

Abstract
Background
Increasing evidence suggests that the effect of HLA-E on Natural Killer (NK) cell activity can be affected by the nature of the peptides bound to this non-classical, MHC class Ib molecule. However, its reduced cell surface expression, and until recently, the lack of specific monoclonal antibodies hinder studying the peptide-binding specificity HLA-E.
Results
An in vitro refolding system was used to assess binding of recombinant HLA-E to either specific peptides or a nonamer random peptide library. Peptides eluted from HLA-E molecules refolded around the nonamer library were then used to determine a binding motif for HLA-E. Hydrophobic and non-charged amino acids were found to predominate along the peptide motif, with a leucine anchor at P9, but surprisingly there was no methionine preference at P2, as suggested by previous studies.
Conclusions
Compared to the results obtained with rat classical class Ia MHC molecules, RT1-A1c and RT1-Au, HLA-E appears to refold around a random peptide library to reduced but detectable levels, suggesting that this molecule's specificity is tight but probably not as exquisite as has been previously suggested. This, and a previous report that it can associate with synthetic peptides carrying a viral sequence, suggests that HLA-E, similar to its mouse counterpart (Qa-1b), could possibly bind peptides different from MHC class I leader peptides and present them to T lymphocytes.
Background
Non-classical MHC class Ib molecules are closely homologous to classical class Ia molecules but are distinguished by their limited polymorphism and low cell surface expression. Contrary to some views expressed in the past, class Ib molecules are not just vestigial evolutionary remnants of classical class Ia molecules: rather some are endowed with important highly specialized roles, as testified by their conservation between different species. In this regard, the trio comprised of HLA-E in human, Qa-1 in mouse and RT.BM1 in rat constitutes the only group of class Ib molecules where clear homologues have been identified in all three species. A major role of this group of molecules has recently emerged in the regulation of Natural Killer (NK) cell activity, through interaction with both the inhibitory CD94-NKG2A receptor and the activatory CD94-NKG2C receptor [1,2,3]. For cell surface expression, these MHC molecules preferentially bind peptides derived from the signal peptides of other MHC class I molecules by a TAP-dependent mechanism [4,5,6]. Hence, expression of other class I heavy chain polypeptides regulate the expression of HLA-E and it is thought that this in turn enables NK cells to monitor the state of the MHC class I-dependent antigen presentation pathway in the cells they inspect. Thus, the level of cell surface HLA-E is critical for NK cell cytotoxicity towards certain tumour and viral-infected cells, and a recent report suggests that viruses that shut down MHC class I expression may evolve mechanisms to maintain HLA-E expression [7].
However, not all leader sequences from human class I MHC molecules contain peptides that are able to bind to HLA-E. For example, sequences derived from certain HLA-B alleles, that contain a threonine for methionine substitution at the P4 position of the leader peptide (P2 position of the processed peptide), were not able to bind to HLA-E in an in vitro binding assay [3]. Furthermore, transfectants of the HLA-B alleles carrying Thr at P4 into 721.221 cells could not inhibit killing by CD94/NKG2A NK clones in contrast to those from other HLA-A, -B, -C and -G alleles with a Met at P4. Analysis of the crystal structure of HLA-E seemed to confirm this stringent peptide requirement since it showed the occupation of all the pockets and the involvement of all the peptide side chains in burying the peptide deep in the groove [8]. However, recent results have shown that the sequence of the bound peptide can influence binding to both the CD94/NKG2A and CD94/NKG2C receptors in both cellular and in vitro binding assays [9,10,11]. In addition, multiple studies (reviewed in [12]) have shown that Qa-1 can carry out antigen presentation to γδ and/or CD8+ T cells. This suggests that HLA-E, similar to its mouse counterpart, could possibly bind antigenic peptides different from MHC class I leader peptides, and present them to T lymphocytes.
In the present study, we aimed to determine the peptide binding specificity of HLA-E via a purely biochemical approach based on an in vitro refolding system. Any such study within a biological system requires not only access to sufficient material, but also the availability of a specific antibody by which the class I molecule can be efficiently purified from all other cellular components. For the classical class Ia molecules, this is usually not a problem since cell surface expression is high and specific antibodies are often available. For HLA-E, low expression and the lack of a truly specific antibody has thus far hindered attempts to obtain a peptide binding motif although a recent report described the production and characterisation of a specific monoclonal antibody called V16 [13].
Results
Cloning and expression of HLA-E
Using PCR, we engineered an expression plasmid to produce a soluble form of the heavy chain of HLA-E (E*0102) with a C-terminal His-Tag sequence. Under IPTG induction, the heavy chain was expressed as inclusion bodies and therefore required urea solubilization before purification could be performed. Solubilized heavy chain was purified on a Ni-NTA agarose column to >95 % purity as determined by SDS-PAGE. The total yield of HLA-E was approximately 88 mg/l of bacterial culture. This eluted off the Ni-NTA column as two species, one in pH 5.9 buffer and the other at pH 4.5. SDS-PAGE in the presence/absence of reducing agent confirmed the mixture eluting in the pH 5.9 buffer to be mainly composed of α chain monomers, whilst protein eluting in the pH 4.5 buffer comprised large molecular weight multimers (results not shown), similar to what we had previously experienced with rat MHC class Ia molecules [14]. Only the monomer fraction was used for refolding work (yield 21 mg/l). Protein sequencing of the monomer fraction revealed that the bacteria had successfully cleaved the initiation methionine to give the expected N-terminus (GSHSLKYFH). Human β2-microglobulin was expressed and purified, as described previously for the rat form [14]. Elution of monomeric human β2-microglobulin from the Q-Sepharose column (>95 % pure by SDS-PAGE) was achieved with 100 mM NaCl with a yield of approximately 23 mg/l of bacterial culture.
