

Abstract
Hypothesizing that rapid estrogen signaling could be modulated from different estrogen receptors with unique localization patterns, a number of groups have attempted to design drug conjugates that target or restrict compounds to specific subcellular compartments. This article will briefly discuss the history of using conjugates to dissect rapid estrogen signaling and different strategies to attempt to target estrogens and antiestrogens to different locations. It will also detail some of the potential issues that can arise with different types of conjugates, using examples drawn from the authors’ own work.
Keywords: Rapid Estrogen Signaling, Drug Conjugates, E2-BSA, 4-hydroxytamoxifen, membrane estrogen receptors
Estrogens, including 17β-estradiol, play an important role in the development and maintenance of female reproductive tissues such as the breast and uterus, but also play an important role in non-reproductive tissues such as bone and brain. Both estrogenic and antiestrogenic drugs are widely used for the treatment of various ailments such as breast cancer and the relief of menopausal symptoms, but both classes of drugs have significant therapeutic limitations [1]. These limitations typically arise from undesired responses in non-target tissues. In some cases, responses in two tissues will be completely different from each other and be at odds with the well-established in vitro structure-activity relationships that have been developed over decades of estrogen receptor-focused medicinal chemistry. In a quest to better understand the mechanisms underlying the multifunctional response of some of these drugs, a complex web of estrogen signaling has been uncovered with multiple estrogen receptors, novel locations for estrogen receptor function and novel rapid crosstalk with other cellular signaling pathways [2] (Figure 1). While the rules for making small molecules that either activate or antagonize the “classic” transcriptional modulation by the nuclear estrogen receptors (ER alpha or ER beta) at a consensus estrogen response element (ERE) are well understood in most cell types [3, 4], structure-activity relationships defining rapid estrogen signaling are poorly understood. Getting a better grasp of the role of drug structure in dictating these crosstalk-based responses could greatly improve our ability to design better drugs for estrogen-related ailments. This article will focus on the strategy of using estrogen and antiestrogen conjugates to attempt to dissect the role of subcellular localization in specific aspects of cellular estrogen signaling.
FIGURE 1.

Possible targets for rapid responses to estrogen and antiestrogens.
Defining the targets of complex estrogen signaling
There are two major hypotheses that could explain why the activity of compounds in rapid estrogen signaling does not seem to always correlate with the activity seen with classic ER activity on ERE-containing promoters. The first hypothesis invokes novel estrogen receptors. As shown in Figure 1, in any given cell type, there can be nuclear receptors, ER alpha and ER beta, which are expressed at differing levels between cell and tissue types. These cells may also have different combinations of other transcription factors, coactivators, corepressors and promoter modifications that can cause unique transcriptional responses depending on the context. Outside of the nucleus, ER alpha and beta can be localized to a number of different locations such as the mitochondria. Finally, there are also a number of other estrogen-binding receptors. These include, but are not limited to, the estrogen receptor related receptors (ERRs)[5], a GPCR known originally as GPR30, but now known as GPER[6], and other less well-characterized receptors. The other hypothesis posits that the rapid responses are due to ER alpha and/or ER beta acting in an extranuclear signaling capacity [7, 8]. The two models are not necessarily mutually exclusive and it is possible that there are a number of receptors, both known and unknown, signaling from multiple locations in cell to give an integrated cellular response to estrogen.
Much of what is known about estrogen signaling has been obtained through the use of selective receptor modulation, either by the development of novel, selective chemical probes or by the use of selective genetic tools such as knockout mice. The strategy behind developing selective compounds for novel estrogen receptors is fairly straightforward– if the receptors are different proteins from ER alpha, they likely have significantly different ligand binding sites that can targeted selectively. Most of these types of unique estrogenic receptors have already been targeted with a selective compound (Figure 2). ER alpha and beta have a number of selective ligands such as PPT for ER alpha and DPN for ER beta [9]. GPER has a selective agonist, G-1 and antagonist, G-36 [10]. Even the relatively uncharacterized Gq-protein coupled membrane estrogen receptor in POMC neurons has been selectively targeted with a small molecule, STX [11]. Some efforts have been made to understand the structural determinants of selectivity. In the case of the nuclear receptors, some pharmacophores for selective binding have been proposed, but the diversity of selective ligands for both ER alpha and ER beta have made finding specific structural features for selective binding difficult [12]. The selectivity of the GPR30 compounds over the nuclear receptor appear to come from the ethanone and isopropyl groups present, which are believed to sterically clash with an arginine in the ER alpha and beta binding pocket [10]. STX has many similar structural components as the nonselective antiestrogen tamoxifen, but the stereochemistry of the alkene and the presence of the amide linker likely diminish its affinity for ER alpha or beta.
