

Abstract
A rapid, selective, and sensitive liquid chromatography-tandem mass spectrometry method was developed and validated for the simultaneous determination of unbound sunitinib and its active metabolite N-desethyl sunitinib in plasma. Plasma and post-dialysis buffer samples were extracted using a liquid-liquid extraction procedure with acetonitrile/n-butylchloride (1:4, v/v). Chromatographic separation was achieved on a Waters X-Terra® MS RP18 column with a mobile phase consisting of acetonitrile and water (60:40, v/v) containing formic acid (0.1 %, v/v) using an isocratic run, at a flow rate of 0.2 mL/min. Analytes were detected by electrospray tandem mass spectrometry in the selective reaction monitoring mode. Linear calibration curves were generated over the range of 0.1–100 ng/mL and 0.02–5 ng/mL for sunitinib and 0.2–200 ng/mL and 0.04–10 ng/mL for N-desethyl sunitinib in plasma and in phosphate buffered solution, respectively. The values for both within day and between day precision and accuracy were well within the generally accepted criteria for analytical methods. The analytical range was sufficient to determine the unbound and total concentrations of both analytes. The method was applied for measurement unbound concentrations in addition to total concentrations of sunitinib and its metabolite in plasma of a cancer patient receiving 50 mg daily dose.
Keywords: Protein binding, tyrosine kinase inhibitor, equilibrium dialysis, LC/MS/MS, pharmacokinetics
1. Introduction
Sunitinib is an oral, small molecule tyrosine kinase inhibitor with potent antitumor and antiangiogenic activity. It was approved by US FDA for first-line treatment of metastatic renal cell carcinomas (RCC) and also for gastrointestinal stromal tumors (GIST) after disease progression (Arora et al., 2005). Sunitinib represents an important treatment option for patients with cytokine–refractory RCC, which are primarily treated with cytokines with limited effectiveness and tolerability issues (Atkins et al., 2006, Adams et al., 2007). Sunitinib inhibits tumor cell proliferation and angiogenesis by inhibiting multiple receptor tyrosine kinase targets such as platelet-derived growth factor (PDGF-Rs) and vascular endothelial growth factor receptors (VEGFRs) (Atkins et al., 2006, Patel et al., 2003, Sun et al., 2003). Due to poor pharmacological properties demonstrated by other tyrosine kinase inhibitors, sunitinib was rationally designed to obtain better pharmacokinetic as well as pharmacodynamic properties demonstrating high oral bioavailability and nanomolar range potency against the receptor tyrosine kinases (Adams et al., 2007, Chow et al., 2007).
Sunitinib is being evaluated in multiple clinical trials including one to determine the safety and to investigate the pharmacological interactions in HIV+ cancer patients on highly active antiretroviral therapy (HAART). Sunitinib is metabolized primarily by CYP3A4 (Schwandt et al., 2009) to an N-desethyl metabolite (SU12662) which exhibits comparable biological activity and potency to sunitinib (Adams et al., 2007). Coadministration of sunitinib with a potent CYP3A4 inhibitor (e.g., ketoconazole) or inducer (e.g., rifampin) has altered the combined area under the concentration time curve (AUC) of sunitinib and the active metabolite in healthy volunteers (Goodman et al., 2007) Sunitinib and its active metabolite extensively bind to circulating plasma proteins (95 and 90%, respectively) and their pharmacological activity is dependent on the concentration of unbound drug (Goodman et al., 2007). Since HAART regimens combine multiple antiretroviral drugs, which are potent inducers and inhibitors of CYP3A4, (Seden et al., 2009) their concomitant administration with sunitinib, may alter its metabolic fate. Additionally co-administration of multiple drugs may result in a drug-drug interaction leading to displacement of sunitinib or its metabolite from the protein binding site. This may cause alterations in available unbound concentrations of the drug leading to clinical consequences. To date, no published reports have addressed the quantitation of the free fraction of sunitinib and its active metabolite in plasma.
Several methodologies have been reported in literature for determination of unbound drug concentrations in plasma include ultrafiltration (Whitlam et al., 1981), ultracentrifugation (Barre et al., 1985), equilibrium dialysis (Jolliet-Riant et al., 1998, Brouwer et al., 2000), high performance frontal analysis (Shibukawa et al., 1999), and capillary electrophoresis (Shibukawa et al., 1994). Each of these methodologies offer several advantages and disadvantages, of which equilibrium dialysis is preferred due to its simplicity as well as its suitability to high throughput assays. Additionally, drug adsorption concerns with this method are minimal provided equilibrium is allowed to be attained and drug concentrations on both sides of the membrane are measurable (Sillen et al., 2011).
Previously published methods involved HPLC/UV (Etienne-Grimaldi et al., 2009), or LC/MS/MS (Minkin et al., 2008, de Bruijn et al., 2010, Rodamer et al., 2011) for detection of sunitinib alone or with the active metabolite. These methods involved detection of the total concentrations of the drug and/or the metabolite, and lacked the ability of reliably detecting the unbound fraction, which has been the main focus of the present study. Minkin and colleagues reported the determination of sunitinib over a wide range of 0.2–500 ng/mL, but lacked the determination of active metabolite and employed a non deutrated internal standard (Minkin et al., 2008). de Bruijn and colleagues quantified both the active and the metabolite total concentrations yet their calibration curve range is narrower (0.2–50 ng/mL) than that reported here (de Bruijn et al., 2010). Rodamer and colleagues employed a simple one step method and have provided a great summary of methods published previously but none of those methods report analysis of unbound fraction (Rodamer et al., 2011). For determination of unbound fraction, both aliquots of plasma (total concentration) and the dialysate need to be analyzed. Accordingly, a method with better sensitivity of the quantification method along with a wide dynamic analytical range is required for detection of both the parent and the metabolite in total and unbound forms to comprehensively characterize the clinical pharmacokinetic profile of this drug in a drug interaction study, and to explore the relationship with pharmacodynamic effects. The method described achieves improved analytical methodology and a wider dynamic range suitable for drug interaction studies, in addition to being able to detect unbound drug concentrations.