Binding assay for specific peptides
We have previously reported the successful use of in vitro refolding of bacterially produced recombinant proteins to study the peptide-binding properties of rat MHC class I molecules [14]. To validate that a similar system could be applied to HLA-E, small scale refolding experiments were performed using recombinant human β2 microglobulin and a peptide derived from the HLA-A2 leader peptide, VMAPRTLVL, which is known to bind to HLA-E [4]. Refolding of HLA-E to produce a monomeric complex of correct molecular weight was assessed by gel filtration, over a range of peptide concentrations. Quantitative comparisons between refolding experiments could not be performed directly because loss of material through precipitation was variable, as judged from the total peak areas observed during gel filtration (results not shown). The extent of refolding was therefore assessed by expressing results as an induction ratio, calculated as the proportion of monomers detected in the presence of peptide (relative to the combined area of monomeric and aggregate material), compared to the proportion of monomers recorded without peptide (see Materials and Methods section for the exact calculation). Results presented in Figure 1 show that a plateau of maximum induction was reached with 10 μM of the HLA-A2 leader peptide, whilst 0.1 μM of this peptide led to 50% maximal induction.
Figure 1.

Effect of peptide concentration on refolding recombinant HLA-E. Truncated heavy chain of HLA-E was refolded by the dilution method [14, 30, 31] in the presence of light chain human β2-microglobulin and varying concentrations of peptides. Gel filtration was used to assess successful refolding by separating monomeric complexes from aggregate species. Refolding levels were calculated as induction ratios (see Materials and Methods). Results presented are representative of three independent experiments.
For a negative control, we used a 13-mer peptide, ILFPSSERLISNR, derived from the rat mitochondrial A chain of ATP synthase, which corresponds to the rat maternally transmitted minor histocompatibility peptide (MTF-E) [15]. As expected, this peptide did not induce any significant refolding (Fig.1). Six other peptides that were identified as potential binders within our laboratory's collection were also tested for binding to HLA-E and compared to the HLA-A2 leader-derived peptide. To simplify binding assays, all these were tested at 10 μM, the concentration required to reach maximum induction with the A2-derived control peptide, VMAPRTLVL. As shown in Figure 2A, refolding efficiency similar to that of the positive control was attained with AMAPRTLLL, the corresponding peptide found in the leader sequence of mouse H2-D and H2-L and in rat RT1-A class Ia molecules. To assess the importance of the position of anchor residues, we used two other nonamer peptides derived from the murine leader sequence, but shifted by either one or two residues. The fact that both peptides were found to bind poorly to HLA-E, (MAPRTLLLL: 5 %, APRTLLLLL: 23 %) suggests that the precise positioning of anchor residues inside the groove is crucial for the efficient binding of peptides derived from class I leader sequences.
Figure 2.

Effect of various peptides on refolding recombinant HLA-E. A Various specific peptides were tested for their ability to induce and stabilize the refolding of recombinant HLA-E. Peptides were added to the binding assay at 10 μM, the concentration required for maximal observed refolding for the known binding peptide, VMAPRTLVL (see Figure 1). The rat class I MHC molecule RT1-A1c was also used as a control to assess the validity of this refolding assay. Results shown are the average of 3 independent experiments. B HLA-E was refolded with 100 μM nonamer random peptide library and its induction ratio was determined and compared to two rat TAP-B associated classical Ia MHC molecules, RT1-A1c and RT1-Au. Results shown are the average of 3 independent experiments.
Within our lab's collection, three other peptides were identified that shared Met at P2 and/or Leu as C terminal anchor residues with the canonical A2 peptide. Somewhat surprisingly, all three peptides induced significant refolding: SMFPVSENR (32 %), NPRKVTAYL (23 %) and VMPTSNDPTL (78%). For the latter one, which is a decamer, we had access to sufficient amounts to carry out a dose-response curve (Fig. 1). Whilst maximum induction was reached at 10 μM, similarly to the of the leader-derived canonical peptide, 10-fold more (1 μM) peptide was required to obtain 50 % refolding of its own maximum refolding value.
As an additional control three of the peptides tested were also used to refold the rat classical class Ia MHC molecule RT1-A1c. Results show that two of the peptides, NPRKVTAYL (a synthetic peptide designed from the published binding motif of RT1-A1c [14]) and APRTLLLLL, which only induced partial refolding with HLA-E, did induce substantial refolding of the rat class I molecule, whilst conversely, the mouse leader derived peptide (AMAPRTLLL) was relatively inefficient.
Determination of an HLA-E binding motif based on binding of HLA-E to random peptide library
Having validated that the extent of HLA-E refolding could be assessed after in vitro refolding, we went on to test the nonamer random peptide library in this system. In doing so, we found that HLA-E could be induced to refold around random nonamer peptides, but this was only seen when 100 μM of the random peptide library was used (see Figure 1). For three rat MHC class Ia molecules, refolding studies with recombinant protein and random peptide libraries have already yielded binding motif information, with a good correlation to the motifs obtained from peptides eluted from naturally expressed molecules [14]. In this previous study, we had found that RT1-A1c and RT1-Au required 10 fold more peptide to achieve refolding efficiencies comparable to those obtained for RT1-Aa with a heavy chain:β2m:peptide ratio of 1:2:10. This was interpreted as an indication that RT1-A1c and RT1-Au had more stringent peptide requirements than RT1-Aa. To ascertain that, under the conditions used here, the refolding seen for HLA-E did correspond to true binding of the peptides in the random library, we therefore used refolding of RT1-A1c and RT1-Au as controls. Results in Figure 2B show that for all three class I molecules, there was a specific increase in production of refolded material, although the induction ratio of the nonamer random peptide library for HLA-E (ratio = 1.14) was reproducibly less than the ratios of both RT1-A1c (ratio = 1.26) and RT1-Au (ratio = 1.28).
