

Abstract
Excessive neutrophil infiltration to the lungs is a hallmark of acute lung injury (ALI). Milk fat globule-EGF factor 8 (MFG-E8) was originally identified for phagocytosis of apoptotic cells. Subsequent studies revealed its diverse cellular functions. However, whether MFG-E8 can regulate neutrophil function to alleviate inflammation is unknown. We therefore aimed to reveal MFG-E8 roles in regulating lung neutrophil infiltration during ALI. To induce ALI, C57BL/6J wild-type (WT) and Mfge8−/− mice were intra-tracheally injected with LPS (5 mg/kg). Lung tissue damage was assessed by histology and the neutrophils were counted by a hemacytometer. Apoptotic cells in lungs were determined by TUNEL, while caspase-3 and MPO activities were assessed spectrophotometrically. CXCR2 and GRK2 expressions in neutrophils were measured by flow cytometry. Following LPS challenge, Mfge8−/− mice exhibited extensive lung damage due to exaggerated infiltration of neutrophils and production of TNF-α, MIP-2 and MPO. Increased number of apoptotic cells was trapped into the lungs ofMfge8−/− mice than WT mice, which may be due to insufficient phagocytosis of apoptotic cells or increased occurrence of apoptosis through the activation of caspase-3. In vitro studies using MIP-2 mediated chemotaxis, revealed higher migration of neutrophils of Mfge8−/− mice than WT mice via increased surface exposures to CXCR2. Administration of recombinant mouse (rm)MFG-E8 reduces neutrophil migration through up-regulation of GRK2, and down-regulation of surface CXCR2 expression. Conversely, these effects could be blocked by anti-αv-integrin antibodies. These studies clearly indicate the importance of MFG-E8 in ameliorating neutrophil infiltration and suggest MFG-E8 as a novel therapeutic potential for ALI.
Keywords: MFG-E8, Neutrophil, LPS, MIP-2, CXCR2, GRK2, αvβ3-integrin
INTRODUCTION
Acute lung injury (ALI) and its more severe form, acute respiratory distress syndrome (ARDS), which are characterized by an excessive inflammatory response, remain as considerable clinical challenges to the intensive care medicine (1). Infectious etiologies, such as sepsis, pneumonia and gut ischemia/reperfusion (I/R) are among the leading causes of ALI/ARDS (2). Despite extensive research in this field, only a few therapeutic strategies for ALI/ARDS have been emerged, and current specific options for treatment remain limited.
Neutrophil recruitment is critical to the pulmonary inflammatory responses associated with ALI (3, 4). Activated neutrophils release proteolytic enzymes, such as elastase and myeloperoxidase, and reactive oxygen species (ROS) including hydrogen peroxide and superoxide. Excessive production of these agents not only kill invaded pathogens, but also engage in disruption of endothelial barrier functions and promote extravascular host tissue damage during uncontrolled inflammation (4, 5). Neutrophil infiltration into the lungs is mediated by a local production of chemokines released by macrophages as well as other cell types in response to inflammation (6, 7). CXC chemokines, such as IL-8 are elevated significantly in the BALF of patients with ARDS, and increased IL-8 levels are associated with increased neutrophil infiltration (8, 9). In rodents, the IL-8 homologue, CINC-1/2 and MIP-2 regulate neutrophil recruitment into the lungs of experimental ALI via chemokine receptor, CXCR2 (10–12). CXCR2 is a seven transmembrane type G protein-coupled receptor (GPCR) whose expression, localization and function in polymorphonuclear leukocytes (PMN) are tightly regulated by the intracellular G protein-coupled receptor kinase 2 (GRK2). Upon activation, GRK2 phosphorylates CXCR2 and causes receptor desensitization and internalization, leading to down-regulation of neutrophil chemotaxis (13–15).
Milk fat globule-EGF factor VIII (MFG-E8), a secretary glycoprotein, is composed of a N-terminal cleavable signal peptide, followed by two EGF-like domains, a proline-threonine (PT) rich motif and two C-terminal discoidin domains resembling the sequences of blood coagulation factors V and VIII (16). The second EGF domain contains an Arginine-Glycine-Aspartate (RGD) motif which enables it to bind αvβ3-integrin of macrophages, while the discoidin domains facilitate opsonization of the apoptotic cells via recognizing phosphatidylserine (PS), thus promoting their engulfment by macrophages (16). Expression of MFG-E8 is ubiquitously found in spleen, lungs, liver, kidneys, intestine and mammary glands (17), while a marked decrease in its content is noted in various inflammatory diseases, causing exaggerated inflammation and abnormal tissue homeostasis (17, 18). The cellular expression pattern of MFG-E8 reveals it’s localization into the macrophages, dendritic, epithelial, and fibroblast cells from various organs (17). In normal lungs, MFG-E8 is present in the alveolar interstitium and pulmonary vasculature which comprise largely of epithelial and endothelial cells and a few resident macrophages (19). In our previous studies, we noticed significant decrease of MFG-E8 expression in the lungs after gut ischemia/reperfusion (I/R) injury (20). This decrease might be due to the exaggerated activation of immune-reactive cells since MFG-E8 expression is negatively regulated by toll-like receptor (TLR)-mediated pathways (21). Among several features, cellular apoptosis is markedly noticed in ALI (7), thus predicting a scavenging role of MFG-E8 to get rid of the deleterious effects of apoptotic cells before undergoing secondary necrosis (18). Phagocytosis of apoptotic cells can indirectly regulate pro-inflammatory milieu by modulating the activation of potent transcription factor, nuclear factor-κB (NF-κB) (22). However, in immune reactive cells we recently demonstrated a direct anti-inflammatory role of MFG-E8 by inhibiting NF-κB-mediated pro-inflammatory cytokine production, regardless of its effect in phagocytosis (23–25). Although the concepts of clearance of apoptotic cells and the down-regulation of NF-κB may greatly improve our understanding of delineating the protective roles of MFG-E8 against ALI, the underlying mechanisms of resolving excessive neutrophil infiltration remain unexplored. Recently, the synthetic peptide RGD has been shown to attenuate lung neutrophil chemotaxis in ALI by recognizing αvβ3-integrin and modulating down-stream signaling events (26). Since MFG-E8 has a binding affinity for αvβ3-integrin through its RGD motif (16), we, therefore consider an additional mechanism by which MFG-E8 may attenuate neutrophil migration during ALI.
While the immune-homeostatic functions of MFG-E8 have been demonstrated in macrophages, dendritic cells and epithelial cells (22–25, 27), its effect in PMN is completely unknown. Considering our initial findings of exaggerated accumulations of neutrophils in lungs of Mfge8−/− mice, we hypothesize MFG-E8 as a crucial factor for controlling neutrophil migration in LPS-induced ALI. Based on our hypothesis, we report that Mfge8−/− mice exhibit detrimental impact in experimental ALI due to excessive neutrophil infiltration, pro-inflammatory cytokine production and extensive tissue damage and apoptosis, which can be resolved by treatment with recombinant murine (rm)MFG-E8. We further clarify the pivotal roles of MFG-E8 as αvβ3-integrin mediated regulation of neutrophil migration by modulating the surface expression of CXCR2 via GRK2-dependent pathways. Importantly, the current research identifies an outstanding additional role by which MFG-E8 decreases neutrophil infiltration into the lungs and ameliorates LPS-induced ALI.