2. Experimental
2.1 Chemicals and reagents
Sunitinib Malate (Lot number 5-ANR-67-1, 73.0% pure by HPLC, free drug), N-desethyl sunitinib (Lot number 1-WHH-36-3, 98.0% pure by HPLC) and the deutrated internal standard, sunitinib-d10 (Lot number 3-THT-158-3), were obtained from Toronto Research Chemicals Inc. (North York, ON, Canada). Formic acid (98%, v/v in water), and acetonitrile (HPLC grade) were obtained from EMD Chemicals Inc. (Gibbstown, NJ, USA). n-Butyl Chloride, was obtained from Burdick & Jackson, (Muckegon, MI, USA). Phosphate Buffer Solution (PBS) 1X (pH 7.4, without Ca and Mg), was obtained from BioSource, (Rockville, MD, USA). Human serum albumin (HSA) and human α1-acid glycoprotein (AAG) were obtained from Sigma-Aldrich Company (St. Louis, Missouri, USA). Deionized water was obtained from a Milli-Q-UF system (Millipore, Milford, MA, USA) and used for all aqueous solutions. Drug-free (blank) EDTA plasma was obtained from Biological Specialty Corp. (Colmar, PA, USA).
2.2 Stock solutions
Stock solutions of sunitinib as well as N-desethyl sunitinib were prepared in duplicates to get a final concentration of 0.73 mg/mL for sunitinib and 1 mg/mL for N-desethyl sunitinib in acetonitrile/water (1:1, v/v). The area counts of the stock solutions were quantified in quintuplicate. If the mean values for the area counts were within 5%, the stock solutions were stored as aliquots in amber colored vials, protected from light, at −20°C until used.
2.3 Preparation of calibration standards and quality controls
Calibration standards of both sunitinib and N-desethyl sunitinib were prepared freshly on each day of analysis. To determine both total and unbound fractions, two sets of calibration standards were prepared for sunitinib and metabolite in human plasma and in PBS. For total drug, stock solutions for each analyte were diluted in acetonitrile/water (1:1, v/v) were spiked in blank human EDTA plasma. The dilutions were then used to prepare eight calibration standards containing sunitinib at concentrations of 0.1, 0.2, 0.5, 1, 5, 10, 50, and 100 ng/mL, and N-desethyl sunitinib at 0.2, 0.4, 1, 2, 10, 20, 100, and 200 ng/mL, in duplicates. Quality control (QC) samples were prepared independently in blank EDTA plasma at five different concentrations for each of the analyte including the lower limit of quantitation (LLOQ), the low QC (LQC), the medium QC (MQC), the high QC (HQC), and the above upper limits of quantitation (AULQ) QC which was 0.1, 0.3, 8, 80, and 800 ng/mL for sunitinib and 0.2,0.6, 16, 160, and 1600 ng/mL for N-desethyl sunitinib. The AULQ QC was diluted 1:10 (v/v) in plasma prior to extraction. For the determination of unbound drug, the calibration standards were prepared in PBS for sunitinib at concentrations of 0.02, 0.05, 0.1, 0.5, 1, and 5 ng/mL and at 0.04, 0.1, 0.2, 1, 2, and 10 ng/mL for N-desethyl sunitinib. Quality control (QC) samples were prepared at the LLOQ, LQC, MQC, and HQC which were 0.02, 0.06, 0.4, and 4 ng/mL for sunitinib and 0.04, 0.12, 0.8, and 8 ng/mL for N-desethyl sunitinib. For long-term and freeze-thaw stability, QC samples were prepared as a batch and stored at −70°C.
2.4 Sample preparation
Frozen samples were thawed in a water bath at ambient temperature prior to extraction. A 50 μL aliquot of plasma (for total drug) and a 100 μL aliquot of PBS (for unbound drug) was added to a borosilicate glass test tube (13×100 mm) to which 2 mL of the extraction solution acetonitrile/n-butylchloride (1:4, v/v) spiked with sunitinib-d10 (0.2 ng/mL) as internal standard (IS) was added to the tube; except for the blank matrix sample, where extraction solution without IS was used. The tube was mixed vigorously for 30 seconds on a vortex-mixer, followed by centrifugation at 913 × g for 10 minutes at ambient temperature. A volume of 1.5 mL of the top layer was transferred to a disposable borosilicate glass culture tube (13×100 mm) and evaporated to dryness under nitrogen at 35±5°C. The residue was reconstituted in 100 μL of acetonitrile: water (60:40, v/v) by vortex mixing for 30 seconds. The sample was transferred to a 300μl polypropylene plastic screw-cap vial with bonded pre-slit PTFE/Silicone septa. A 10 μL volume was injected into the LC/MS/MS instrument using an auto-sampling device operating at 10±5°C.
2.5 Chromatographic and mass-spectroscopic conditions
Chromatographic analysis was performed using Waters Acquity UPLC system (Waters Corporation, Milford, MA, USA). Separation of the analyte from potentially interfering material was achieved at ambient temperature using Waters X-Terra® MS RP18 column (150 × 2.1mm i.d.) packed with a 3.5 μm C18 stationary phase, protected by a Waters X-Terra® MS guard column (20 × 2.1 mm i.d.) packed with 3.5 μm RP18 material (Milford, MA, USA). The mobile phase used for the chromatographic separation was composed of acetonitrile: water (60:40, v/v) containing 0.1% formic acid, and was delivered isocratically at a flow rate of 0.2 mL/min with a total run time of 5 min. The column effluent was monitored using an AB Sciex triple quadrapole ™ 5500 mass-spectrometric detector (Applied Biosystems, Foster City, CA, USA). The instrument was equipped with an electrospray interface in positive ion mode, and controlled by the Analyst version 1.2 software (Applied Biosystems). Samples were introduced to the interface through a Turbo Ion Spray with the temperature setting at 450°C. A high positive voltage of 5.5 kV was applied to the ion spray. Nitrogen was used as the nebulizer gas, curtain gas, and collision gas with the settings of 30, 40 and 7, respectively. Other optimal parameters including declustering potential (DP), entrance potential (EP), collision energy (CE), and collision cell exit potential (CXP) are reported in Table 1. The spectrometer was programmed to allow the ions of sunitinib at m/z 399.0, N-desethyl sunitinib at m/z 371.2, and sunitinib-d10 at m/z 409.1 to pass through the first quadrupole (Q1) and into the collision cell (Q2). The major fragments observed for sunitinib (m/z 283.2), N-desethyl sunitinib (m/z 283.2), and sunitinib-d10 at (m/z 283.2), were monitored in the third quadrupole (Q3).