Whilst the reduced efficiency of refolding and the high amounts of peptide library required suggest an even more stringent specificity than that of rat classical class Ia MHC molecules we had studied previously, we could nevertheless proceed to the determination of a binding motif for HLA-E. Successfully refolded monomeric complexes were purified by gel filtration chromatography, then acidified to disrupt the complexes and release the bound peptides, which were subjected to further purification by reversed phase chromatography over a 0-90 % acetonitrile gradient (see Figure 3). For the binding motif determination of rat class Ia molecules [14, 16], we had used fractions collected between 6.75 % and 40.5 % of the acetonitrile gradient, but for HLA-E, we found that more stringent conditions were necessary for the elution of the peptides, probably because of their increased hydrophobicity. Fractions covering the large peak eluting at 5.45 ml (fractions 33-35) were submitted for pool sequencing separately. Results (not shown) revealed that the peak contained little or no peptidic material. Sequencing of the peptides released in all other fractions pooled together yielded the results and motif presented in Figure 4. Dominant anchor residues (boxed values) as determined previously [14, 17], were observed at positions 4 (leucine), 7 (asparagine) and 9 (leucine). The amino acids glutamine and phenylalanine were also seen as significant increases at positions 2 and 3, respectively, but were not large enough increases (compared to the previous cycle) to be considered as anchor residues. Along the length of the binding motif all except one of the observed increases (lysine at P6) were uncharged or hydrophobic amino acids. This is in contrast to the binding motif reported recently for the mouse homologue Qa-1b, and obtained with an approach similar to ours, which revealed basic preferences (Lys/Arg) at positions 3, 4 and 5 in the motif and acidic preferences (Asp/Glu) at 3,4 and 8 [12].
Figure 3.

Elution profile of HLA-E bound peptides by reverse phase chromatography. Buffers and elution conditions are as described in Experimental Procedures. Sixty fractions (42 μl) were collected, pooled and subjected to protein sequence analysis. Any fractions covering any large peaks were omitted from the pool and submitted separately to protein sequence analysis.
Figure 4.

Peptide binding motif obtained by refolding recombinant HLA-E around a nonamer random peptide library. Denatured recombinant HLA-E heavy chain was refolded around a 100-fold molar excess of nonamer random peptides in the presence of a 2-fold excess of β2-microglobulin. Complexes were purified by gel filtration and their bound peptides released by acid elution. This population was purified further by reversed-phase chromatography and sequenced by Edman degradation. A Results show the yield of each amino acid at each cycle. Anchor residues were determined as described in the "Materials and Methods" and are boxed whilst other increases are underlined. B The peptide motif of the pool sequence data from A. Values next to residues are the % increases compared to the previous sequencing cycle.
Discussion
In this study we have successfully used a previously described recombinant system [14] to assess the peptide binding specificity of the human non-classical class Ib MHC molecule, HLA-E. Truncated soluble HLA-E heavy chain, containing only the α1, α2 and α3 domains, was cloned, expressed and purified from E. coli. Engineering a His-Tag at the C-terminal end of the heavy chain and use of the Ni-NTA matrix permitted the enrichment of monomers from aggregates (which was only 24 % of the total yield of heavy chain protein), whilst leaving the protein denatured in 8 M urea and ready for refolding. Human β2-microglobulin was purified by urea solubilization, renaturation by dialysis and subsequent ion exchange chromatography to purify monomers from aggregates.
Gel filtration chromatography was used to test whether the protein could refold properly using a control peptide derived from the HLA-A2 leader sequence, which has previously been shown to bind to HLA-E [4, 5]. In the presence of peptide, results showed a peptide-specific increase in monomeric complexes eluting at the correct molecular weight (approx. 45 kDa). By varying the concentration of test peptide, the affinity of HLA-E for the peptide could be measured. As seen in Figure 1, the A2-leader derived peptide refolded to a maximum induction with 10 μM peptide and 50 % induction was observed with 0.1 μM peptide. When a decamer synthetic peptide carrying the 2 anchor residues M2 and L9 was used for comparison, the maximum level of induction, seen with 10 μM, was 50% of that attained with the A2-derived nonamer. This suggests that although successful refolding is seen with this peptide, its affinity for HLA-E is not optimal. This reduction in affinity with a peptide longer than nine residues is in agreement with the results and conclusions of a previous study [4].
Other peptides were also tested for their ability to bind to HLA-E. Three overlapping peptides of the mouse H2-D and H2-L leader peptide were tested along with two synthetic peptides, SMFPVSENR and NPRKVTAYL. The former has the reported preferred methionine at P2 and what should be a non-binding arginine at P9 and the latter a non-binding proline at P2 and reported preferred leucine a P9. As shown in Figure 2A, binding comparable to that of the A2-leader derived peptide was only observed with AMAPRTLLL, the equivalent peptide found in mouse and rat class Ia leader sequences. The use of the rat class Ia MHC molecule, RT1-A1c, served to compare the binding of the same peptides to a different classical class I molecule. Results in Figure 2A show that successful refolding in this system is dependent on the specificity of the peptide for the class I molecule being tested. AMAPRTLLL, which bound to give the same induction ratio as the A2-leader derived peptide, bound with a much reduced efficiency to RT1-A1c, whereas the converse was true for two other peptides, NPRKVTAYL and APRTLLLLL, which produced a higher induction ratio for RT1-A1c than HLA-E.