MATERIALS AND METHODS
Experimental Model
Male weight (25–30 g) and age-matched WT C57BL/6J (Taconic, Albany, NY) and Mfge8−/− mice (a kind gift of Dr. Shigekazu Nagata, Kyoto University, Japan) were anesthetized with isoflurane and then instilled with 40 µl of sterile saline (PBS) without or with 5 mg/kg body weight (BW) of LPS (Escherichia coli serotype O55:B5; Sigma-Aldrich, St. Louis, MO) via intratracheal (i.t.) injection using 28G U-100 insulin syringe. Although the current model of LPS-induced ALI is non-lethal, it can significantly induce lung injury. The rmMFG-E8 (R&D Systems, Minneapolis, MN) was administered intraperitoneally (i.p.) at 20 µg/kg BW dose, 2 h before i.t. LPS instillation. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of The Feinstein Institute for Medical Research.
Lung Tissue Histology
Lung tissues were fixed in 10% formalin and embedded in paraffin. Tissue blocks were sectioned at a thickness of 5 µm and stained with hematoxylin/eosin (H&E). Morphological changes were scored by an independent pathologist as absent (0), mild (+1), moderate (+2), or severe injury (+3) based on the presence of exudates, hyperemia/congestion, neutrophilic infiltrates, intraalveolar hemorrhage/debris, and cellular hyperplasia (20). The sum of scores of different animals was averaged.
In situ TUNEL assay
DNA breaks occur late in the apoptotic pathway and can be determined by performing the terminal deoxynucleotide transferase dUTP nick end labeling (TUNEL) assay. The presence of apoptotic cells in lung tissues was determined using a TUNEL staining kit (Roche Diagnostics, Indianapolis, IN). Briefly, lung tissues were fixed in 10% phosphate buffered formalin and were then embedded into paraffin and sectioned at 5 µm following the standard histology procedures. Lung sections were dewaxed, rehydrated and equilibrated in Tris buffered saline (TBS). The sections were then digested with 20 µg/mL proteinase K for 20 min at room temperature. Following this, the sections were washed and incubated with a mixture containing terminal deoxynucleotidyl transferase and fluorescence labeled nucleotides and examined under a fluorescence microscope (Nikon Eclipse Ti-S, Melville, NY).
Caspase-3 enzyme activity assay
The caspase-3 activity in lung tissues was assessed using a fluorimetric assay kit (Sigma, Saint Louis, MO), which is based on the principles of hydrolysis of the peptide substrate acetyl-Asp-Glu-Val-Asp-7-amido-4-methylcoumarin (Ac-DEVD-AMC) by caspase-3, resulting in the release of the fluorescent 7-amino-4-methylcoumarin (AMC) moiety. In brief, lung tissues were homogenized in liquid nitrogen, and equal weight of powdered tissues (~50 mg) were dissolved in 500 µl of lysis buffer (10 mM Hepes, pH 7.4, 5 mM MgCl2, 1 mM DTT, 1% Triton X-100, and 2 mM each of EDTA and EGTA), and subjected for sonication in ice. Protein concentration was determined by the Bio Rad protein assay reagent (Hercules, CA). Equal amounts of proteins in a 5 µl volume were added to the 100 µl assay buffer (20 mM Hepes, pH 7.4, 5 mM DTT, 2 mM EDTA, and 0.1% CHAPS) containing 10 µM DEVD-AMC substrate and the changes of fluorescence intensity with time at 37°C were measured at excitation: 370 nm and emission: 450 nm in a fluorometer (Synergy H1, BioTek, Winooski, VT). A standard curve was generated using various concentrations of 7-Amino-4-methylcoumarin (AMC) as standard. The results are expressed as mM AMC/min/g protein.
Isolation of Bronchoalveolar Lavage Fluid (BALF) and Cell Counts
Mice were euthanized and the trachea was cannulated by using PE50 catheter. PBS (1 ml) was infused to the lungs to collect the lavage fluid for 5 times. The number of total cells in BALF was counted by a hemacytometer. To identify cell population, BALF aliquot was subjected to cytospin and stained with Differential Quik Stain Kit (Polysciences, Warrington, PA). Alternatively, the percentage of neutrophils in BALF was determined by gating with cell size and positive staining of antibodies APC-Ly6G and FITC-CD11b (BD Biosciences, San Jose, CA) in flow cytometric analysis.
Isolation of Bone Marrow Derived Neutrophils (BMDN)
BMDN were isolated as described by Boxio et al (28). Mice were euthanized and femurs from both hind legs were removed. The distal tip of each edge was cut off and bone marrow cells were isolated by flushing the femur with Hanks Balanced Salt Solution (HBSS). Cell suspension was filtered through a nylon membrane and centrifuged at 1,000 rpm for 10 min. Cell pellet was resuspended in HBSS and laid on a three layer Percoll gradient of 78%, 69%, and 52% (Amersham Pharmacia, Uppsala, Sweden), followed by centrifugation at 3,000 rpm for 30 min without braking. Cells at the 69%/78% interface were carefully removed and washed with cold PBS. The identification and purity of isolated BMDN were examined by flow cytometric analysis stained with antibodies APC-Ly6G and FITC-CD11b.
Myeloperoxidase (MPO) Staining and Activity Assay
Paraffin-embedded lung tissue sections were incubated with rabbit anti-MPO antibodies (Abcam, Cambridge, MA), followed by incubation with biotinylated anti-rabbit IgG. Staining was developed by Vectastain ABC reagent and DAB kit (Vector, Burlingame, CA). For the negative control, the primary antibody was substituted with normal rabbit IgG. To determine MPO activity, tissues were homogenized in KPO4 buffer containing 0.5% hexa-decyl-trimethyl-ammonium bromide and incubated at 60°C for 2 h, followed by centrifugation. The supernatant was diluted in a reaction solution, and ΔOD was measured at 460 nm to calculate MPO activity.
Quantitative Real-time PCR Analysis
Total RNA was extracted from lung tissues by the Trizol reagent (Invitrogen, Carlsbad, CA) and reverse-transcribed into cDNA by using murine leukemia virus reverse-transcriptase (Applied Biosystems, Foster City, CA). A PCR reaction was carried out in a 25 µl of final volume containing 0.08 µmol of each forward and reverse primer, cDNA, and 12.5 µl SYBR Green PCR Master Mix (Applied Biosystems). Amplification was conducted in an Applied Biosystems 7300 real-time PCR machine under the thermal profile of 50°C for 2 min, 95°C for 10 min and followed by 45 cycles of 95°C for 15 sec and 60°C for 1 min. The level of mouse β-actin mRNA was used for normalization. Relative expression of mRNA was calculated by the 2−ΔΔCt method and results were expressed as fold change in comparison to control group. The sequence of primers used for this study are: TNF-α (NM_013693): forward: 5’-AGACCCTCACACTCAGATCATCTTC-3’, reverse: 5’-TTGCTACGACGTGGGCTACA-3’; MIP-2 (NM_009140): forward: 5’-CATCCAGAGCTTGATGGTGA-3’, reverse: 5’-CTTTGGTTCTTCCGTTGAGG-3’; MFG-E8 (NM_008594): forward: 5’-CGGGCCAAGACAATGACATC-3’, reverse: 5’-TCTCTCAGTCTCATTGCACACAAG-3’; and β-actin (NM_007393): forward: 5’-CGTGAAAAG ATG ACCCAGATCA-3’, reverse: 5’-TGGTACGACCAGAGGCATACAG-3’.