Table 1.
Optimization parameters for sunitinib, N-desethyl sunitinib and sunitinib-d10
| DP (V) | EP (V) | CE (eV) | CXP (V) | |
|---|---|---|---|---|
| Sunitinib | 231 | 10 | 33 | 28 |
| N-Desethyl Sunitinib | 186 | 10 | 27 | 26 |
| Sunitinib-d10 | 231 | 10 | 33 | 28 |
2.6 Calibration curves
Calibration curves for sunitinib were computed using the ratio of the peak area of analyte to the internal standard by using a least-squares linear regression analysis with 1/[nominal concentration] weight. For N-desethyl sunitinib, the calibration curve was computed using quadratic regression with 1/[nominal concentration]2 weight. The parameters of each calibration curve were used to back-calculate concentrations and to obtain values for the QC samples and unknown samples by interpolation.
2.7 Method validation
2.7.1 Specificity
The method specificity was tested using visual inspection of extracted human plasma, from six different healthy donors for the presence of endogenous or exogenous interfering peaks. The interfering peak area needed to be less than 20% of the peak area for sunitinib as well as N-desethyl sunitinib at the lower limit of quantitation in plasma.
2.7.2. Calibration curves and quality controls
Method validation runs for calibration standards and QCs were performed on three consecutive days. This included a calibration curve processed in duplicate, and QC samples at five different concentrations in quintuplicate; a single blank plasma and zero-level standard (blank with internal standard). Estimates of the between-run precision were obtained by one-way analysis of variance (ANOVA) as previously described (Rosing et al., 2000). The extraction efficiency of the assay was measured by comparison of the peak area ratio of sunitinib and N-desethyl sunitinib extracted from plasma and an aqueous solution in triplicate at concentrations of the low, medium, and high QCs.
The stability of sunitinib and N-desethyl sunitinib in plasma was tested at concentrations of the low and high QCs and AULQ QC (1:10 dilution), in triplicate after 3 freeze-thaw cycles. The short-term stability of sunitinib and the metabolite in plasma was assessed in triplicate at room temperature (on the bench-top) to 6.0 hrs. The long-term stability of sunitinib and N-desethyl sunitinib in plasma at −70°C, and stock at −20°C were assessed at 3 month intervals. Stability of drug in neutral extracts was assessed on the autosampler.
2.8 Patient samples analysis for total and unbound concentrations
The patient participated in a phase I study and received a 50 mg dose of sunitinib administered orally, once daily for 4 weeks every 6 weeks. The protocol AMC061 (ClinicalTrials.gov Identifier NCT00890747) was approved by the Institutional Review Board at the lead institution Georgetown University Medical Center (Washington, DC, USA) as well as other AIDS Malignancy Consortium sites. The patient was provided written informed consent. Blood samples were collected in EDTA tubes at baseline (pre-dose), 1, 2, 3, 4, 5, 6, 7, 8, and 24 hr, following administration of a 50 mg dose. Samples were processed immediately by refrigerated (4°C) centrifugation for 10 minutes at 1800 × g. The resultant plasma was stored at −70°C until analysis.
The total concentrations of sunitinib and N-desethyl sunitinib in patient plasma samples were determined using the validated method described above. The unbound fraction of the drug and the metabolite were obtained using equilibrium dialysis on a 96-Well Equilibrium DIALYZER™ with a 5-KDa cut-off regenerated cellulose membrane (Harvard Apparatus Holliston, MA). Preliminary experiments were performed to optimize the equilibrium dialysis with equilibration achieved by 24 hrs and concentration independent binding observed (data not shown). Briefly, equilibrium dialysis was conducted with a 200 μL aliquot of patient plasma against an equal volume of 1X PBS (pH 7.4, without Ca and Mg), by incubating for 24 hrs at 37°C and 5.0% CO2. The degree of sunitinib protein binding was assessed in isolated protein solutions containing either human serum albumin or human α1-acid glycoprotein as described above. The unbound concentration of sunitinib and N-desethyl sunitinib in the post-dialysis buffer solution was determined using the developed LC/MS/MS method. The pharmacokinetic parameters for the total and unbound sunitinib and N-desethyl sunitinib were estimated using noncompartmental analysis as implemented in WinNonlin (Version 5.3, Pharsight Corp., Mountain View, California). Maximum plasma concentration (Cmax) was the observed value, as was the time to Cmax (Tmax). Area under the concentration-time curve (AUC) was calculated using the log/linear trapezoidal rule through the dosing interval (τ). Unbound fraction (fu) was calculated as fu=Cu/Cp × 100%.
3. Results and Discussion
3.1 Detection and chromatography
The mass spectrums of sunitinib and N-desethyl sunitinib showed protonated molecular ions [MH+] at m/z 399.01 and m/z 371.2 respectively (fig. 1). The major fragments selected for subsequent monitoring in the third quadrapole for sunitinib as well as N-desethyl sunitinib was at m/z 283.2. The mass spectrum of the internal standard, Sunitinib-d10 showed a protonated ion [MH+] at m/z 409.1 and the most abundant product ion monitored at the third quadrapole was 283.2 (fig. 1).
Fig. 1.

The mass spectra of daughter scan for (a) sunitinib at m/z 399 → 283.2 (b) N-desethyl sunitinib at m/z 371.2 → 283.2, and (c) sunitinib-d10 m/z 409.1→283.2. Asterisks represent the deuterium atoms in the deutrated internal standard.