Next, the same system was used to test the affinity of a nonamer random peptide library to bind to HLA-E [14]. Results show that HLA-E did refold with the library to produce monomeric complexes (Figure 2B). However, the observed induction was repeatedly less (approximately 13 % compared to the A2 leader derived nonamer peptide, VMAPRTLVL) but significantly higher than the level observed with another closely related nonamer peptide, MAPRTLLLL (5 %). In comparison with the rat molecules tested previously [14], the observed induction ratio was lower, suggesting that HLA-E may have an even more stringent peptide requirement than both the rat molecules, RT1-Au and RT1-A1c. However, the fact that binding to this random library could be detected at all suggests the HLA-E peptide binding groove might not impose as high a peptide specificity as has been suggested by other workers [4, 8].
Having observed a specific increase in refolding with the random peptide library, refolding experiments were scaled up (from 2 ml to 40 ml) in an attempt to obtain sufficient material for binding motif determination. Initial experiments using the same gradient as used previously for the binding motif determination of rat class Ia molecules [14, 16] failed to yield a motif. However, by changing the gradient conditions to collect fractions across a 0-90 % acetonitrile gradient (Figure 3), peptides were successfully recovered, that could then be sequenced to yield a binding motif for HLA-E. This motif is striking in a number of respects. Firstly, along the length of the motif, all except one of the observed increases (a lysine at P6) consisted in uncharged or hydrophobic amino acids, thus explaining the reduced elution of peptides early in the acetonitrile gradient. In addition, such a stringent preference for uncharged/hydrophobic residues at almost every position would reduce the availability of suitable peptides within the library, thus reducing the refold efficiency observed in experiments (Figure 2B). Anchor residues were observed at P4 (leucine), P7 (asparagine) and P9 (leucine) of the binding motif and possible additional secondary anchors at P2 and P3. Assuming the peptide needs to satisfy most, if not all, of these preferences for stable binding in the ER and subsequent processing to the cell surface, then the peptide length preference of this class I MHC molecule should be highly restrictive. Previous binding studies using longer variants of the HLA-B8 leader-derived peptide did find reduced binding of longer peptides to HLA-E compared to the nonamer peptide [4]. Such stringent length considerations could also partly explain the reduced binding efficiency of the decamer peptide used in our study, although care must be exercised since this decamer carries an Asp at position 7, which may also be detrimental to its binding efficiency.
Previous studies have also suggested that HLA-E binding peptides require the presence of a methionine anchor residue at P2 of the peptide [3,4,5]. Other studies have shown that other leader-derived peptides from the HLA-B alleles, which have a threonine at P2 can still bind to HLA-E although these were shown neither to activate nor inhibit NK cell lysis and to be less stable than their methionine counterparts [2, 10]. On the other hand, more recent in vitro data has shown that the HLA-B58 leader derived peptide (VTAPRTVLL) not only binds to HLA-E but has the same thermal stability as the HLA-B7 leader derived peptide (VMAPRTVLL) and both interact with activatory and inhibitory CD94/NKG2 receptors with similar kinetics [11]. Our data show that when HLA-E was refolded around a random peptide library, it did not appear to have a strong preference for either methionine or threonine at the P2 position. This cannot be due to a deficiency of the system used here, since this same approach with the same library revealed a clearly detectable methionine anchor at P2 in RT1-Aa [14]. For HLA-E, instead of methionine, there was a glutamine increase, although this was not significant enough to be considered an anchor (by the criteria used during data analysis).
Whilst we would not have expected to detect the exact sequence derived from a leader peptide (since any one given peptide would only be represented once for every 919 peptides in the random mix, i.e. never), one could have predicted (and we were in fact expecting) to identify anchor residues matching those found in the sequence of the VMAPRTLVL leader peptide. We do not have an explanation for the divergence of our findings from those of others. It is interesting to note that Ulbrecht et al. [18] reported on virally-derived HLA-E stabilising peptides with glutamine or leucine at the P2 position. These authors suggested that HLA-E may have a lower than average affinity for β2-microglobulin, so requiring tighter peptide binding to achieve overall tri-molecular stability in vivo.
Is there a special mechanism required to bring about the preferential association of class I leader peptides with HLA-E/Qa-1b?
Our results raise two important points which must be addressed before the pathways involved in antigen processing and association to HLA-E can be fully understood. Firstly, other workers have suggested that the peptide binding groove of HLA-E has evolved to bind only a tightly defined set of hydrophobic leader-derived peptides [4, 8]. However, from our present studies and others [18], it is obvious that HLA-E has affinity for peptides other than those derived from class I leader peptides. Secondly, our results show that the rat classical class Ia molecule RT1-A1c exhibits low affinity for binding the mouse leader-derived peptide, AMAPRTLLL, but has a higher affinity for the overlapping peptide, APRTLLLLL (See Figure 2A). The possibility that other, more highly expressed classical class Ia molecules could compete with HLA-E to bind to leader-derived peptides, increases the conceptual requirement for specific delivery of these peptides to the HLA-E assembly complexes. In addition, these results also draw attention to the possibility that class I leader-peptides could be processed by other routes to derive peptides such as the overlapping peptide, APRTLLLLL, of the mouse H2-D and H2-L leader sequence used here. If this occurs, then even less material will be available for processing and binding to HLA-E.
Assembly of HLA-E has been shown to be TAP-dependent [6], although it has not been unequivocally proven to require the TAP-transporter for delivery of peptides from the cytoplasm. If such peptides are supplied by the TAP transporter, then they represent only a very small fraction of the total pool of peptides transported, of which a much bigger fraction will be susceptible to bind to classical class Ia molecules than to HLA-E. Thus, whether the high rate of association of class I leader derived peptides with HLA-E, and its counterparts in mouse (Qa-1b, [19]) and presumably rat (RT.BM1), could be brought about in vivo in free competition with cytoplasmically derived peptides is still open to question. Specialized mechanisms might therefore be operating to favour the association of HLA-E with these peptides. Three possible mechanisms can be envisaged:
1) The incident concentration of these peptides could be effectively increased if the proximity of the translocon (site of translation of the heavy chain and possible processing of the signal peptide) is close to the site of assembly of HLA-E. However, there is no evidence to suggest that the site of MHC class I heavy chain translation is in close proximity to the TAP assembly complex and that the translocon complex is capable of processing leader peptides to yield the mature HLA-E binding peptide.