FIGURE 2.

Selective compounds for different types of estrogen binding receptors
Selective targeting of receptors in different sub-cellular localizations has proven more challenging. While it is possible that the ligand binding site of ER alpha or ER beta is significantly altered due to the biochemical context of different subcellular environments, such as the lipid membrane, the more commonly used approach to selectively target localized receptors is to selectively target the ligand to a desired location. This targeting has been mainly accomplished by attaching the estrogenic or antiestrogenic ligand to another molecule that dictates localization. The rest of this article will briefly review the history of this strategy and offer some insights and caveats with this approach.
The use of drug conjugates to dissect estrogen signaling
The general strategy in bioconjugate chemistry is to attach a potent ligand to a scaffold through a covalently attached linker. A number of estrogen and antiestrogen conjugates have been synthesized for various applications, including drug delivery and targeting, combination drug therapy including the PROTACS-based approach of targeting estrogen receptor for proteosomal degradation, immunotherapy, imaging and induction of protein multimerization [13-15]. This article will focus on those conjugates that have been used to probe mechanisms of complex estrogen signaling. A summary of these conjugates is shown in Table 1. Most of these conjugates have been developed with the intent of restricting the access of the small molecule to either the plasma membrane or the cytoplasm. Most estrogen-receptor modulating molecules are highly permeable with respect to lipid membranes, so most of the conjugates have been attached to scaffolds with increased size and/or polarity to limit that permeability. The scaffolds have ranged from proteins such as bovine serum albumin (BSA) and horseradish peroxidase [16] to synthetic scaffolds such as dendrimers, fluorescent dyes and polymers. Equally important, but often overlooked by non-chemists, is the linker that attaches the ligand to the scaffold. Both the attachment point of the linker to the drug and its chemical nature can play key roles in determining the overall biological activity and localization properties of the conjugate.
TABLE 1.
Summary of conjugates used to probe rapid estrogen responses organized by nature of ligand, linker and scaffold.
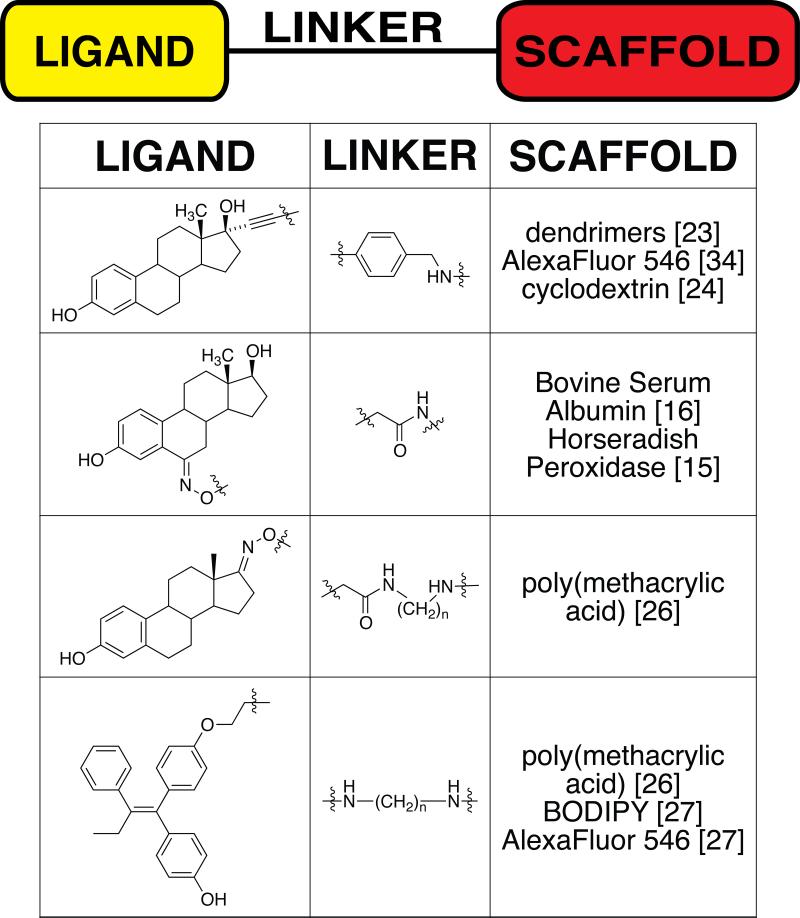
|
Most of the early work done with identifying the location of rapid estrogen signaling used the estrogen-BSA conjugates [17]. The conjugates are commercially available, have fluorescently labeled derivatives and have found widespread use as probes of a number of different types of estrogen-induced responses [18-20]. In these studies, the BSA conjugates are used primarily to probe whether the estrogen-induced signal is originating from the plasma membrane, since BSA is