3.2 Linearity of detector responses
Calibration curves for sunitinib standards were constructed from the peak area ratio of the analyte to the internal standard using linear regression with a weighting factor of 1/[nominal concentration] over the range of 0.1–100 ng/mL in plasma and 0.02–5 ng/mL in PBS. Linear regression of the back-calculated concentrations versus the nominal values provided a unit slope and an intercept not significantly different from zero. For sunitinib plasma samples, the mean ± standard deviation of the equation was 0.0025 ± 0.0001 for the intercept and 0.0436 ± 0.0039 for the slope with a correlation coefficient 0.9994±0.0002. For PBS samples, the mean ± standard deviation was −0.0007 ± 0.0014 for the intercept and 2.30E-04± 9.07E-06 for the slope, with a correlation coefficient 0.998±0.001. For N-desethyl sunitinib, standards were similarly constructed using a quadratic standard curve over the range of 0.2–200 ng/mL in plasma or 0.04–10 ng/ml in PBS with a weighting factor (1/[nominal concentration]2) calibration curves. For the plasma samples, the equation for N-desethyl sunitinib was 0.0012 ± 0.0004 for the a0 parameter, 0.0309 ± 0.003 for the a1 parameter, 7.50E-06± 1.52E-05 for the a2 parameter with a correlation coefficient 0.997±0.002. For PBS samples, the equation for N-desethyl sunitinib was −5.72E-04± 2.31E-04 for the a0 parameter, 9.84E-05± 4.08E-06 for the a1 parameter, 8.59E-10± 9.11E-10 for the a2 parameter with a correlation coefficient 0.997±0.002.
In plasma, for each point on the calibration curves for the analytes, the concentrations back-calculated from the equation of the regression analysis were always within 3.3% for sunitinib and 2.4% for N-desethyl sunitinib of the nominal value (Table 2). The LLOQ for sunitinib was established at 0.1 ng/mL, and for N-desethyl sunitinib at 0.2 ng/mL which was associated with a mean (± standard deviation) signal-to-noise ratio for 15 observations of 68.9±20.8 and 35.6±12.0 respectively. The relative standarad deviation (RSD%) serves as a measure of precision, whereas percent deviation from the nominal concentration (bias%) serves as a measure of accuracy. As shown in the table 2, the between day RSD% and bias ranged from 1.67 to 9.47% and −3.33 to 1.43 % respectively for sunitinib and 3.55 to 7.34% and −2.4 to 1.68 % respectively for N-desethyl sunitinib in plasma. In PBS, the concentrations back-calculated from the equation of the regression analysis were always within 8.5% for sunitinib and 6.6% for N-desethyl sunitinib, of the nominal value (Table 3). The between day RSD% and bias ranged from 3.2 to 8.15 % and −7.87 to 8.52 % respectively for sunitinib and 1.06 to 8.04 % and −6.63 to 6.33 % respectively for N-desethyl sunitinib in PBS. The LLOQ for sunitinib was established at 0.02 ng/mL, and for N-desethyl sunitinib at 0.04 ng/mL, associated with a mean (± standard deviation) and signal-to-noise ratio for 15 observations of 37.8±11.2 and 23.1±13.8 respectively.
Table 2.
Back-calculated concentrations from calibration curves for sunitinib (0.1–100 ng/mL) and N-desethyl sunitinib (0.2–200 ng/mL) in plasmaa
| Nominal Concentration (ng/mL) | Calculated Concentration (ng/mL)b | Accuracy (%) | Bias | Precision (%)
|
RSD% Between Run | |
|---|---|---|---|---|---|---|
| Within Run | Between Run | |||||
| Sunitinib | ||||||
| 0.1 | 0.10 ± 0.01 | 100.98 | 0.98 | 11.8 | -c | 9.47 |
| 0.2 | 0.20 ± 0.02 | 97.83 | −2.17 | 9.56 | -c | 8.07 |
| 0.5 | 0.48 ± 0.02 | 96.67 | −3.33 | 5.66 | -c | 4.51 |
| 1 | 1.03 ± 0.06 | 102.87 | 2.87 | 5.86 | -c | 5.76 |
| 5 | 5.00 ± 0.17 | 100.00 | 0 | 3.45 | -c | 3.41 |
| 10 | 10.1 ± 0.2 | 101.08 | 1.08 | 1.68 | -c | 1.67 |
| 50 | 50.7 ± 2.5 | 101.43 | 1.43 | 3.69 | 3.66 | 4.94 |
| 100 | 99.3 ± 2.5 | 99.28 | −0.72 | 1.48 | 2.28 | 2.52 |
| N-desethyl sunitinib | ||||||
| 0.2 | 0.20 ± 0.02 | 100.08 | 0.08 | 9.41 | -c | 7.34 |
| 0.4 | 0.39 ± 0.03 | 99.17 | −0.83 | 6.28 | 0.25 | 6.29 |
| 1 | 1.02 ± 0.06 | 101.68 | 1.68 | 5.08 | 3.33 | 5.89 |
| 2 | 2.02 ± 0.14 | 100.75 | 0.75 | 7.68 | -c | 6.90 |
| 10 | 9.97 ± 0.51 | 99.70 | −0.3 | 6.05 | 5.63 | 5.09 |
| 20 | 20.1 ± 0.7 | 100.50 | 0.5 | 3.75 | -c | 3.55 |
| 100 | 97.6 ± 5.8 | 97.60 | −2.4 | 4.15 | 4.87 | 6.01 |
| 200 | 201.8 ± 14.9 | 100.92 | 0.92 | 9.14 | -c | 7.42 |
Performed in duplicate on 3 separate days.
Values are mean ± standard deviation.
No significant variation was observed as a result of performing the assay in different runs.
Table 3.