2) Leader-derived peptides might be released from the translocon complex into the ER lumen and processed through the ER directly. Signal-derived peptides have been shown to be delivered in a TAP-independent manner for binding to HLA-A2 [20]. It is possible that some as yet unknown mechanisms for specific enrichment are operating, such as chaperones which could shuttle the peptides from the translocon complex to the TAP transporter. If the peptides are processed and delivered via the ER lumen, calreticulin could act as such a chaperone, as it is known to bind antigenic peptides and elicit tumour immunity [21]. ER-resident proteases might also be present to process signal peptides correctly, since such activity has been reported [22].
3) Alternatively, the peptides could in fact be released from the translocon complex and associated with chaperones on the cytoplasmic side of the ER membrane. Recent work by Paz et al. revealed that antigenic peptides were not free in the cytoplasm, but bound to high molecular weight material which were different from the previously described heat shock proteins [22]. A distinct cytoplasmic chaperone might therefore perform the role of delivering leader-derived peptides preferentially to a transporter complex with HLA-E molecules primed for peptide loading, perhaps via specific recognition of a unique binding motif in the cytoplasmic tail of HLA-E.
Any peptide binding study of a non-classical class Ib MHC molecule is technically limited by the amount of material that can be purified due to its low surface expression, which is often less than 10 % compared to class Ia MHC molecules. For example previous studies on HLA-E required 20-50 grams of cell pellets for immunoprecipitation experiments using cell lines deficient in class Ia expression, in order to detect bound leader peptides derived from constructs transfected into the cell lines under strong promoters to drive expression [5]. Availability of specific antibodies poses an additional problem, since even weak cross-reactivity to a class Ia molecule, expressed at much higher levels (10-20x), would result in a contaminated purified peptide pool, thus affecting the motif.
Therefore, binding motif determination of a non-classical MHC class I molecule using a recombinant protein and a fixed length random peptide library has some clear advantages over immunoprecipitation of the same molecule expressed in mammalian cells. Unlike class II MHC molecules, peptides are usually locked into the groove of class I molecules at their N- and C-termini. In a natural system, peptides of different lengths can still bind to class I molecules [15, 23,24,25] and when eluted and subjected to pool sequencing, the yield of a C-terminal anchor can be 'diluted' due to the different lengths of the population. This should occur much less significantly with a random peptide library of a given length. Having access to large amounts of purified recombinant protein also removes the need for large amounts of biological material, and for a specific antibody, to purify the class I molecule of interest. Finally, an in vitro derived binding motif is free from the influences of cellular machinery which might operate to restrict the supply of peptides available to the class I molecule under study and select only for high affinity species. The present motif was obtained from a range of high and low affinity peptides and is therefore closer to the true biochemical preference of the molecule under study. For the three rat classical MHC class I molecules studied [14], in vivo and in vitro derived motifs were very similar and, for RT1-A1c, natural peptides were identified with the conserved anchors [26]. By contrast, the known, natural HLA-E peptide ligands do not match closely our in vitro motif. This finding may suggest an as yet unrecognised and specialised in vivo mechanism for peptide selection to be operating, rather than being an issue of peptide affinities in the selection process.
Conclusions
Using recombinant bacterially produced HLA-E, we have shown that it is possible to obtain a binding motif of the non-classical class Ib MHC molecule. The same system can also be used to test the binding affinity of specific peptides when no cell-based assay is available. Whilst the motif obtained confirmed a strong preference of HLA-E for hydrophobic residues at most positions, the fact that a library of random peptides could bind at all demonstrates that this molecules requirements are not as exquisite as previously suggested. If, as suggested by these results and those of others [18], HLA-E has the capacity to present a range of different peptides, then presentation of non-self antigens may have to be considered as an important accessory role to its function in regulating NK function.
Materials and Methods
Peptides
The HLA-A2 leader derived peptide (VMAPRTLVL) was synthesized by Sigma-Genosys (Pampisford, UK). The 13-mer peptide, ILFPSSERLISNR, was synthesized by Alta Biosciences, (The University of Birmingham, UK). Among the collection of synthetic peptides available in the laboratory, we identified the following which had suitable anchor residues at positions P2 and/or the C-terminus; VMPTSNDPTL, SMFPVSENR and NPRKVTAYL. The overlapping mouse H2-D and H2-L leader-derived peptides (AMAPRTLLL, MAPRTLLLL and APRTLLLLL) and the synthetic nonamer random peptide library used in this study were all purchased from ECHAZ Microcollections (Tübingen, Germany). The synthetic nonapeptide library was prepared by fully automated solid phase peptide synthesis using Fmoc/tBu chemistry. The randomness of the peptide library was ascertained by pool sequencing [27], electrospray mass spectrometry [28] and amino acid analysis[29]. Prior to use, each of the specific peptides and the library was dissolved in DMSO (Pierce, packed under nitrogen) at a concentration of 10 mg/ml.
Cloning, expression and purification of HLA-E
The region coding for amino acids 1-276 of the HLA-E heavy chain was amplified from a human T cell cDNA library (a kind gift from Dr. Martin Turner, Lymphocyte Signalling and Development Laboratory, The Babraham Institute) by polymerase chain reaction (PCR) using the oligonucleotide prime pair: forward 5'-CGGGATCCCCATATGGGTTCACACTCCTTGAAGTA-TTTCCACACTT-3' and reverse 5'-GAAGATCTCGAGCGGCTTCCATCTCAGGGTGACGG-GCT-3' (restriction sites used are underlined). The resulting product was digested with NdeI/XhoI, ligated into the T7 expression plasmid, pET-22b(+) (Novagen Inc., WI, USA) and transformed/ selected in XL2-Blue (Stratagene). DNA sequences from these constructs were checked (found to be identical to E*0102) and plasmids were re-transformed into the Escherichia coli strain BL21(DE3) (Novagen Inc., WI, USA).