assumed to membrane-impermeable. The use of the BSA-estrogen conjugates should be cautiously monitored. Albumins have high binding affinities for steroid and the commercially purchased conjugates have been reported to contain a high amount of unconjugated ligand [21, 22]. In addition, they exhibit slow binding kinetics to the receptor and stimulate pathways that are not stimulated by estradiol alone [21, 22]. Albumin cannot be assumed to be a pharmacologically inert molecule– BSA has been shown to induce oxidative stress in renal tubule cells [23] and apoptosis in fibril form in different cell lines [24]. Albumins can also extract other molecules from membranes such as arachidonic acid and significantly alter membrane fluidity and signaling [25]. These conjugates can still be useful tools to study estrogen signaling, but careful attention should be paid to the purity of the conjugates and nature of the control experiments.
Attempting to improve on the estrogen-BSA conjugates, a number of labs have made non-proteinaceous estrogen and antiestrogen conjugates. The first molecules were estradiol or tamoxifen analogs modified into membrane-impermeable molecules by adding a charge to the ligand, as in the case of Q-Tam, a quaternary ammonium salt of tamoxifen. [26]. Confirming the localization of these molecules has proven to be difficult and the fact that these molecules have effects on transcription strongly suggest that these charged molecules are not entirely extracellular. Recent HPLC analysis of cell lysates suggest significant intracellular accumulation of quaternary ammonium tamoxifen derivatives, which raises questions as to the overall membrane-targeting utility of these compounds [27]. Later attempts to generate localization-specific conjugates involved coupling compounds to highly polar, fluorescent dyes or to large macromolecules such as dendrimers, cyclodextrins or polymers [28-32]. The multifunctional potential of these conjugates enabled easier tracking of the conjugates and showed that they all localize to the cytoplasm, although the kinetics of cellular uptake differed significantly. The difference in uptake kinetics and in the effect of unconjugated drug on uptake suggests that there might be multiple mechanisms of uptake, ranging from a ligand-dependent endocytosis to passive diffusion [27, 28, 33]. These conjugates, especially the estrogenderivatized dendrimer, are starting to be used more frequently to probe questions regarding extranuclear estrogen signaling[34-36].
Challenges of using conjugates to study estrogen signaling
As mentioned above, both proteinaceous and non-proteinaceous estrogen conjugates are being used extensively to probe the location of specific estrogen signaling. While these molecules have the potential to be useful to people with varying degrees of expertise with conjugate chemistry, there are a number of issues related to conjugate preparation that can complicate their use.