Back-calculated concentrations from calibration curves for sunitinib (0.02–5 ng/mL)and N-desethyl sunitinib (0.04–10.0 ng/mL) in phosphate buffer solution (PBS)a
| Nominal Concentration (ng/mL) | Calculated Concentration (ng/mL)b | Accuracy (%) | Bias | Precision (%)
|
RSD% Between Run | |
|---|---|---|---|---|---|---|
| Within Run | Between Run | |||||
| Sunitinib | ||||||
| 0.02 | 0.021 ± 0.001 | 104.33 | 4.33 | 2.78 | 8.56 | 8.15 |
| 0.05 | 0.046 ± 0.001 | 92.13 | −7.87 | 2.50 | 2.22 | 3.20 |
| 0.10 | 0.100 ± 0.007 | 100.72 | 0.72 | 4.31 | 6.44 | 7.20 |
| 0.50 | 0.48 ± 0.03 | 95.70 | −4.3 | 2.33 | 5.10 | 5.12 |
| 1.0 | 1.09 ± 0.07 | 108.52 | 8.52 | 6.79 | -c | 6.23 |
| 5.0 | 4.94 ± 0.24 | 98.80 | −1.2 | 6.30 | -c | 4.97 |
| N-desethyl sunitinib | ||||||
| 0.04 | 0.041 ± 0.003 | 101.42 | 1.42 | 7.93 | -c | 6.32 |
| 0.1 | 0.094 ± 0.007 | 94.23 | −5.77 | 3.95 | 7.01 | 7.41 |
| 0.2 | 0.211 ± 0.017 | 105.25 | 5.25 | 6.82 | 4.75 | 8.04 |
| 1.0 | 0.933 ± 0.039 | 93.37 | −6.63 | 4.19 | -c | 4.13 |
| 2.0 | 2.12 ± 0.02 | 106.33 | 6.33 | 1.24 | -c | 1.06 |
| 10.0 | 9.93 ± 0.67 | 99.20 | −0.8 | 8.65 | -c | 6.70 |
Performed in duplicate on 3 separate days.
Values are mean ± standard deviation.
No significant variation was observed as a result of performing the assay in different runs.
3.3 Selectivity and specificity
No interfering peaks were observed in the chromatograms of blank plasma from 6 donors when monitored for sunitinib and N-desethyl sunitinib. Representative chromatograms of blank human plasma, plasma spiked with internal standard and the two analytes, as well as patient plasma sample obtained collected at 3 hr after oral administration are shown in fig. 2, 3, and 4, respectively. The mean (± standard deviation) retention times for sunitinib and N-desethyl sunitinib under the optimal conditions were both 1.40±0.02 and 1.40±0.02 min, respectively, with an overall chromatographic run time of 5 min. The selectivity for the analysis is shown by symmetrical resolution of the peaks, with no significant chromatographic interference around the retention times of the analyte and internal standard in drug-free specimens. Representative chromatograms of the blank PBS, PBS spiked with internal standard and the two analytes, as well as the post-dialysis buffer solution sample from patient plasma sample collected at 3 hr after the oral administration of sunitinib are shown in fig. 2, 3, and 4, respectively.
Fig. 2.

Chromatograms of blank human plasma (a,b,c,) and PBS (d,e,f,) for monitoring of (a,d) sunitinib, (b,e) N-desethyl sunitinib, and (c,f,) internal standard sunitinib-d10.
Fig. 3.

Chromatograms of plasma (a,b,c,) and post-dialysis buffer solution (d,e,f,) spiked with (a,d) sunitinib 0.1 ng/mL (LLOQ), (b,e) the N-desethyl sunitinib 0.2 ng/mL (LLOQ), and (c,f) the internal standard sunitinib-d10.
Fig. 4.

Representative chromatograms from a select cancer patient 3 hrs after receiving a 50 mg dose of sunitinib of plasma (a,b,c,) and post-dialysis buffer solution (d,e,f,) of (a, d) sunitinib, (b,e) N-desethyl sunitinib, and (c,f) the internal standard sunitinib-d10.
3.4 Accuracy, precision, and recovery
The intra- and inter-day precision and accuracy were assessed in plasma and PBS for both sunitinib and N-desethyl sunitinib at the LLOQ, the low QC, medium QC, high QC, and AULQ QC (plasma only) after 1:10 dilution over 3 days. For QC samples prepared by spiking human plasma with the two analytes, the within run and the between-run variability (precision) were within 6.6% for sunitinib and 6.5% for N-desethyl sunitinib (Table 4). Likewise the mean predicted concentration accuracy ranged from 91.9% to 102.6% for sunitinib and 95.5% to 103.5% for N-desethyl sunitinib. The relative recovery that was calculated by spiking the internal standard and analyte in plasma followed by extraction or directly into the mobile phase was >90% for sunitinib as well as its metabolite. The between day RSD (%) and bias ranged from 3.31 to 7.04% and −8.06 to 2.55%, respectively, for sunitinib and 3.69 to 6.92% and −4.5 to 3.5%, respectively, for N-desethyl sunitinib in plasma. For QC samples prepared in PBS with the two analytes, the within run and the between-run variability (precision) were within 7.2% for sunitinib and 6.5% for N-desethyl sunitinib (Table 5). Likewise the mean predicted concentration accuracy ranged from 100.3 to 102.8% for sunitinib and 99.4% to 102.4% for N-desethyl sunitinib. The between day RSD (%) and bias ranged from 4.13 to 7.49% and 0.79 to 2.8%, respectively, for sunitinib and 5.53 to 6.84% and −0.59 to 1.06%, respectively, for N-desethyl sunitinib in PBS.
Table 4.