Bacteria were grown in LB containing 100 μg/ml ampicillin. Protein expression was induced at mid-log phase for 3 hours with 1 mM IPTG and the heavy chain protein was found to be overexpressed as inclusion bodies. Bacterial pellets were resuspended in 10 mM Tris-HCl; pH 7.5, 1 mM EDTA, 100 μg/ml PMSF, 0.1 % (v/v) Triton-X100, disrupted by sonication and centrifuged (25,000 g). The cell pellet was washed twice with resuspension buffer and then solubilized by resuspending and mixing for 1 hour at room temperature in 8 M urea, 0.1 M NaH2PO4, 0.01 M Tris-HCl; pH 8.0 (buffer A; pH 8.0). The mixtures were then centrifuged (25,000 g) to remove any insoluble material. Urea-solubilized heavy chain was purified in a denatured state using a Ni-NTA agarose column (Qiagen Inc., CA, USA) according to the manufacturer's instructions. Briefly, the urea-solubilized protein was mixed with the nickel-charged matrix for 1 hour. The matrix was washed with buffer A; pH 8.0, followed by buffer A; pH 6.3. Pure protein (>95% as determined by SDS-PAGE; results not shown) was eluted from the column by washing with buffer A; pH 5.9 and buffer A; pH 4.5. Protein concentration was determined by the BCA assay (Pierce) and the sample was concentrated to approximately 1 mg/ml and stored at -70°C.
Expression of human β2-microglobulin
Bacteria expressing human β2-microglobulin [30] were a kind gift of Professor Don Wiley. The bacteria were grown and induced as for the heavy chain. β2-microglobulin was also overexpressed as inclusion bodies and was isolated and solubilized and clarified as described above. Urea-solubilized β2-microglobulin was refolded by extensive dialysis against 10 mM Tris-HCl; pH 7.5 at room temperature. The dialysed mixture was centrifuged and any precipitate formed was re-solubilized and re-dialysed. Solutions were pooled and applied to a Q-Sepharose column (Pharmacia Biotech) equilibrated with 10 mM Tris-HCl; pH 7.5. Pure monomeric human β2-microglobulin (>95 % purity as determined by SDS-PAGE; results not shown) eluted from this column with 10 mM Tris-HCl, 100 mM NaCl; pH 7.5. Protein concentration was determined by the BCA assay (Pierce).
Expression and purification of rat class I molecules and β2-microglobulin
Expression and purification of both rat heavy and light chains were performed as described previously [14].
Assembly assays and purification of complexes
Refolding was performed by the dilution method [14, 30, 31]. Small scale refolds with specific peptides were performed in a total volume of 2 ml. β2-microglobulin (48 μg, 4 nmol) and peptide (20 μg, 20 nmol) were added to 1.5 ml of refold buffer (50 mM Tris-HCl; pH 8.0, 400 mM arginine, 0.1 mM EDTA, 0.1 mM PMSF). Denatured heavy chain (62 μg; 2 nmol) was added in aliquots with mixing and refold buffer was added to give a final volume of 2 ml in each tube and final molar ratio of 1:2:10 (heavy chain: β2-microglobulin:peptide). The amount of peptide added was varied from the value stated above when studies on the effect of peptide concentration on refolding were performed. After 24-48 hours at 4°C the refolding mixture was concentrated down to 100 μl using a Centricon-10 unit (Millipore), with the temperature maintained at 18°C during the concentration step and refolded class I complex was purified by gel filtration on an FPLC Superdex 75 10/30 column (Pharmacia Biotech) equilibrated in 20 mM Tris-HCl; pH 8.0, 100 mM NaCl. Results were calculated as the ratio of the peak area of monomeric soluble class I complex in the presence of a peptide compared to a control sample without added peptide. Due to variable sample loss from increased precipitation of high multimeric forms in the absence of peptide (or presence of low binding peptide), induction of refolding was calculated by comparing proportionate changes according to the following equation:
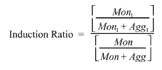
where Mon1 = peak area of monomer with peptide; Agg1 = peak area of aggregate with peptide; Mon = peak area of monomer without peptide; Agg = peak area of aggregate without peptide.
Peptide elution and pool sequencing
For binding motif determination, refolds were performed in 40 ml incubations. Briefly, β2-microglobulin (960 μg, 80 nmol) and nonamer random peptide (4 mg, 4 μmol) were added to the refolding buffer and denatured heavy chain (1.24 mg; 40 nmol) was added dropwise with constant mixing.
After 24-48 hours at 4°C the refolding mixture was concentrated down to 1 ml using Centriprep-10 and Centricon-10 units (Millipore) and refolded class I complexes were purified by gel filtration on an FPLC Superdex 75 16/60 Hi-load column (Pharmacia Biotech) equilibrated in 20 mM Tris-HCl; pH 8.0, 100 mM NaCl.