Purity of the conjugates
Compounds that modulate estrogen receptor activity are typically hydrophobic with relatively poor water solubility. They also show high binding affinities for serum carrier proteins such as sex hormone binding globulin (SHBG) and different albumins. It is possible that these same compounds would also have a high propensity for binding nonspecifically to the scaffolds that are used with these conjugates [21, 31]. For macromolecular conjugates, such as the estrogen-BSA and tamoxifen-polymer conjugate, a small amount of free drug associating with the conjugate in a noncovalent manner could significantly hinder the utility of the conjugate as a specific extranuclear or membrane probe (unpublished observation). In addition, even though dialysis is likely the simplest method to remove contaminants, the dissociation equilibrium of free drug from a carrier protein complex could be highly unfavorable in dilute aqueous solutions. During the development of 4-hydroxytamoxifen conjugates with poly (methacrylic acid) scaffolds, we discovered using a small, but persistent, amount of free drug still associated with the conjugate even after extensive dialysis (unpublished observation). Ultimately reverse phase preparative HPLC was required to remove the contaminant [31]. Cursory analysis of work with the BSA-estradiol conjugates, which have already been documented to have some degree of contaminating free ligand [21], suggest that some groups pay great attention to ensuring the purity of the conjugates [37] while many others do not appear to have performed any purification at all. Dialysis can still be an effective method to purify conjugates and is likely sufficient for most studies, but ideally, the purity of the material should be assessed before using. There are a number of good examples of purity assessment in the literature with both synthetic conjugates and the BSA conjugates. The most thorough example was reported for testing the purity of the estrogen dendrimer conjugate after a methanol washing process [37]. Prior to purification, the authors spiked the estrogen-dendrimer mixture with a free, radiolabeled version of the ligand prior to dialysis and then monitored the amount of radioactivity that was still left bound to the dendrimer.
Nature of the ligand on a conjugate
In a typical rapid signaling experiment using an estrogen conjugate, the signaling elicited by the conjugate is compared directly to the signaling elicited by estradiol. While this comparison will likely still yield an answer that will mostly reflect what portion of the response is dictated by receptors in the location targeted by the conjugate, free, unmodified hormone is not exactly the best comparison to make. The better comparison would be to compare the conjugate to the hormone attached to its linker. The linker and scaffold attached to the drug could also be influencing the signaling by the receptor. It is well known that subtle changes in ligand structure can have dramatic effects on estrogen signaling, although it is more pronounced with selective estrogen receptor modulators (SERMs) like tamoxifen than with full estrogen agonists such as ethinyl estradiol. Even so, some estrogen signaling pathways have been reported to be activated by only subsets of agonists that were previously believed to all modulate identically [38]. It is reasonable to believe that the attachment point and nature of the linker can have at least some influence on some aspect of the overall signaling modulated by the conjugate. In addition, the scaffold also needs to be tested independently of ligand or linker to determine whether the chemical nature of the scaffold is playing a role in the signaling, especially in highly generalized kinase activation assays. Using this logic, the proper control experiments for an estrogen-BSA conjugate would be estradiol, BSA and an estrogen derivatized with the same linker as that used in the conjugate.
Uptake and localization of the conjugate
The third challenge regarding the use of conjugates to study rapid estrogen signaling is the extent of uptake and localization of the conjugate. Drugs targeting estrogen receptor are generally hydrophobic and can easily pass through the cell membrane. It would seem straightforward to turn a molecule that was previously membrane-permeable into a molecule that is membrane-impermeable. There is extensive experience in medicinal chemistry with compounds with poor membrane permeability, so mimicking the properties of some of those molecules would seem to be a successful strategy. Permanently charged derivatives of estradiol and tamoxifen have been synthesized with the objective of creating compounds that are too polar to cross the plasma membrane. When the compounds were actually tested, however, all of the estradiol-derived compounds except for a quaternary ammonium salt derivative of ethinyl estradiol were cell-permeable as judged by their ability to inhibit radiolabeled estradiol binding by ER alpha-transfected COS7 cells [30]. The verdict concerning the membrane permeability of the tamoxifen analogs has been mixed, but recent HPLC-MS extraction studies with HeLa cells suggest that all of the permanently charged tamoxifen derivatives show significant intracellular accumulation [27].