Assessment of accuracy and precision of sunitinib and N-desethyl sunitinib in plasmaa
| Nominal Concentration (ng/mL) | Calculated Concentration (ng/mL) | Accuracy (%) | Bias | Precision (%)
|
RSD (%) Between Run | Recovery (%) | |
|---|---|---|---|---|---|---|---|
| Within Run | Between Run | ||||||
| Sunitinib | |||||||
| 0.1 (LLOQ) | 0.092 ±0.003 | 91.94 | −8.06 | 3.08 | 3.31 | 3.31 | -b |
| 0.3 | 0.306 ± 0.021 | 102.11 | 2.11 | 5.66 | 7.04 | 7.04 | 93.81 |
| 8 | 8.20 ± 0.49 | 102.55 | 2.55 | 2.74 | 6.01 | 6.01 | 97.17 |
| 80 | 80.6 ± 4.7 | 100.78 | 0.78 | 1.88 | 5.86 | 5.86 | 97.39 |
| 80.0 (1:10)c | 81.2 ± 3.9 | 101.50 | 1.5 | 4.16 | 4.79 | 4.79 | -b |
| N-desethyl sunitinib | |||||||
| 0.2 (LLOQ) | 0.191 ± 0.011 | 95.50 | −4.5 | 5.64 | 5.90 | 5.90 | -b |
| 0.6 | 0.609 ± 0.042 | 101.58 | 1.58 | 6.49 | 6.92 | 6.92 | 90.34 |
| 16.0 | 16.6 ± 0.7 | 103.50 | 3.5 | 3.11 | 4.32 | 4.32 | 101.1 |
| 160.0 | 154.0 ± 5.7 | 96.25 | −3.75 | 3.02 | 3.69 | 3.69 | 98.13 |
| 160.0 (1:10)c | 160.8 ± 9.2 | 100.50 | 0.5 | 5.08 | 5.75 | 5.75 | -b |
Performed in quintuplicate on three separate days
Not determined
Sample diluted 1:10 (v/v) prior to analysis
Table 5.
Assessment of accuracy and precision for sunitinib and N-desethyl sunitinib in phosphate buffered solution (PBS)a
| Nominal Concentration (ng/mL) | Calculated Concentration (ng/mL) | Accuracy (%) | Bias | Precision (%)
|
RSD (%) Between Run | |
|---|---|---|---|---|---|---|
| Within Run | Between Run | |||||
| Sunitinib | ||||||
| 0.02 (LLOQ) | 0.020 ± 0.002 | 101.00 | 1 | 6.92 | 3.38 | 7.49 |
| 0.06 | 0.060 ± 0.004 | 100.79 | 0.79 | 7.18 | -b | 6.90 |
| 0.40 | 0.411 ± 0.017 | 102.80 | 2.8 | 3.44 | 2.72 | 4.13 |
| 4.0 | 4.01 ± 0.186 | 100.32 | 0.32 | 2.75 | 4.40 | 4.63 |
| N-desethyl sunitinib | ||||||
| 0.04 (LLOQ) | 0.040 ± 0.002 | 100.78 | 0.78 | 5.64 | 2.08 | 6.75 |
| 0.12 | 0.121 ± 0.007 | 101.06 | 1.06 | 6.49 | 2.86 | 6.14 |
| 0.80 | 0.819 ± 0.045 | 102.40 | 2.4 | 3.11 | 3.54 | 5.53 |
| 8.0 | 7.95 ± 0.54 | 99.41 | −0.59 | 3.02 | 2.51 | 6.84 |
Performed in quintuplicate on three separate days
Not determined
3.5 Analyte stability
QC samples prepared in human plasma undergoing three freeze-thaw cycles showed no significant degradation (< 15%) for sunitinib and N-desethyl sunitinib (Table 6). Sunitinib was stable up to 4.4 hrs on the autosampler without any significant degradation, allowing for 43 samples to be analyzed simultaneously within a single chromatographic run. The long-term stability tests suggested that sunitinib and N-desethyl sunitinib was stable in stock solutions of acetonitrile/water (1:1, v/v) at −20°C for 192 days, and stable in plasma at −70°C for at least 406 days, with degradation less than 15%.
Table 6.
Assessment of stability for sunitinib and N-desethyl sunitinib in human plasmaa
| Conditions | sunitinib (ng/mL) | N-desethyl sunitinib (ng/mL) | ||||
|---|---|---|---|---|---|---|
| 0.30 | 80 | 80 (1:10)b | 0.60 | 160 | 160 (1:10)b | |
| Freeze-thaw stability (−70°C)b | ||||||
| Cycle 1 | 92.56 | 99.29 | 107.71 | 98.72 | 102.71 | 109.17 |
| Cycle 2 | 91.00 | 99.54 | 109.17 | 104.89 | 103.33 | 101.92 |
| Cycle 3 | 90.67 | 106.58 | 101.92 | 99.50 | 103.33 | 111.00 |
| Autosampler stability (4°C)c | ||||||
| Time = 4.4 h | 100.0 | 103.9 | -d | 99.3 | 106.3 | -d |
| Short-term stability (RT)e | ||||||
| Time = 0.5 h | 99.3 | 101.3 | 105.6 | 97.4 | 99.2 | 101.93 |
| Time = 1.0 h | 106.4 | 98.45 | 105.6 | 107.6 | 98.5 | 95.7 |
| Time = 2.0 h | 101.8 | 97.8 | 98.47 | 112.6 | 104.8 | 97.88 |
| Time = 4.0 h | 99.01 | 103.9 | 101.1 | 103.6 | 114.1 | 108.3 |
| Time = 6.0 h | 105.6 | 98.6 | 101.2 | 105.2 | 108.2 | 105.2 |
| Long-term stability (−70°C)e | ||||||
| Time = 299 days | 98.7 | 101.5 | 111.9 | 92.2 | 97.3 | 100.2 |
Expressed as the mean percentage change from time zero (nominal concentration)
Sample diluted 1:10 (v/v) prior to analysis
Performed repeatedly for 4.4 h with 1 sample
Not done.
