DOI] [PubMed] [Google Scholar]
- 18.Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E, Piccirillo CA, Salomon BL, Bluestone JA. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity. 2008;28:687–697. doi: 10.1016/j.immuni.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shin HJ, Baker J, Leveson-Gower DB, Smith AT, Sega EI, Negrin RS. Rapamycin and IL-2 reduce lethal acute graft-versus-host disease associated with increased expansion of donor type CD4+CD25+Foxp3+ regulatory T cells. Blood. 118:2342–2350. doi: 10.1182/blood-2010-10-313684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP, 3rd, Armand P, Cutler C, Ho VT, Treister NS, Bienfang DC, Prasad S, Tzachanis D, Joyce RM, Avigan DE, Antin JH, Ritz J, Soiffer RJ. Interleukin-2 and regulatory T cells in graft-versus-host disease. N. Engl. J. Med. 365:2055–2066. doi: 10.1056/NEJMoa1108188. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.