Fractions containing refolded monomeric complexes were pooled and concentrated to 900 μl using Centricon-10 units. The concentrated HLA-E complexes were acidified by the addition of 100 μl acetic acid. After 5 minutes incubation at room temperature, the mixture was spun through Centricon-3 units. The flow through was then subjected to reversed phase chromatography using an Applied Biosystems Aquapore (250 mm x 1 mm) Brownlee C18 column with an acetonitrile gradient (0-90 %) in 0.025 % TFA. Material eluting in the acetonitrile gradient was pooled, concentrated and submitted for protein sequencing by Technix™ (The Babraham Institute, UK). One major peak (fractions 33-35) was submitted for sequencing separately. As described previously [14, 17], a pmol yield >150 % of the previous cycle was considered significant and is presented as boxed values in the results (Figure 4). For cycles 6-10, this cut-off value was reduced to 120 % to allow for cycle-to-cycle sample loss.
Acknowledgments
Acknowledgements
This work was supported by a BBSRC Project Grant to JT/GWB, a BBSRC Project Grant to EJ and BBSRC CSG funding to the Laboratory of Functional Immunogenetics, Babraham. The authors would like to thank Professor Don Wiley for kindly providing the bacterial strain expressing human β2-microglobulin and Dr. Martin Turner for providing the human cDNA library.
Contributor Information
James Stevens, Email: jstevens@scripps.edu.
Etienne Joly, Email: atn@cict.fr.
John Trowsdale, Email: jt233@cam.ac.uk.
Geoffrey W Butcher, Email: geoff.butcher@bbsrc.ac.uk.
References
- Lee N, Llano M, Carretero M, Ishitani A, Navarro F, López-Botet M, Geraghty DE. HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc Natl Acad Sci U S A. 1998;95:5199–5204. doi: 10.1073/pnas.95.9.5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrego F, Ulbrecht M, Weiss EH, Coligan JE, Brooks AG. Recognition of human histocompatibility leukocyte antigen (HLA)-E complexed with HLA class I signal sequence-derived peptides by CD94/NKG2 confers protection from natural killer cell-mediated lysis. J Exp Med. 1998;187:813–818. doi: 10.1084/jem.187.5.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braud VM, Allan DS, O'Callaghan CA, Soderstrom K, D'Andrea A, Ogg GS, Lazetic S, Young NT, Bell JI, Phillips JH, Lanier LL, McMichael AJ. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature. 1998;391:795–799. doi: 10.1038/35869. [DOI] [PubMed] [Google Scholar]
- Braud V, Jones EY, McMichael A. The human major histocompatibility complex class Ib molecule HLA-E binds signal sequence-derived peptides with primary anchor residues at positions 2 and 9. Eur J Immunol. 1997;27:1164–1169. doi: 10.1002/eji.1830270517. [DOI] [PubMed] [Google Scholar]
- Lee N, Goodlett DR, Ishitani A, Marquardt H, Geraghty DE. HLA-E surface expression depends on binding of TAP-dependent peptides derived from certain HLA class I signal sequences. J Immunol. 1998;160:4951–4960. [PubMed] [Google Scholar]
- Braud VM, Allan DS, Wilson D, McMichael AJ. TAP- and tapasin-dependent HLA-E surface expression correlates with the binding of an MHC class I leader peptide. Curr Biol. 1998;8:1–10. doi: 10.1016/s0960-9822(98)70014-4. [DOI] [PubMed] [Google Scholar]
- Tomasec P, Braud VM, Rickards C, Powell MB, McSharry BP, Gadola S, Cerundolo V, Borysiewicz LK, McMichael AJ, Wilkinson GW. Surface expression of HLA-E, an inhibitor of natural killer cells, enhanced by human cytomegalovirus gpUL40. Science. 2000;287:1031–1033. doi: 10.1126/science.287.5455.1031. [DOI] [PubMed] [Google Scholar]
- O'Callaghan CA, Tormo J, Willcox BE, Braud VM, Jakobsen BK, Stuart DI, McMichael AJ, Bell JI, Jones EY. Structural features impose tight peptide binding specificity in the nonclassical MHC molecule HLA-E. Mol Cell. 1998;1:531–541. doi: 10.1016/s1097-2765(00)80053-2. [DOI] [PubMed] [Google Scholar]
- Llano M, Lee N, Navarro F, Garcia P, Albar JP, Geraghty DE, López-Botet M. HLA-E-bound peptides influence recognition by inhibitory and triggering CD94/NKG2 receptors: preferential response to an HLA-G- derived nonamer. Eur J Immunol. 1998;28:2854–2863. doi: 10.1002/1521-4141(199809)28:09<2854::AID-IMMU2854>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Brooks AG, Borrego F, Posch PE, Patamawenu A, Scorzelli CJ, Ulbrecht M, Weiss EH, Coligan JE. Specific recognition of HLA-E, but not classical, HLA class I molecules by soluble CD94/NKG2A and NK cells. J Immunol. 1999;162:305–313. [PubMed] [Google Scholar]
- Valés-Gómez M, Reyburn HT, Erskine RA, López-Botet M, Strominger JL. Kinetics and peptide dependency of the binding of the inhibitory NK receptor CD94/NKG2-A and the activating receptor CD94/NKG2-C to HLA-E. EMBO J. 1999;18:4250–4260. doi: 10.1093/emboj/18.15.4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft JR, Vance RE, Pohl J, Martin AM, Raulet DH, Jensen PE. Analysis of Qa-1b peptide binding specificity and the capacity of CD94/NKG2A to discriminate between Qa-1-peptide complexes. J Exp Med. 2000;192:613–623. doi: 10.1084/jem.192.5.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacasova R, Martinozzi S, Boulouis HJ, Szpak Y, Ulbrecht M, Sigaux F, Weiss EH, Pla M. Cell surface detection of HLA-E gene products with a specific monoclonal antibody. J Reprod Immunol. 1999;43:195–201. doi: 10.1016/S0165-0378(99)00033-9. [DOI] [PubMed] [Google Scholar]