A few of the conjugates have been reported to membrane-impermeable. The most commonly used conjugates for selective membrane targeting of estrogen have been the protein-based conjugates, particularly the estrogen-BSA conjugate. Fluorescently labeled estrogen-BSA conjugates have repeatedly been shown to localize on the plasma membrane of cells and this binding appears to be estrogen-dependent [22, 39]. Another non-proteinaceous conjugate; an ethinyl estradiol analog conjugated to a highly polar fluorescent dye, was also used to determine the possible intracellular localization of GPER and showed no uptake in cells after 15 minutes of dosing [40]. All of the other conjugates reported thus far, including estradiol and tamoxifen-derived polymers with a high degree of polar side chains, showed cytoplasmic localization.
An important issue to consider when using membrane-impermeable conjugates to probe specific signaling from membrane receptors is the kinetics of uptake. Some molecules, such as the ethinyl estradiol fluorescent dye conjugate mentioned in the previous paragraph, show no apparent uptake after short periods of dosing, but do show detectable uptake after longer periods of dosing [32]. Therefore, one must be cautious in using these reagents to explore membrane-specific responses under longer-term dosing regimens than those typically used for rapid estrogen signaling experiments. In addition, some rapid estrogen signaling responses are stimulated at sub-nanomolar concentrations of drug [41]. Most of these fluorescent conjugates will be undetectable at those concentrations.
One relatively unexplored question regarding these compounds concerns the mechanism of uptake for these conjugates, particularly the molecules that would be predicted to be membrane-impermeable based on polarity. These predictions are based on a model of a single free molecule passing across the lipid bilayer and it is likely that this model is not appropriate for some of these molecules. One possible reason for this is that the addition of highly polar groups to hydrophobic molecules such as steroids makes them amphiphilic and potentially susceptible to aggregation. The cellular uptake of large aggregates of undefined size and charge is more difficult to predict. In addition, carrier proteins such as SHBG have also been hypothesized to facilitate steroid entry into cells[42] and the increased polarity of the conjugates may not significantly affect SHBG binding or uptake or the protein-drug complex. Efforts to understand whether these uptake mechanisms play a role in the cytoplasmic localization of estradiol and 4-hydroxytamoxifen-polymer conjugates are currently underway.
Conclusions
Overall, the use of estrogenic and antiestrogenic conjugates to dissect estrogen signaling has yielded some valuable insights into the role of extranuclear and membrane-initiated rapid estrogen signaling. At the same time, misplaced trust in the properties of these conjugates, especially the purity, presumed localization and equivalence to other compounds, could muddle our understanding and potentially explain why some of the data regarding rapid estrogen signaling seem contradictory. Better understanding of the chemical properties of these conjugates with new ligands, linkers and scaffolds should expand the capabilities of these probes to explore all facets of estrogen signaling. In addition, some of these conjugates also could have beneficial therapeutic properties. For instance, conjugates of 4-hydroxytamoxifen have been found to be effective at inhibiting the proliferation of ER-positive breast cancer cells that are resistant to tamoxifen and the estrogen-dendrimer conjugates appear to have cardioprotective effects in mice. [31, 32, 34] In addition, the biologically active conjugates could be modified to have other scaffolds, either anticancer compounds or inducers of proteosomal degradation [43]. It is likely that even more useful applications will be found in the future for this interesting class of steroid hormone receptor modulators.
Highlights.
Estrogen conjugates are used to study localization of rapid responses.
Conjugates are useful, but they should be used very carefully.
Conjugate purity, uptake and equivalence are potential problems
Issues can be overcome with careful handling and proper controls.