Measurements of TNF-α and MIP-2 Proteins
Levels of TNF-α and MIP-2 in the lung tissues, BALF, and cell culture supernatants were measured by using an enzyme-linked immunosorbent assay (ELISA) kit specific for mouse TNF-α (BD Biosciences) and MIP-2 (R&D systems). The assay was carried out according to the instructions provided by the manufacturer.
Western Blot Analysis
Lung tissues were homogenized and lysed in RIPA buffer (10 mM Tris–HCl pH 7.5, 120 mM NaCl, 1% NP-40, 1% sodium deoxycholate, and 0.1% SDS) containing a protease inhibitor cocktail (Roche Diagonstics, Indianapolis, IN). Protein concentration was determined by the Bio Rad protein assay reagent. Lysates were electrophoresed on SDS-polyacrylamide gels and transferred onto nitrocellulose membranes. Membranes were blocked with 5% nonfat dry milk in TTBS buffer (0.1% Tween-20, 20 mM Tris–HCl pH 7.5, and 140 mM NaCl) and incubated with primary antibody against MFG-E8 (MBL International, Nagoya, Japan), followed by secondary antibody–horseradish peroxidase conjugate and detected using chemiluminescence (GE Healthcare, Buckinghamshire, UK) and autoradiography. The immunoblot was reprobed with anti-β-actin antibodies as loading control.
Flow Cytometric Analysis
To examine surface CXCR2 expression, BMDN with various treatments were stained with propidium iodide (PI), antibodies FITC-CD11b, APC-Ly6G, PerCP/Cy5.5-CXCR2 (BioLegend, San Diego, CA) and subjected to FACSCalibur (BD Biosciences). CXCR2 levels were analyzed in the cell population with PI−CD11b+Ly6G+. To examine intracellular GRK2 expression, cells were first stained with appropriate fluorescence antibodies to detect cell surface markers, and then fixed and permeabilized with Intraprep (Beckman Coulter, Fullerton, CA), followed by staining with PE-GRK2 antibodies (Abcam). After washing, the stained cells were subjected to FACSCalibur. Data were analyzed by Flowjo software (Ashland, OR) with 15,000 events per sample. Isotype controls were used for all the samples.
In vitro Neutrophil Migration Assay
The migration assays was conducted in a modified 24-well (3.0 µm) Boyden chamber (BD Biosciences). Cells (3×105) were plated in the upper well and medium with 1 ng/ml rmMIP-2 (R&D Systems) was placed in the lower well as chemotactic stimulus. After 2 h of incubation, the upper surface of the filter was swabbed with cotton to remove non-migratory cells. Migrated cells were fixed with 10% formalin and stained with PI. Five random microscopic fields per well were counted. For blocking experiments, cells were incubated with rmMFG-E8, anti-MFG-E8 (Santa Cruz Biotechnology, Santa Cruz, CA) and anti-αv neutralizing antibodies (EMD Biosciences, La Jolla, CA) for 2 h prior to plating.
Statistical Analysis
All data are expressed as means ± SE and compared by one-way ANOVA and Student-Newman-Keuls (SNK) test. Student t test was used when only two groups were compared. Differences in values were considered significant if P <0.05.
RESULTS
MFG-E8 Deficiency Augments Pulmonary Inflammation and Injury Induced by LPS
To identify whether MFG-E8 played a role in ALI, we compared the inflammatory parameters between WT and Mfge8−/− mice subjected to i.t. injection of LPS. Within 4 h after LPS instillation, robust induction of TNF-α was noted in the lungs of WT and Mfge8−/− mice (Figures 1A and B). However, the TNF-α mRNA and protein levels in Mfge8−/− mice after 4 h of LPS-treatment were found to be 1.50- and 1.45-fold higher than those in WT mice, respectively. Similar trends were also noted at 24 h post LPS-exposure, where the Mfge8−/− mice showed 1.62- and 1.86-fold higher amounts of TNF-α mRNA and proteins than WT mice, respectively (P <0.05). Moreover, the TNF-α levels in the BALF collected from Mfge8−/− mice were significantly higher than those from WT mice at 4 and 24 h (Figure 1C). The histological images of the lung tissues at 24 h after LPS instillation represented increased alveolar congestion, exudates, interstitial and alveolar neutrophilic infiltrates, intraalveolar capillary hemorrhages, and extensive damage of their epithelial architecture in Mfge8−/− mice as compared to the WT counterpart (Figure 2A). These changes were reflected in a higher lung tissue injury score in Mfge8−/− mice than WT mice (10.9 vs. 7.6; P <0.05, Figure 2B).
Figure 1.

Assessment of TNF-α in lungs and BALF after ALI. WT and Mfge8−/− mice were subjected to i.t. injection of LPS (5 mg/kg). At 4 and 24 h after LPS instillation, lung tissues and BALF were collected. (A) The levels of TNF-α mRNA in lungs were determined by real-time PCR. Results are normalized by β-actin as an internal control and are expressed as fold induction in comparison to sham WT mice. The protein levels of TNF-α in (B) lungs and (C) BALF were determined by ELISA. Data are expressed as means ± SE (n=5 mice/group). *P < 0.05 vs. WT sham; #P < 0.05 vs. WT LPS.
Figure 2.

Histological assessment of lung tissue damage after ALI. WT and Mfge8−/− mice were subjected to i.t. injection of LPS (5 mg/kg). At 24 h after LPS instillation, lung tissues were fixed and stained with H&E. (A) Representative histological images at 100× and 400× (insert) magnification. Scale bar, 100 µm. (B) Tissue injury was scored based on the presence of exudates, hyperemia/congestion, neutrophilic infiltrates, intra-alveolar hemorrhage/debris, and cellular hyperplasia. Data are expressed as means ± SE (n=5 mice/group). *P < 0.05 vs. WT LPS.
MFG-E8 Deficiency Leads to Increased Neutrophil Infiltration to the Lungs
To further validate the neutrophil infiltration as observed in the histological analysis (Figure 2), we first carried-out qualitative assessment of MPO, a marker of infiltrating granulocytes in lung tissues by immunostaining, which revealed a stronger intensity of MPO staining in Mfge8−/− mice than that in WT mice (Figure 3A). After quantitation by MPO activity assay, we noticed its activity in Mfge8−/− mice was 1.85-fold higher than that in WT mice after 24 h of LPS instillation (Figure 3B). We next analyzed the cells isolated from BALF, which revealed no significant increase in their numbers in both WT and Mfge8−/− mice at the 4 h after LPS instillation (Figure 3C). However, at 24 h, the numbers of total cell counts from WT mice reached to 3.1 ± 0.27 × 106 cells, whereas it was 5.8 ± 0.45 × 106 cells in Mfge8−/− mice (P <0.05, Figure 3C). The major cell-type in BALF of sham mice was alveolar macrophages, while neutrophils were predominantly found in both WT and Mfge8−/− mice after ALI (Figure 3D). Consistent with the cytospin results, BALF isolated from WT and Mfge8−/− mice after 24 h of LPS instillation contained about 80% of neutrophils, as determined by flow cytometry (data not shown). The number of neutrophil counts in Mfge8−/− mice was 2.0-fold higher than that in WT mice at 24 h after LPS instillation (Figure 3E). Furthermore, we detected a significant increase in total protein levels in BALF, which emerged to be comparatively higher in Mfge8−/− mice than WT with 24 h of ALI (16.4 ± 0.86 vs. 11.6 ± 0.85 mg/ml; P <0.05, Figure 3F).