- Stevens J, Wiesmüller K-H, Barker PJ, Walden P, Butcher GW, Joly E. Efficient generation of MHC class I-peptide complexes using synthetic peptide libraries. J Biol Chem. 1998;273:2874–2884. doi: 10.1074/jbc.273.5.2874. [DOI] [PubMed] [Google Scholar]
- Bhuyan PK, Young LL, Lindahl KF, Butcher GW. Identification of the rat maternally transmitted minor histocompatibility antigen. J Immunol. 1997;158:3753–3760. [PubMed] [Google Scholar]
- Powis SJ, Young LL, Joly E, Barker PJ, Richardson L, Brandt RP, Melief CJ, Howard JC, Butcher GW. The rat cim effect - TAP allele-dependent changes in a class-I MHC anchor motif and evidence against C-terminal trimming of peptides in the ER. Immunity. 1996;4:159–165. doi: 10.1016/s1074-7613(00)80680-9. [DOI] [PubMed] [Google Scholar]
- Falk K, Rötzschke O, Stevanovic S, Jung G, Rammensee HG. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature. 1991;351:290–296. doi: 10.1038/351290a0. [DOI] [PubMed] [Google Scholar]
- Ulbrecht M, Modrow S, Srivastava R, Peterson PA, Weiss EH. Interaction of HLA-E with peptides and the peptide transporter in vitro: implications for its function in antigen presentation. J Immunol. 1998;160:4375–4385. [PubMed] [Google Scholar]
- Aldrich CJ, Decloux A, Woods AS, Cotter RJ, Soloski MJ, Forman J. Identification of a TAP-dependent leader peptide recognized by alloreactive T-cells specific for a class Ib antigen. Cell. 1994;79:649–658. doi: 10.1016/0092-8674(94)90550-9. [DOI] [PubMed] [Google Scholar]
- Wei ML, Cresswell P. HLA-A2 molecules in an antigen-processing mutant cell contain signal sequence-derived peptides. Nature. 1992;356:443–446. doi: 10.1038/356443a0. [DOI] [PubMed] [Google Scholar]
- Basu S, Srivastava PK. Calreticulin, a peptide-binding chaperone of the endoplasmic reticulum, elicits tumor- and peptide-specific immunity. J Exp Med. 1999;189:797–802. doi: 10.1084/jem.189.5.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz P, Brouwenstijn N, Perry R, Shastri N. Discrete proteolytic intermediates in the MHC class I antigen processing pathway and MHC I-dependent peptide trimming in the ER. Immunity. 1999;11:241–251. doi: 10.1016/s1074-7613(00)80099-0. [DOI] [PubMed] [Google Scholar]
- Guo HC, Jardetzky TS, Garrett TPJ, Lane WS, Strominger JL, Wiley DC. Different length peptides bind to HLA-Aw68 similarly at their ends but bulge out in the middle. Nature. 1992;360:364–366. doi: 10.1038/360364a0. [DOI] [PubMed] [Google Scholar]
- Joyce S, Kuzushima K, Kepecs G, Angeletti RH, Nathenson SG. Characterization of an incompletely assembled major histocompatibility class-I molecule (H-2Kb) associated with unusually long peptides - implications for antigen-processing and presentation. Proc Natl Acad Sci U S A. 1994;91:4145–4149. doi: 10.1073/pnas.91.10.4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban RG, Chicz RM, Lane WS, Strominger JL, Rehm A, Kenter MJH, UytdeHaag FGCM, Ploegh H, Uchanska-Ziegler B, Ziegler A. A subset of HLA-B27 molecules contains peptides much longer than nonamers. Proc Natl Acad Sci U S A. 1994;91:1534–1538. doi: 10.1073/pnas.91.4.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens J, Jones RC, Bordoli RS, Trowsdale J, Gaskell SJ, Butcher GW, Joly E. Specificity of RT1-A1c, an Inhibitory Rat Major Histocompatibility Complex Class I Natural Killer Cell Ligand. J Biol Chem. 2000;275:29217–29224. doi: 10.1074/jbc.M002565200. [DOI] [PubMed] [Google Scholar]
- Stevanovic S, Wiesmüller K-H, Metzger J, Beckickinger AG, Jung G. Natural and synthetic peptide pools - characterization by sequencing and electrospray mass-spectrometry. Bioorg Med Chem Lett. 1993;3:431–436. doi: 10.1016/S0960-894X(01)80226-2. [DOI] [Google Scholar]
- Metzger JW, Kempter C, Wiesmüller K-H, Jung G. Electrospray mass-spectrometry and tandem mass-spectrometry of synthetic multicomponent peptide mixtures - determination of composition and purity. Anal Biochem. 1994;219:261–277. doi: 10.1006/abio.1994.1266. [DOI] [PubMed] [Google Scholar]
- Wiesmüller K-H, Feiertag S, Fleckenstein B, Kienle S, Stoll D, Herrmann M, Jung G. Peptide and Cyclopeptide Libraries: Automated Synthesis, Analysis and Receptor Binding Assays. Combinatorial Peptide and Nonpeptide Libraries-A Handbook for the Search of Lead Structures. Edited by Jung G. pp. Weinheim, FRG: Verlag Chemie; 1996. pp. 203–246.
- Garboczi DN, Hung DT, Wiley DC. HLA-A2-peptide complexes - refolding and crystallization of molecules expressed in Escherichia coli and complexed with single antigenic peptides. Proc Natl Acad Sci U S A. 1992;89:3429–3433. doi: 10.1073/pnas.89.8.3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid SW, Smith KJ, Jakobsen BK, O'Callaghan CA, Reyburn H, Harlos K, Stuart DI, McMichael AJ, Bell JI, Jones EY. Production and crystallization of MHC class-Ib allele single peptide complexes. FEBS Lett. 1996;383:119–123. doi: 10.1016/0014-5793(96)00226-8. [DOI] [PubMed] [Google Scholar]