Performed in triplicate
3.6 Concentration-time profiles
The LC/MS/MS method was successfully applied to study the pharmacokinetics of sunitinib and its active metabolite N-desethyl sunitinib in an adult patient receiving oral sunitinib as a dose of 50 mg daily. A representative chromatogram from a patient receiving 50 mg of sunitinib is shown in Fig. 4. Fig. 5 represents the plasma concentration time profile for total and unbound sunitinib and N-desethyl sunitinib. The maximum total plasma concentration (Cmax) for sunitinib and N-desethyl sunitinib was 30.2 ng/mL and 3.4 ng/mL which occurred at 3.1 h and 4.1 h with an AUCτ of 408.4 and 55.4 ng*h/mL, respectively. The maximum unbound concentration for sunitinib and N-desethyl sunitinib was 1.34 and 0.415 ng/mL, occurred at 3.1 h and had an AUCτ of 18.8 and 5.8 ng*h/mL, respectively. This represents 4.6% (95.4% bound) and 10.4% (89.6% bound) unbound drug for sunitinib and N-desethyl sunitinib, respectively, which is consistent with reported values (Goodman et al., 2007) In isolated protein solutions, approximately 76.7% of sunitinib and 63.8% of N-desethyl sunitinib was bound to human serum albumin (4 g/dL) while 59.7% of sunitinib 37.8% of N-desethyl sunitinib was bound to α1-acid glycoprotein (0.14 g/dL).
Fig. 5.
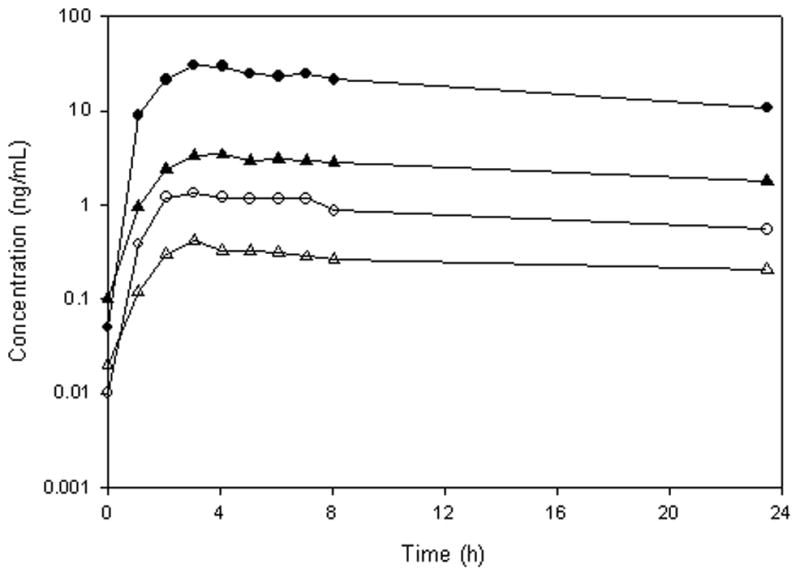
Plasma concentration–time profile of total (●) and unbound (○) sunitinib and total (▲) and unbound (△) N desethyl sunitinib after an oral dose of 50 mg/day.
4. Conclusion
In conclusion, we have developed and validated an assay for measuring total and unbound sunitinib and its metabolite in human plasma. The current method is the first to report the quantification of unbound drug and metabolite which is a key determinant of drug action. In comparison to the published methods, which only assessed total drug concentrations (Minkin et al., 2008, de Bruijn et al., 2010), the current assay is more sensitive (0.1 ng/mL) for sunitinib and similar (0.2 ng/mL) for N-desethyl sunitinib using 2 times less sample volume. The sample preparation procedure is simple and fast with a chromatographic run time of 5 min. These characteristics allow this assay to be easily applied to the quantitation of sunitinib and N-desethyl sunitinib in a large number of plasma samples. The described method for quantitation for sunitinib and N-desethyl sunitinib in plasma and in PBS is sufficient to allow plasma pharmacokinetic monitoring for total and unbound drug concentrations during daily, continuous administration. The method has been successfully applied to the study of the total and unbound plasma pharmacokinetics of sunitinib and its metabolite after its oral administration in a patient, and is being used to support clinical pharmacology projects in addition to the ones with potential drug-drug protein binding interactions
Acknowledgments
Funding: The project described was supported by a grant from the National Center Institute (NCI) U01 CA121947 to the AIDS Malignancy Consortium and by the Analytical Pharmacology Core of the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins (NIH grants P30 CA006973 and UL1 RR025005, and the Shared Instrument Grant (1S10RR026824-01)). The project described was supported in part by Grant Number UL1 RR 025005 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH) and NIH Roadmap for Medical Research, and its contents are solely the responsibility of the authors and do not necessarily represent the official view of NCRR or NIH.