Figure 3.

Neutrophil infiltration to the lungs after ALI. WT and Mfge8−/− mice were injected via i.t with LPS (5 mg/kg). After LPS instillation, lung tissues and BALF were collected. (A) Representative images of lung tissues (24 h after LPS instillation) immuostained with MPO at 100× and 400× (insert) magnification. Scale bar, 100 µm. (B) MPO enzymatic activity in lungs was determined at 24 h after LPS instillation. (C) Total cell numbers in BALF isolated at 4 and 24 h after LPS instillation were counted by hemacytometer. (D) Representative cytospin images of BALF stained with Differential Quik. Yellow arrow, alveolar macrophages; white arrow, neutrophils. (E) The number of neutrophils in BALF isolated at 24 h after LPS instillation was determined by the ratio of cell population in cytospin. N.D., not detectable. (F) The protein content in BALF isolated at 24 h after LPS instillation was measured by Bio-Rad DC protein assay reagent. Data are expressed as means ± SE (n=5 mice/group). *P < 0.05 vs. WT sham; #P < 0.05 vs. WT LPS.
Accumulation of Apoptotic Cells in Lungs after ALI
MFG-E8 was initially identified as a factor for engulfment of apoptotic cells by professional phagocytes and its deficiency led to the development of autoimmune disease (16, 27). In this study, we carried-out TUNEL assay in lung tissues which revealed significant increase in the number of apoptotic cells in Mfge8−/− mice as compared to the WT mice after ALI (Figures 4A and B), reflecting inadequate clearance of apoptotic cells by the phagocytes. Besides this, following ALI, we further noticed increased activation of caspase-3 in the lungs of Mfge8−/− mice, which led to increased occurrence of cellular apoptosis in Mfge8−/− mice than that of WT counterpart (Figure 4C). Collectively, these findings demonstrated that the higher amounts of apoptotic cells in the lungs of Mfge8−/− mice were due to the reduced phagocytosis and/or increased rate of apoptosis mediated by the activation of caspase-3.
Figure 4.

Infiltration of apoptotic cells into the lungs after ALI. LPS (5 mg/kg)-induced experimental ALI was generated in WT and Mfge8−/− mice, and then at 24 h post LPS exposure lung tissues were harvested. (A) Formalin fixed and paraffin embedded lung tissue sections were stained with FITC-TUNEL (green), and counterstained with DAPI (blue). Representative images at 100× original magnifications are shown. Scale bar, 100 µm. (B) Green fluorescent TUNEL positive apoptotic cells were counted at five random fields in a blinded fashion, and the average numbers of cells per field are shown. (C) Lung tissue homogenates were used for caspase-3 fluorometric assay using Ac-DEVD-AMC as substrate. The results were generated by plotting the sample values to a multi-point standard curve. Data are expressed as means ± SE (n=3). *P < 0.05 vs. WT sham; #P < 0.05 vs. WT LPS.
Supplement of rmMFG-E8 Attenuates Neutrophil Infiltration to the Lungs during ALI
At 4 h after LPS instillation, MFG-E8 mRNA and protein levels in the lungs decreased by 42% and 57%, respectively, compared to the WT sham controls (Figures 5A and B). Although at 24 h, MFG-E8 expression was rebounded, it was still significantly lower than the WT sham controls (Figures 5A and B). Considering this fact, we aimed to determine the effect of rmMFG-E8 pre-treatment to WT mice prior to induction of ALI; as a therapeutic regimen to salvage the deficits of endogenous MFG-E8 which occurs during ALI. Interestingly, the number of total cells and neutrophil counts in BALF of rmMFG-E8 pre-treated WT mice were significantly lower than those in WT mice without rmMFG-E8 treatment after LPS instillation (Figures 6A and B). Based on this finding, we proposed that the excess in neutrophil infiltration into the lungs was due to decreased production of endogenous MFG-E8 during ALI, which could be ameliorated by exogenous treatment of rmMFG-E8.
Figure 5.

Endogenous expression of MFG-E8 in lungs following ALI. WT mice were injected with LPS (5 mg/kg) i.t., and then after 4 and 24 h, lung tissues were collected. (A) Total RNA isolated from lung tissues was subjected to real-time PCR to determine MFG-E8 mRNA levels. (B) Total protein extracts isolated from lung tissues were subjected to Western blotting to determine MFG-E8 protein levels. Results are normalized by β-actin as an internal control and are expressed as fold induction in comparison to sham. Data are expressed as means ± SE (n=3 mice/group). *P < 0.05 vs. sham.
Figure 6.

Effects of rmMFG-E8 administration on neutrophil infiltration in lungs after ALI. WT mice were i.p. injected with rmMFG-E8 (20 µg/kg) 2 h prior to LPS (5 mg/kg) instillation. At 24 h after LPS instillation, BALF were collected. (A) Total cell numbers in the BALF were counted by hemacytometer. (B) The number of neutrophils in the BALF was determined by the ratio of cell population in cytospin. Data are expressed as means ± SE (n=5 mice/group). *P < 0.05 vs. sham; #P < 0.05 vs. LPS. N.D., not detectable.
Exaggerated Production of MIP-2 and TNF-α in Lungs, BALF and Alveolar Macrophages of Mfge8−/− Mice during Inflammation
After observing the excessive neutrophil infiltration in Mfge8−/− mice, we then examined the levels of MIP-2, a critical chemokine responsible for neutrophil chemotaxis. MIP-2 mRNA and protein levels in Mfge8−/− mice were 2.2- and 2.1-fold higher than those in WT mice at 4 h, respectively, and 2.4- and 2.3-fold at 24 h, respectively (P <0.05; Figure 7A and B). Similarly, the MIP-2 levels in BALF from Mfge8−/− mice were significantly higher than those from WT mice at 4 and 24 h (Figure 7C). Since macrophages are one of the major cell types for producing chemokines and cytokines, we therefore, harvested alveolar macrophages from BALF of sham mice and assessed MIP-2 and TNF-α levels in LPS-treated conditions. After 4 h exposure to LPS, the MIP-2 mRNA and protein levels were increased significantly in alveolar macrophages from Mfge8−/− mice with 1.8- and 1.9-fold higher than those from WT mice (P <0.05; Figures 7D and E). Similarly, TNF-α mRNA and protein levels in alveolar macrophages from Mfge8−/− mice were also significantly higher than WT mice after LPS stimulation (Figures 7F and G).
Figure 7.