Acknowledgements
The Army Breast Cancer Research Program (BC030507) and the National Institutes of Health (R01 DK075376) have supported the authors’ conjugate studies described in this article.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jordan VC. SERMs: meeting the promise of multifunctional medicines. J Natl Cancer Inst. 2007;99:350–6. doi: 10.1093/jnci/djk062. [DOI] [PubMed] [Google Scholar]
- 2.Weatherman RV. Untangling the estrogen receptor web: Tools to selectively study estrogen binding receptors. In: Ottow E, Weinmann H, editors. Nuclear Receptors as Drug Targets. Wiley-VCH; Weinhem: 2008. pp. 47–64. [Google Scholar]
- 3.Rosano C, Stec-Martyna E, Lappano R, Maggiolini M. Structure-based approach for the discovery of novel selective estrogen receptor modulators. Curr Med Chem. 2011;18:1188–94. doi: 10.2174/092986711795029645. [DOI] [PubMed] [Google Scholar]
- 4.McDonnell DP, Wardell SE. The molecular mechanisms underlying the pharmacological actions of ER modulators: implications for new drug discovery in breast cancer. Curr Opin Pharmacol. 2010;10:620–8. doi: 10.1016/j.coph.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giguere V. Transcriptional control of energy homeostasis by the estrogen-related receptors. Endocr Rev. 2008;29:677–96. doi: 10.1210/er.2008-0017. [DOI] [PubMed] [Google Scholar]
- 6.Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ. Estrogen signaling through the transmembrane G Protein-Coupled Receptor GPR30. Annu Rev Physiol. 2008;70:165–90. doi: 10.1146/annurev.physiol.70.113006.100518. [DOI] [PubMed] [Google Scholar]
- 7.Levin ER. Minireview: Extranuclear steroid receptors: roles in modulation of cell functions. Mol Endocrinol. 2011;25:377–84. doi: 10.1210/me.2010-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moriarty K, Kim KH, Bender JR. Minireview: estrogen receptor-mediated rapid signaling. Endocrinology. 2006;147:5557–63. doi: 10.1210/en.2006-0729. [DOI] [PubMed] [Google Scholar]
- 9.Nilsson S, Gustafsson JA. Estrogen receptors: therapies targeted to receptor subtypes. Clin Pharmacol Ther. 2011;89:44–55. doi: 10.1038/clpt.2010.226. [DOI] [PubMed] [Google Scholar]
- 10.Dennis MK, Field AS, Burai R, Ramesh C, Petrie WK, Bologa CG, et al. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J Steroid Biochem Mol Biol. 2011;127:358–66. doi: 10.1016/j.jsbmb.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qiu J, Bosch MA, Tobias SC, Krust A, Graham SM, Murphy SJ, et al. A G-protein-coupled estrogen receptor is involved in hypothalamic control of energy homeostasis. J Neurosci. 2006;26:5649–55. doi: 10.1523/JNEUROSCI.0327-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Agatonovic-Kustrin S, Turner JV. Molecular structural characteristics of estrogen receptor modulators as determinants of estrogen receptor selectivity. Mini Rev Med Chem. 2008;8:943–51. doi: 10.2174/138955708785132747. [DOI] [PubMed] [Google Scholar]
- 13.Moore TW, Gunther JR, Katzenellenbogen JA. Probing the topological tolerance of multimeric protein interactions: evaluation of an estrogen/synthetic ligand for FK506 binding protein conjugate. Bioconjug Chem. 2010;21:1880–9. doi: 10.1021/bc100266v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peng KW, Wang H, Qin Z, Wijewickrama GT, Lu M, Wang Z, et al. Selective estrogen receptor modulator delivery of quinone warheads to DNA triggering apoptosis in breast cancer cells. ACS Chem Biol. 2009;4:1039–49. doi: 10.1021/cb9001848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rai S, Paliwal R, Vaidya B, Gupta PN, Mahor S, Khatri K, et al. Estrogen(s) and analogs as a non-immunogenic endogenous ligand in targeted drug/DNA delivery. Curr Med Chem. 2007;14:2095–109. doi: 10.2174/092986707781368432. [DOI] [PubMed] [Google Scholar]