References
- Arora A, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. Journal of Pharmacology and Experimental Therapeutics. 2005;315:971–979. doi: 10.1124/jpet.105.084145. [DOI] [PubMed] [Google Scholar]
- Atkins M, Jones CA, Kirkpatrick P. Sunitinib maleate. Nature Review Drug Discovery. 2006;5:279–280. doi: 10.1038/nrd2012. [DOI] [PubMed] [Google Scholar]
- Adams VR, Leggas M. Sunitinib malate for the treatment of metastatic renal cell carcinoma and gastrointestinal stromal tumors. Clinical Therapeutics. 2007;29:1338–1353. doi: 10.1016/j.clinthera.2007.07.022. [DOI] [PubMed] [Google Scholar]
- Patel N, Sun L, Moshinsky D, Chen H, Leahy KM, Le P, Moss KG, Wang X, Rice A, Tam D, Laird AD, Yu X, Zhang Q, Tang C, McMahon G, Howlett A. A selective and oral small molecule inhibitor of vascular epithelial growth factor receptor (VEGFR)-2 and VEGFR-1 inhibits neovascularization and vascular permeability. Journal of Pharmacology and Experimental Therapeutics. 2003;306:838–845. doi: 10.1124/jpet.103.052167. [DOI] [PubMed] [Google Scholar]
- Sun L, Liang C, Shirazian S, Zhou Y, Miller T, Cui J, Fukuda JY, Chu JY, Nematalla A, Wang X, Chen H, Sistla A, Luu TC, Tang F, Wei J, Tang C. Discovery of 5-[5-fluoro-2-oxo-1,2- dihydroindol-(3Z)-ylidenemethyl]-2,4- dimethyl-1H-pyrrole-3-carboxylic acid (2-diethylaminoethyl)amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet-derived growth factor receptor tyrosine kinase. Journal of Medicinal Chemistry. 2003;46:1116–1119. doi: 10.1021/jm0204183. [DOI] [PubMed] [Google Scholar]
- Chow LQ, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. Journal of Clinical Oncology. 2007;25:884–896. doi: 10.1200/JCO.2006.06.3602. [DOI] [PubMed] [Google Scholar]
- Schwandt A, Wood LS, Rini B, Dreicer R. Management of side effects associated with sunitinib therapy for patients with renal cell carcinoma. Oncology Targets and Therapy. 2009;2:51–61. doi: 10.2147/ott.s4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman VL, Rock EP, Dagher R, Ramchandani RP, Abraham S, Gobburu JV, Booth BP, Verbois SL, Morse DE, Liang CY, Chidambaram N, Jiang JX, Tang S, Mahjoob K, Justice R, Pazdur R. Approval summary: sunitinib for the treatment of imatinib refractory or intolerant gastrointestinal stromal tumors and advanced renal cell carcinoma. Clinical Cancer Research. 2007;13:1367–1373. doi: 10.1158/1078-0432.CCR-06-2328. [DOI] [PubMed] [Google Scholar]
- Seden K, Back D, Khoo S. Antiretroviral drug interactions: often unrecognized, frequently unavoidable, sometimes unmanageable. Journal of Antimicrobial Chemotherapeutics. 2009;64:5–8. doi: 10.1093/jac/dkp152. [DOI] [PubMed] [Google Scholar]
- Whitlam JB, Brown KF. Ultrafiltration in serum protein binding determinations. Journal of Pharmaceutical Sciences. 1981;70:146–150. doi: 10.1002/jps.2600700208. [DOI] [PubMed] [Google Scholar]
- Barre J, Chamouard JM, Houin G, Tillement JP. Equilibrium dialysis, ultrafiltration, and ultracentrifugation compared for determining the plasma-protein-binding characteristics of valproic acid. Clinical Chemistry. 1985;31:60–64. [PubMed] [Google Scholar]
- Jolliet-Riant P, Boukef MF, Duche JC, Simon N, Tillement JP. The genetic variant A of human alpha 1-acid glycoprotein limits the blood to brain transfer of drugs it binds. Life Science. 1998;62:PL219–226. doi: 10.1016/s0024-3205(98)00061-7. [DOI] [PubMed] [Google Scholar]
- Brouwer E, Verweij J, de Bruijn P, Loos WJ, Pillay M, Buijs D, Sparreboom A. Measurement of fraction unbound paclitaxel in human plasma. Drug Metabolism and Disposition. 2000;28:1141–1145. [PubMed] [Google Scholar]
- Shibukawa A, Kuroda Y, Nakagawa T. High-performance frontal analysis for drug-protein binding study. Journal of Pharmaceutical and Biomedical Analysis. 1999;18:1047–1055. doi: 10.1016/s0731-7085(98)00201-5. [DOI] [PubMed] [Google Scholar]
- Shibukawa A, Yoshimoto Y, Ohara T, Nakagawa T. High-performance capillary electrophoresis/frontal analysis for the study of protein binding of a basic drug. Journal of Pharmaceutical Sciences. 1994;83:616–619. doi: 10.1002/jps.2600830503. [DOI] [PubMed] [Google Scholar]
- Sillen H, Cook M, Davis P. Determination of unbound ticagrelor and its active metabolite (AR-C124910XX) in human plasma by equilibrium dialysis and LC-MS/MS. Journal of Chromatography B Analytical Technology and Biomedical Life Sciences. 2011;23:2315–2322. doi: 10.1016/j.jchromb.2011.06.023. [DOI] [PubMed] [Google Scholar]
- Etienne-Grimaldi MC, Renee N, Izzedine H, Milano G. A routine feasible HPLC analysis for the anti-angiogenic tyrosine kinase inhibitor, sunitinib, and its main metabolite, SU12662, in plasma. Journal of Chromatography B Analytical Technology and Biomedical Life Sciences. 2009;877:3757–3761. doi: 10.1016/j.jchromb.2009.09.011. [DOI] [PubMed] [Google Scholar]
- Minkin P, Zhao M, Chen Z, Ouwerkerk J, Gelderblom H, Baker SD. Quantification of sunitinib in human plasma by high-performance liquid chromatography-tandem mass spectrometry. Journal of Chromatography B Analytical Technology and Biomedical Life Sciences. 2008;874:84–88. doi: 10.1016/j.jchromb.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruijn P, Sleijfer S, Lam MH, Mathijssen RH, Wiemer EA, Loos WJ. Bioanalytical method for the quantification of sunitinib and its n-desethyl metabolite SU12662 in human plasma by ultra performance liquid chromatography/tandem triple-quadrupole mass spectrometry. Journal of Pharmaceutical and Biomedical Analysis. 2010;51:934–941. doi: 10.1016/j.jpba.2009.10.020. [DOI] [PubMed] [Google Scholar]
- Rodamer M, Elsinghorst PW, Kinzig M, Gutschow M, Sorgel F. Development and validation of a liquid chromatography/tandem mass spectrometry procedure for the quantification of sunitinib (SU11248) and its active metabolite, N-desethyl sunitinib (SU12662), in human plasma: application to an explorative study. Journal of Chromatography B Analytical Technology and Biomedical Life Science. 2011;879:695–706. doi: 10.1016/j.jchromb.2011.02.006. [DOI] [PubMed] [Google Scholar]
- Rosing H, Man WY, Doyle E, Bult A, Beijnen JH. Bioanalytical liquid chromatographic method validation. A review of current practices and procedures. Journal of Liquid Chromatography and Related Technology. 2000;23:329–354. [Google Scholar]
- Pfizer Inc. Investigator’s Brochure SU011248 (Pfizer Inc) July 2004. Pfizer Global Research and Development; New York, NY: 2004. (unpublished) [Google Scholar]