Effects of LPS on MIP-2 and TNF-α expression in lungs, BALF and alveolar macrophages. (A-C) WT and Mfge8−/− mice were subjected to i.t. injection of LPS (5 mg/kg). At 4 and 24 h after LPS instillation, lung tissues and BALF were collected. (A) The levels of MIP-2 mRNA in lungs were determined by real-time PCR. Results are normalized by β-actin as an internal control and are expressed as fold induction in comparison to sham WT mice. The protein levels of MIP-2 in (B) lungs and (C) BALF were determined by ELISA. Data are expressed as means ± SE (n=5 mice/group). *P < 0.05 vs. WT sham; #P < 0.05 vs. WT LPS. (D-G) Alveolar macrophages isolated from normal WT and Mfge8−/− mice were cultured and stimulated with LPS (10 ng/ml). After 4 h, total RNA isolated from cells and culture supernatants were used to determine (D, E) MIP-2 and (F, G) TNF-α mRNA and extracellular protein levels by real-time PCR and ELISA, respectively. Data are expressed as means ± SE (n=5 independent experiments). *P < 0.05 vs. WT control; #P < 0.05 vs. WT + LPS.
MFG-E8 Inhibits Neutrophil Migration through Regulating CXCR2 Expression
We further examined whether MFG-E8 had a direct effect on the neutrophil migration by isolating neutrophils from bone marrow of WT and Mfge8−/− mice. The number of migrated neutrophils of Mfge8−/− mice was 1.7-fold higher than that of WT mice (Figure 8A). CXCR2 is the putative receptor expressed on neutrophils for MIP-2-dependent chemotaxis (10). By using flow cytometric analysis, we observed that CXCR2 surface levels of MFG-E8-deficient neutrophils (CD11b+Ly6G+) were 30% higher than those of neutrophils from WT mice (Figure 8B). To validate the role of MFG-E8 in regulating neutrophil migration, BMDN were treated with rmMFG-E8 before applying to migration assay. As shown in Figure 8C, the number of migrated cells in rmMFG-E8-treated BMDN was reduced by 40%, compared to vehicle-treated BMDN. Furthermore, co-treatment of anti-MFG-E8 neutralizing antibodies with rmMFG-E8 abrogated the functions of rmMFG-E8 for reducing the migration of BMDN, hence become comparable to the vehicle control (Figure 8C). Correspondingly, the rmMFG-E8-treated BMDN had a lower CXCR2 expression compared to the vehicle control and the reduction of CXCR2 expression was rescued by co-treatment of anti-MFG-E8 antibodies (Figure 8D). Intracellular GRK2 is a major determinant for surface CXCR2 flip-flop in neutrophils, whose activation leads to desensitization of CXCR2, resulting in its intracellular translocation, and negative regulation of the neutrophil migration (29–31). GRK2 expression in BMDN was increased by 44% after rmMFG-E8 treatment, compared to the vehicle control, while co-treatment of anti-MFG-E8 antibodies diminished such induction of GRK2 expression (Figure 8E).
Figure 8.

Inhibition of BMDN migration by MFG-E8 is mediated through regulation of CXCR2 and GRK2. (A and B) BMDN were isolated from WT and Mfge8−/− mice. (A) BMDN were placed in the upper well of a modified Boyden chamber for migration assays. The media containing rmMIP-2 (1 ng/ml) was placed in the lower wells as chemotactic stimulus. After 2 h, migrated cells were fixed and stained with PI. Representative images of migrated BMDN are shown. Five random microscopic fields per well were counted. (B) CXCR2 expression in BMDN was analyzed by flow cytometry. Granulocytes were gated according to forward and side scatter dot plots, followed by identification of neutrophils with staining of FITC-CD11b and APC-Ly6G antibodies. Representative histograms of expression intensity of CXCR2 in CD11b+Ly6G+ cells stained with PerCP/Cy5.5-CXCR2 antibody are shown. The average of mean fluorescent intensities (MFI) for CXCR2 in BMDN is plotted. Data are expressed as means ± SE (n=5 mice/group). *P < 0.05 vs. WT. (C-E) BMDN isolated from WT mice were treated with isotype antibody (Ab) (1 µg/ml) + rmMFG-E8 (500 ng/ml) or anti-MFG-E8 Ab (1 µg/ml) + rmMFG-E8 (500 ng/ml) for 2 h before analysis. (C) BMDN with various treatments were subjected to migration assay. Representative images of migrated BMDN are shown. Five random microscopic fields per well were counted. (D) CXCR2 expression in BMDN with various treatments was analyzed by flow cytometry. (E) Intracellular GRK2 expression in BMDN with various treatments was analyzed by flow cytometry. Representative histograms of expression intensity of GRK2 in CD11b+Ly6G+ cells stained with PE-GRK2 antibody are shown. The average of MFI for GRK2 in BMDN is plotted. Data are expressed as means ± SE (n=5 independent experiments). *P < 0.05 vs. PBS control; #P < 0.05 vs. isotype Ab + rmMFG-E8.
MFG-E8 Interacts with αvβ3-Integrin for Inhibition of Neutrophil Migration
It has been well characterized that MFG-E8 can bind αvβ3-integrin in immune cells through its N-terminal domain (16). To examine the utilization of αvβ3-integrin in MFG-E8-mediated suppression of neutrophil migration, anti-αv-integrin neutralizing antibodies were applied for the study (32). As shown in Figure 9A, BMDN pre-treated with anti-αv-integrin antibodies abrogated the functions of MFG-E8-mediated inhibition of their migration. We further demonstrated that the pre-treatment of BMDN with anti-αv-integin antibodies blocked the effects of rmMFG-E8-mediated down-regulation of CXCR2 and up-regulation of GRK2 (Figures 9B and C). Collectively, these features clearly demonstrated the critical roles of MFG-E8 for GRK2-dependent down-regulation of surface CXCR2 expression in neutrophils through αvβ3-integrin-mediated pathway.
Figure 9.
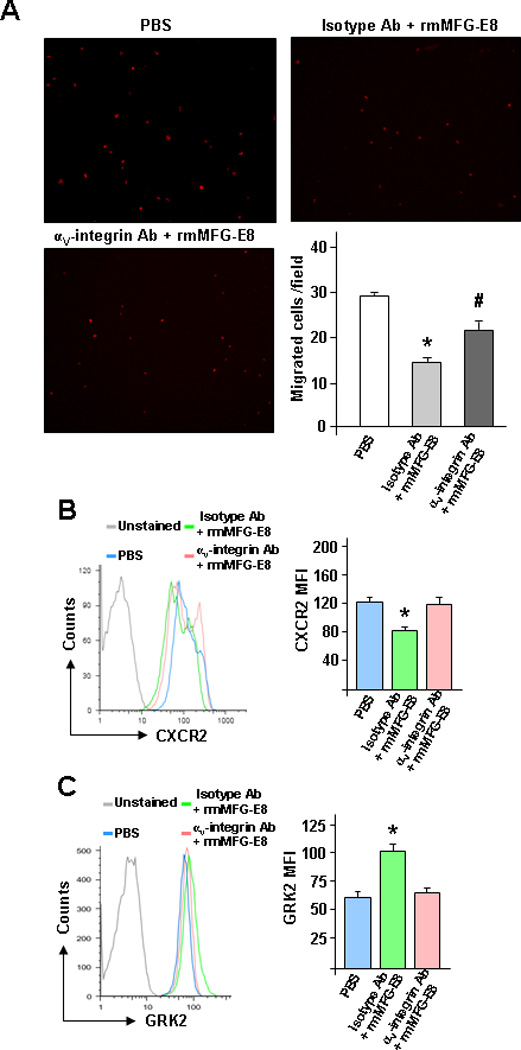
MFG-E8-mediated inhibition of BMDN migration is through αvβ3-integrin. BMDN isolated from WT mice were treated with isotype Ab (1 µg/ml) + rmMFG-E8 (500 ng/ml) or anti-αv-integrin Ab (1 µg/ml) + rmMFG-E8 (500 ng/ml) for 2 h before analysis. (A) BMDN with various treatments was subjected to migration assay. Representative images of migrated BMDN are shown. Five random microscopic fields per well were counted. (B) CXCR2 and (C) GRK2 expression in BMDN with various treatments was analyzed by flow cytometry. Representative histograms of expression intensity of CXCR2 and GRK2 in CD11b+Ly6G+ cells are shown. The averages of MFI for CXCR2 and GRK2 in BMDN are plotted. Data are expressed as means ± SE (n=5 independent experiments). *P < 0.05 vs. PBS control; #P < 0.05 vs. isotype Ab + rmMFG-E8.