- 16.Nadal A, Ropero AB, Laribi O, Maillet M, Fuentes E, Soria B. Nongenomic actions of estrogens and xenoestrogens by binding at a plasma membrane receptor unrelated to estrogen receptor alpha and estrogen receptor beta. Proc Natl Acad Sci U S A. 2000;97:11603–8. doi: 10.1073/pnas.97.21.11603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berthois Y, Pourreau-Schneider N, Gandilhon P, Mittre H, Tubiana N, Martin PM. Estradiol membrane binding sites on human breast cancer cell lines. Use of a fluorescent estradiol conjugate to demonstrate plasma membrane binding systems. J Steroid Biochem. 1986;25:963–72. doi: 10.1016/0022-4731(86)90330-4. [DOI] [PubMed] [Google Scholar]
- 18.Davis TL, Whitesell JD, Cantlon JD, Clay CM, Nett TM. Does a nonclassical signaling mechanism underlie an increase of estradiol-mediated gonadotropin-releasing hormone receptor binding in ovine pituitary cells? Biol Reprod. 2011;85:770–8. doi: 10.1095/biolreprod.111.091926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kampa M, Pelekanou V, Gallo D, Notas G, Troullinaki M, Pediaditakis I, et al. ERalpha17p, an ERalpha P295 -T311 fragment, modifies the migration of breast cancer cells, through actin cytoskeleton rearrangements. J Cell Biochem. 2011;112:3786–96. doi: 10.1002/jcb.23309. [DOI] [PubMed] [Google Scholar]
- 20.Zhang C, Kelly MJ, Ronnekleiv OK. 17Beta-estradiol rapidly increases K(ATP) activity in GnRH via a protein kinase signaling pathway. Endocrinology. 2010;151:4477–84. doi: 10.1210/en.2010-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fischermann K, Bech I, Foged P, Pedersen JH, Lauritzen JB. Relation between cystic fibroadenomatosis and cancer of the breast. Acta Chir Scand. 1969;135:671–4. [PubMed] [Google Scholar]
- 22.Taguchi Y, Koslowski M, Bodenner DL. Binding of estrogen receptor with estrogen conjugated to bovine serum albumin (BSA). Nucl Recept. 2004;2:5. doi: 10.1186/1478-1336-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee YJ, Suh HN, Han HJ. Effect of BSA-induced ER stress on SGLT protein expression levels and alpha-MG uptake in renal proximal tubule cells. American journal of physiology Renal physiology. 2009;296:F1405–16. doi: 10.1152/ajprenal.90652.2008. [DOI] [PubMed] [Google Scholar]
- 24.Huang CY, Liang CM, Chu CL, Liang SM. Albumin fibrillization induces apoptosis via integrin/FAK/Akt pathway. BMC biotechnology. 2009;9:2. doi: 10.1186/1472-6750-9-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beck R, Bertolino S, Abbot SE, Aaronson PI, Smirnov SV. Modulation of arachidonic acid release and membrane fluidity by albumin in vascular smooth muscle and endothelial cells. Circ Res. 1998;83:923–31. doi: 10.1161/01.res.83.9.923. [DOI] [PubMed] [Google Scholar]
- 26.Jarman M, Leung OT, Leclercq G, Devleeschouwer N, Stoessel S, Coombes RC, et al. Analogues of tamoxifen: the role of the basic side-chain. Applications of a whole-cell oestrogen-receptor binding assay to N-oxides and quaternary salts. Anticancer Drug Des. 1986;1:259–68. [PubMed] [Google Scholar]
- 27.Rivera-Guevara C, Perez-Alvarez V, Garcia-Becerra R, Ordaz-Rosado D, Morales-Rios MS, Hernandez-Gallegos E, et al. Genomic action of permanently charged tamoxifen derivatives via estrogen receptor-alpha. Bioorg Med Chem. 2010;18:5593–601. doi: 10.1016/j.bmc.2010.06.039. [DOI] [PubMed] [Google Scholar]
- 28.Harrington WR, Kim SH, Funk CC, Madak-Erdogan Z, Schiff R, Katzenellenbogen JA, et al. Estrogen dendrimer conjugates that preferentially activate extranuclear, nongenomic versus genomic pathways of estrogen action. Mol Endocrinol. 2006;20:491–502. doi: 10.1210/me.2005-0186. [DOI] [PubMed] [Google Scholar]