DISCUSSION
In this current study, we demonstrated the novel mechanism of MFG-E8 for regulating the chemokine-mediated neutrophil migration in LPS-induced murine model of ALI, which revealed severe lung injury in MFG-E8 deficient mice than WT counterparts. This observation is consistent with our previous study showing the beneficial effects of MFG-E8 for attenuating lung injury induced by intestinal I/R (20). Herein, we noticed exaggerated infiltration of neutrophils into the lungs of Mfge8−/− mice, as confirmed by increased levels of MPO in interstitial tissues, and neutrophil numbers and protein contents in BALF in comparison to WT mice. We further identified the potential roles of MFG-E8 in inhibiting migration of neutrophils by comparing the BMDN isolated from WT and Mfge8−/− mice, and the effect of rmMFG-E8 administration by modulating the surface exposures of CXCR2 via GRK2 dependent pathways. Since, αvβ3-integrin is a putative receptor of MFG-E8 (16) we finally revealed the novel mechanism of MFG-E8 for regulating neutrophil migration through recognizing αvβ3-integrin.
We previously demonstrated the roles of MFG-E8 in promoting phagocytosis of apoptotic cells as one of the mechanisms of ameliorating inflammation in animal models of sepsis, renal, and gut I/R (18, 20, 24). We therefore monitored the well characterized function of MFG-E8 for engulfment of apoptotic cells to maintain the homeostatic balance, and observed increased number of apoptotic cells in lung tissues of Mfge8−/− mice after ALI (Figure 4). We further noticed significant induction of caspase-3 activity in Mfge8−/− mice as compared to the WT animals (Figure 4), suggesting the increased occurrences of apoptosis in lung tissues following ALI. However, exaggerated infiltration of neutrophils into the lungs is a hallmark property of ALI. The initial cascade of the development of ALI is the promotion of neutrophils into the lung tissues, which release inflammatory mediators, proteases, as well as ROS to cause inflammation and epithelial cell apoptosis mediated lung damage (33). Moreover, substantial research demonstrated that increased rate of epithelial cell death and decreased rate of activated neutrophil apoptosis in lungs are the two potentially important pathological mechanisms of ALI (33). Uncontrolled migration of neutrophils due to ALI deteriorates the disease status, while reducing their contents improves lung integrity. Hence, blocking the neutrophil migration to the lungs by MFG-E8 to attenuate ALI precedes phagocytosis of apoptotic cells. Correspondingly, we recently demonstrated reduced contents of MPO, a marker of granulocyte infiltration into the lung tissues of rmMFG-E8-treated animals with gut I/R injuries (20). Hence, delineating the direct effects of MFG-E8 on regulation of neutrophil migration could be the dominant mechanism for attenuating ALI.
The therapeutic strategy for ALI is attained by attenuating neutrophil chemotaxis towards the inflammatory sites. CXCR2 expressed on the surface of neutrophils functions as a sensor to lead neutrophils at the inflamed sites (7, 10). The significant role of CXCR2 in contributing to neutrophil infiltration to the inflamed lungs has been well demonstrated by using CXCR2 knockout mice (11). Inhibition or knockout of this chemokine receptor diminished neutrophil influx into the lungs and improved mortality associated with ALI (11, 34, 35). Considering the putative roles of CXCR2 for neutrophil migration, several selective CXCR2 inhibitors have been developed to treat various lung diseases caused by acute or chronic inflammation (36). In this study, we demonstrated that MFG-E8 can regulate surface expression of CXCR2 in neutrophils, which led to inhibition of neutrophil migration and subsequent infiltration to the lungs. Thus, MFG-E8 may serve as a potentially therapeutic agent for attenuating ALI.
We further elucidated the signaling pathway mediated by MFG-E8 for suppressing surface CXCR2 expression. Several studies have demonstrated that phosphorylation and internalization of CXCR2 is tightly controlled by GRK2 in leukocytes (37, 38). Activity of GRK2 is further regulated though its subcellular localization, kinase activity, and expression levels (15). Other than the chemokine receptors, under inflammatory conditions, GRK2 expression can be regulated through activation of Toll-like recptor-2 or -4 signaling (30, 31, 39). Likewise, our findings led to identify the MFG-E8-mediated up-regulation of GRK2 which can be correlated with reduction of surface CXCR2. Next, we focused on identifying the surface receptors that can transmit extracellular MFG-E8 signaling to regulate GRK2/CXCR2 expression in neutrophils. MFG-E8 has two functional parts: N-terminal EGF domains that bind to αvβ3-integrin of mostly hematopoietic cells, while the C-terminal discoidin domains can recognize the PtdSer exposed in apoptotic cells (16). Herein, we considered αvβ3-integrin as focal receptor for MFG-E8-mediated signal transduction. Structurally αvβ3-integrin is a heterodimeric transmembrane receptor formed by non-covalent association of α and β subunits (40, 41). Herein, we demonstrated that the blocking αv-integrin in neutrophils could effectively diminish the rmMFG-E8 effects on GRK2/CXCR2 expression, indicating the involvement of αv-integrin in mediating MFG-E8 activity. Furthermore, these findings add the integrin signaling pathway as another critical factor in controlling GRK2.
Our previous study has demonstrated that MFG-E8 can attenuate cytokine release from the peritoneal macrophages after LPS stimulation (25), which is consistent with the present study showing the higher levels of TNF-α expression and release in MFG-E8 deficient mice. In addition to demonstrating the roles of MFG-E8 in regulating neutrophil migration, we also observed that the induction of MIP-2 by LPS stimulation in the lungs and alveolar macrophages were augmented by the deficiency of MFG-E8. Since it has been previously established that the role of MFG-E8 in down-regulating the pro-inflammatory cytokines has been mediated by modulating the intra-cellular signaling cascade (22, 25), it is conceivable that MFG-E8 has an additional role in protecting the inflammatory consequences in ALI. Future studies are required for further confirmation.
In summary, we have identified another novel function of MFG-E8 in attenuating lung injury induced by LPS through inhibiting neutrophil infiltration to the lungs. The MFG-E8 effect is mediated by αvβ3-integrin to up-regulate GRK2 expression and results in down-regulation of surface CXCR2 levels in neutrophils, leading to decrease of neutrophil migration (Figure 10). With this regulatory function on neutrophils, MFG-E8 possesses as potentially therapeutic regimen for treating ALI.
Figure 10.

Hypothesis scheme. Intra-tracheal LPS exposure triggers MIP-2 expression in lungs, which serves as a potent chemoattractant for neutrophil migration into the lungs by recognizing CXCR2, causing widespread inflammation and tissue damage. In neutrophils, rmMFG-E8 recognizes αvβ3-integrin and then generates down-stream signaling for intracellular GRK2 activation, which ultimately down-regulates the surface CXCR2 expression, hence attenuating the MIP-2 dependent neutrophil migration during LPS-induced ALI.
Acknowledgements
We thank Mian Zhou, Weifeng Dong and Cletus Cheyuo for technical helps and valuable discussion.
This study was supported by the National Institutes of Health (NIH) grants, R01 GM 057468 and R33 AI 080536 (P.W.).
List of abbreviations
- MFG-E8
milk fat globule-epidermal growth factor factor 8
- ALI
acute lung injury
- ARDS
acute respiratory distress syndrome
- I/R
ischemia and reperfusion
- NF-κB
nuclear factor-κB
- MIP-2
macrophage inflammatory protein 2
- GPCR
G protein-coupled receptor
- GRK2
G protein-coupled receptor kinase 2
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- ROS
reactive oxygen species
Footnotes
Author Contributions
MA, AM, WLY, PW conceived and designed the experiments. MA, AM, WLY performed the experiments. MA, AM, WLY, AJ analyzed the data. MA, AM, WLY, AJ wrote the paper. PW supervised the whole project. All authors read and approved the final manuscript.
This work was presented partially as abstract at the 41st Critical Care Congress of the Society of Critical Care Medicine (SCCM) in Houston, Texas in 2012.
REFERENCES
- 1.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N. Engl. J. Med. 2005;353:1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 2.Ware LB. Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Semin. Respir. Crit. Care Med. 2006;27:337–349. doi: 10.1055/s-2006-948288. [DOI] [PubMed] [Google Scholar]
- 3.Ayala A, Chung CS, Lomas JL, Song GY, Doughty LA, Gregory SH, Cioffi WG, LeBlanc BW, Reichner J, Simms HH, Grutkoski PS. Shock-induced neutrophil mediated priming for acute lung injury in mice: divergent effects of TLR-4 and TLR-4/FasL deficiency. Am. J. Pathol. 2002;161:2283–2294. doi: 10.1016/S0002-9440(10)64504-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abraham E. Neutrophils and acute lung injury. Crit. Care Med. 2003;31:S195–S199. doi: 10.1097/01.CCM.0000057843.47705.E8. [DOI] [PubMed] [Google Scholar]
- 5.Lee WL, Downey GP. Neutrophil activation and acute lung injury. Curr. Opin. Crit. Care. 2001;7:1–7. doi: 10.1097/00075198-200102000-00001. [DOI] [PubMed] [Google Scholar]
- 6.Zarbock A, Ley K. Mechanisms and consequences of neutrophil interaction with the endothelium. Am. J. Pathol. 2008;172:1–7. doi: 10.2353/ajpath.2008.070502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol. Med. 2011;17:293–307. doi: 10.2119/molmed.2010.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donnelly SC, Strieter RM, Kunkel SL, Walz A, Robertson CR, Carter DC, Grant IS, Pollok AJ, Haslett C. Interleukin-8 and development of adult respiratory distress syndrome in at-risk patient groups. Lancet. 1993;341:643–647. doi: 10.1016/0140-6736(93)90416-e. [DOI] [PubMed] [Google Scholar]
- 9.Goodman RB, Strieter RM, Martin DP, Steinberg KP, Milberg JA, Maunder RJ, Kunkel SL, Walz A, Hudson LD, Martin TR. Inflammatory cytokines in patients with persistence of the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1996;154:602–611. doi: 10.1164/ajrccm.154.3.8810593. [DOI] [PubMed] [Google Scholar]
- 10.Olson TS, Ley K. Chemokines and chemokine receptors in leukocyte trafficking. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002;283:R7–R28. doi: 10.1152/ajpregu.00738.2001. [DOI] [PubMed] [Google Scholar]
- 11.Reutershan J, Morris MA, Burcin TL, Smith DF, Chang D, Saprito MS, Ley K. Critical role of endothelial CXCR2 in LPS-induced neutrophil migration into the lung. J. Clin. Invest. 2006;116:695–702. doi: 10.1172/JCI27009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belperio JA, Keane MP, Burdick MD, Londhe V, Xue YY, Li K, Phillips RJ, Strieter RM. Critical role for CXCR2 and CXCR2 ligands during the pathogenesis of ventilator-induced lung injury. J. Clin. Invest. 2002;110:1703–1716. doi: 10.1172/JCI15849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chuang TT, Iacovelli L, Sallese M, De Blasi A. G protein-coupled receptors: heterologous regulation of homologous desensitization and its implications. Trends. Pharmacol. Sci. 1996;17:416–421. doi: 10.1016/s0165-6147(96)10048-1. [DOI] [PubMed] [Google Scholar]
- 14.Aragay AM, Ruiz-Gómez A, Penela P, Sarnago S, Elorza A, Jiménez-Sainz MC, Mayor F., Jr G protein-coupled receptor kinase 2 (GRK2): mechanisms of regulation and physiological functions. FEBS Lett. 1998;430:37–40. doi: 10.1016/s0014-5793(98)00495-5. [DOI] [PubMed] [Google Scholar]
- 15.Penn RB, Pronin AN, Benovic JL. Regulation of G protein-coupled receptor kinases. Trends Cardiovasc. Med. 2000;10:81–89. doi: 10.1016/s1050-1738(00)00053-0. [DOI] [PubMed] [Google Scholar]
- 16.Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature. 2002;417:182–187. doi: 10.1038/417182a. [DOI] [PubMed] [Google Scholar]
- 17.Aziz M, Jacob A, Matsuda A, Wang P. Review: milk fat globule-EGF factor 8 expression, function and plausible signal transduction in resolving inflammation. Apoptosis. 2011;16:1077–1086. doi: 10.1007/s10495-011-0630-0. [DOI] [PubMed] [Google Scholar]