- 29.Kim HY, Sohn J, Wijewickrama GT, Edirisinghe P, Gherezghiher T, Hemachandra M, et al. Click synthesis of estradiol-cyclodextrin conjugates as cell compartment selective estrogens. Bioorg Med Chem. 2010;18:809–21. doi: 10.1016/j.bmc.2009.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Revankar CM, Mitchell HD, Field AS, Burai R, Corona C, Ramesh C, et al. Synthetic estrogen derivatives demonstrate the functionality of intracellular GPR30. ACS Chem Biol. 2007;2:536–44. doi: 10.1021/cb700072n. [DOI] [PubMed] [Google Scholar]
- 31.Rickert EL, Trebley JP, Peterson AC, Morrell MM, Weatherman RV. Synthesis and characterization of bioactive tamoxifen-conjugated polymers. Biomacromolecules. 2007;8:3608–12. doi: 10.1021/bm070413t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rickert EL, Oriana S, Hartman-Frey C, Long X, Webb TT, Nephew KP, et al. Synthesis and characterization of fluorescent 4-hydroxytamoxifen conjugates with unique antiestrogenic properties. Bioconjug Chem. 2010;21:903–10. doi: 10.1021/bc900461h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sugano K, Kansy M, Artursson P, Avdeef A, Bendels S, Di L, et al. Coexistence of passive and carrier-mediated processes in drug transport. Nat Rev Drug Discov. 2010;9:597–614. doi: 10.1038/nrd3187. [DOI] [PubMed] [Google Scholar]
- 34.Chambliss KL, Wu Q, Oltmann S, Konaniah ES, Umetani M, Korach KS, et al. Non-nuclear estrogen receptor alpha signaling promotes cardiovascular protection but not uterine or breast cancer growth in mice. J Clin Invest. 2010;120:2319–30. doi: 10.1172/JCI38291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Madak-Erdogan Z, Kieser KJ, Kim SH, Komm B, Katzenellenbogen JA, Katzenellenbogen BS. Nuclear and extranuclear pathway inputs in the regulation of global gene expression by estrogen receptors. Mol Endocrinol. 2008;22:2116–27. doi: 10.1210/me.2008-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wong WP, Tiano JP, Liu S, Hewitt SC, Le May C, Dalle S, et al. Extranuclear estrogen receptor-alpha stimulates NeuroD1 binding to the insulin promoter and favors insulin synthesis. Proc Natl Acad Sci U S A. 2010;107:13057–62. doi: 10.1073/pnas.0914501107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davis TL, Whitesell JD, Cantlon JD, Clay CM, Nett TM. Does a nonclassical signaling mechanism underlie an increase of estradiol-mediated gonadotropin-releasing hormone receptor binding in ovine pituitary cells? Biol Reprod. 2011 doi: 10.1095/biolreprod.111.091926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saijo K, Collier JG, Li AC, Katzenellenbogen JA, Glass CK. An ADIOL-ERbeta-CtBP transrepression pathway negatively regulates microglia-mediated inflammation. Cell. 2011;145:584–95. doi: 10.1016/j.cell.2011.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu TW, Chen S, Brinton RD. Membrane estrogen receptors mediate calcium signaling and MAP kinase activation in individual hippocampal neurons. Brain Res. 2011;1379:34–43. doi: 10.1016/j.brainres.2011.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–30. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- 41.Watson CS, Bulayeva NN, Wozniak AL, Alyea RA. Xenoestrogens are potent activators of nongenomic estrogenic responses. Steroids. 2007;72:124–34. doi: 10.1016/j.steroids.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hammes A, Andreassen TK, Spoelgen R, Raila J, Hubner N, Schulz H, et al. Role of endocytosis in cellular uptake of sex steroids. Cell. 2005;122:751–62. doi: 10.1016/j.cell.2005.06.032. [DOI] [PubMed] [Google Scholar]
- 43.Rodriguez-Gonzalez A, Cyrus K, Salcius M, Kim K, Crews CM, Deshaies RJ, et al. Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer. Oncogene. 2008;27:7201–11. doi: 10.1038/onc.2008.320. [DOI] [PMC free article] [PubMed] [Google Scholar]