- 18.Matsuda A, Jacob A, Wu R, Zhou M, Nicastro JM, Coppa GF, Wang P. Milk fat globule-EGF factor VIII in sepsis and ischemia-reperfusion injury. Mol Med. 2011;17:126–133. doi: 10.2119/molmed.2010.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Atabai K, Jame S, Azhar N, Kuo A, Lam M, McKleroy W, Dehart G, Rahman S, Xia DD, Melton AC, Wolters P, Emson CL, Turner SM, Werb Z, Sheppard D. Mfge8 diminishes the severity of tissue fibrosis in mice by binding and targeting collagen for uptake by macrophages. J Clin Invest. 2009;119:3713–3722. doi: 10.1172/JCI40053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cui T, Miksa M, Wu R, Komura H, Zhou M, Dong W, Wang Z, Higuchi S, Chaung W, Blau SA, Marini CP, Ravikumar TS, Wang P. Milk fat globule epidermal growth factor 8 attenuates acute lung injury in mice after intestinal ischemia and reperfusion. Am. J. Respir. Crit. Care Med. 2010;181:238–246. doi: 10.1164/rccm.200804-625OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jinushi M, Nakazaki Y, Dougan M, Carrasco DR, Mihm M, Dranoff G. MFG-E8-mediated uptake of apoptotic cells by APCs links the pro- and antiinflammatory activities of GM-CSF. J Clin Invest. 2007;117:1902–1913. doi: 10.1172/JCI30966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miksa M, Wu R, Dong W, Das P, Yang D, Wang P. Dendritic cell-derived exosomes containing milk fat globule epidermal growth factor-factor VIII attenuate proinflammatory responses in sepsis. Shock. 2006;25:586–593. doi: 10.1097/01.shk.0000209533.22941.d0. [DOI] [PubMed] [Google Scholar]
- 23.Aziz MM, Ishihara S, Mishima Y, Oshima N, Moriyama I, Yuki T, Kadowaki Y, Rumi MA, Amano Y, Kinoshita Y. MFG-E8 attenuates intestinal inflammation in murine experimental colitis by modulating osteopontin-dependent alphavbeta3 integrin signaling. J. Immunol. 2009;82:7222–7232. doi: 10.4049/jimmunol.0803711. [DOI] [PubMed] [Google Scholar]
- 24.Matsuda A, Wu R, Jacob A, Komura H, Zhou M, Wang Z, Aziz MM, Wang P. Protective effect of milk fat globule-epidermal growth factor-factor VIII after renal ischemia-reperfusion injury in mice. Crit. Care Med. 2011;39:2039–2047. doi: 10.1097/CCM.0b013e3182227a3d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aziz M, Jacob A, Wu R, Matsuda A, Zhou M, Dong P, Wang W. MFG-E8 attenuates LPS-induced TNF-α production in macrophages via STAT3-mediated SOCS3 activation. PLoS ONE. 2011;6:e27685. doi: 10.1371/journal.pone.0027685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moon C, Han JR, Park HJ, Hah JS, Kang JL. Synthetic RGDS peptide attenuates lipopolysaccharide-induced pulmonary inflammation by inhibiting integrin signaled MAP kinase pathways. Respir Res. 2009;10:18. doi: 10.1186/1465-9921-10-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, Nagata S. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science. 2004;304:1147–1150. doi: 10.1126/science.1094359. [DOI] [PubMed] [Google Scholar]
- 28.Boxio R, Bossenmeyer-Pourié C, Steinckwich N, Dournon C, Nüsse O. Mouse bone marrow contains large numbers of functionally competent neutrophils. J. Leukoc. Biol. 2004;75:604–611. doi: 10.1189/jlb.0703340. [DOI] [PubMed] [Google Scholar]
- 29.Prado GN, Suzuki H, Wilkinson N, Cousins B, Navarro J. Role of the C terminus of the interleukin 8 receptor in signal transduction and internalization. J. Biol. Chem. 1996;271:19186–19190. doi: 10.1074/jbc.271.32.19186. [DOI] [PubMed] [Google Scholar]
- 30.Alves-Filho JC, Sônego F, Souto FO, Freitas A, Verri WA, Jr, Auxiliadora-Martins M, Basile-Filho A, McKenzie AN, Xu D, Cunha FQ, Liew FY. Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat. Med. 2010;16:708–712. doi: 10.1038/nm.2156. [DOI] [PubMed] [Google Scholar]
- 31.Alves-Filho JC, Freitas A, Souto FO, Spiller F, Paula-Neto H, Silva JS, Gazzinelli RT, Teixeira MM, Ferreira SH, Cunha FQ. Regulation of chemokine receptor by Toll-like receptor 2 is critical to neutrophil migration and resistance to polymicrobial sepsis. Proc. Natl. Acad. Sci. U. S. A. 2009;106:4018–4023. doi: 10.1073/pnas.0900196106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Motegi S, Garfield S, Feng X, Sárdy M, Udey MC. Potentiation of platelet-derived growth factor receptor-β signaling mediated by integrin-associated MFG-E8. Arterioscler. Thromb. Vasc. Biol. 2011;31:2653–2664. doi: 10.1161/ATVBAHA.111.233619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perl M, Lomas-Neira J, Chung CS, Ayala A. Epithelial cell apoptosis and neutrophil recruitment in acute lung injury-a unifying hypothesis? What we have learned from small interfering RNAs. Mol Med. 2008;14:465–475. doi: 10.2119/2008-00011.Perl. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zarbock A, Allegretti M, Ley K. Therapeutic inhibition of CXCR2 by Reparixin attenuates acute lung injury in mice. Br. J. Pharmacol. 2008;155:357–364. doi: 10.1038/bjp.2008.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao X, Town JR, Yang A, Zhang X, Paur N, Sawicki G, Gordon JR. A novel ELR-CXC chemokine antagonist reduces intestinal ischemia reperfusion-induced mortality, and local and remote organ injury. J. Surg. Res. 2010;162:264–273. doi: 10.1016/j.jss.2009.04.047. [DOI] [PubMed] [Google Scholar]
- 36.Chapman RW, Phillips JE, Hipkin RW, Curran AK, Lundell D, Fine JS. CXCR2 antagonists for the treatment of pulmonary disease. Pharmacol. Ther. 2009;121:55–68. doi: 10.1016/j.pharmthera.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 37.Aragay AM, Mellado M, Frade JM, Martin AM, Jimenez-Sainz MC, Martinez-A C, Mayor F., Jr Monocyte chemoattractant protein-1-induced CCR2B receptor desensitization mediated by the G protein-coupled receptor kinase 2. Proc. Natl. Acad. Sci. U. S. A. 1998;95:2985–2990. doi: 10.1073/pnas.95.6.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oppermann M, Mack M, Proudfoot AE, Olbrich H. Differential effects of CC chemokines on CC chemokine receptor 5 (CCR5) phosphorylation and identification of phosphorylation sites on the CCR5 carboxyl terminus. J. Biol. Chem. 1999;274:8875–8885. doi: 10.1074/jbc.274.13.8875. [DOI] [PubMed] [Google Scholar]
- 39.Fan J, Malik AB. Toll-like receptor-4 (TLR4) signaling augments chemokine-induced neutrophil migration by modulating cell surface expression of chemokine receptors. Nat. Med. 2003;9:315–321. doi: 10.1038/nm832. [DOI] [PubMed] [Google Scholar]
- 40.Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science. 1994;264:569–571. doi: 10.1126/science.7512751. [DOI] [PubMed] [Google Scholar]
- 41.Wilder RL. Integrin alpha V beta 3 as a target for treatment of rheumatoid arthritis and related rheumatic diseases. Ann. Rheum. Dis. 2002;61:ii96–ii99. doi: 10.1136/ard.61.suppl_2.ii96. [DOI] [PMC free article] [PubMed] [Google Scholar]