

Abstract
The voltage dependent anion channel (VDAC) provides exchange of metabolites, anions, and cations across the outer mitochondrial membrane. VDAC provides substrates and adenine nucleotides necessary for electron transport and therefore plays a key role in regulating mitochondrial bioenergetics. VDAC has also been suggested to regulate the response to cell death signaling. Emerging data show that VDAC is regulated by protein-protein interactions as well as by post-translational modifications. This review will focus on the regulation of VDAC and its potential role in regulating cell death in cardiac ischemia-reperfusion.
1.0 Background
Mitochondria are thought to originate from gram negative bacteria, and like gram negative bacteria, mitochondria have an outer and inner membrane. The inner mitochondrial membrane has a large number of channels and transporter, whereas the voltage dependent anion channel (VDAC) is the primary channel providing the exchange of anions, cations and metabolites across the outer mitochondrial membrane. VDAC is open to anions and cations at low voltage, but at high potentials it remains open to cations but is less permeable to anions (referred to as a closed state) [1, 2]. Thus, metabolites and anions cross the outer mitochondrial membrane in a voltage dependent manner while allowing cations to pass in a voltage independent manner [1]. Mammalian mitochondria have three VDAC isoforms, VDAC 1, 2 and 3 [3]. The different VDAC isoforms appear to have different localization [4] and different roles. The structure of VDAC1 has been solved and it shows that VDAC has a 19-strand beta-barrel fold [5]. All beta-strands are anti-parallel except for beta1 and beta19 that form a parallel interface.
2.0 VDAC regulation
VDAC is the primary channel for metabolite transfer across the outer mitochondrial membrane. VDAC has also been shown to be a bottleneck for Ca2+ transfer between the mitochondria and the endoplasmic reticulum [6]. Rapizzi et al showed that with loss of VDAC, there was less mitochondrial Ca2+ uptake following addition of an IP3R stimulus [6]. VDAC has also been reported to regulate cell death. Given the large number of roles for VDAC, it is not surprising that there are numerous mechanisms that regulate VDAC, such as protein-protein interactions and post-translational modifications.
2.1 Tubulin
Bernier-Valentin et al. [7], reported that tubulin, a cytoskeletal protein and a subunit of microtubules, can attach to the outer mitochondrial membrane of rat liver. Carre et al. [8] demonstrated that tubulin binds to VDAC. Tubulin, a heterodimer, has two subunits, a and b, and both subunits bind with high affinity to intact mitochondria [7] through its negatively charged extended C-terminal tail (CTT) [9]. Recently, it was shown that this anionic C-terminal tail (CTT) is the key site for the formation of the tubulin-VDAC complex. In an in-vitro system, Rostovtseva et al showed that tubulin promotes voltage sensitive closure of VDAC1 [10]. It has been proposed that tubulin regulation of VDAC might explain the difference in the Km for ADP stimulation of respiration in vivo versus that observed in isolated mitochondria [11]. Tubulin, which would be present in situ, would result in partial closure of VDAC and thereby increase the apparent Km for ADP stimulation of respiration. Thus, alteration in tubulin and the cytoskeleton are likely to alter the kinetics of ADP and ATP exchange which could alter cell energetics and mitochondrial electron transport and mitochondrial membrane potential. Consistent with a role for VDAC in regulating mitochondrial function, Maldonado et al [12] recently reported that modulators of VDAC activity such as tubulin destabilizing agents and GSK-3beta inhibitors can modulate mitochondrial membrane potential.
2.2 Hexokinase
Hexokinase I (HK) is the predominant isoform in brain, whereas hexokinase II (HK II) is more common in heart and muscle. HKI is typically bound to VDAC where it is suggested to promote breakdown of glucose (glycolysis) which will supply pyruvate to the mitochondria for utilization by the TCA cycle. HKII can be either cytosolic or mitochondrial, where it binds to VDAC. HKII has been found to bind to VDAC in cardiac and skeletal muscle [13, 14] and also in HeLa cells [15]. It is suggested that cytosolic HK enhances glycogen synthesis whereas mitochondrial targeted HK promotes glycolysis [16]. Glucose-6-phosphate, the metabolite produced by HK metabolism of glucose, is reported to lead to release of HKII from VDAC, thereby inhibiting glycolysis. The detachment of HKII from VDAC has also been shown to promote apoptosis. Pastorino et al showed that phosphorylation of VDAC1 at Thr 51 by GSK resulted in the dissociation of HKII from VDAC and an increase in apoptosis.
Several recent studies have shown that VDAC1 interacts with either HKI and/or HKII [17-19], through its N-terminal region [19]. The N-terminal 29 AA residue domain for both HKI and II (HKII-VDB; VDAC-binding domain) has high affinity for VDAC [20, 21], whereas HKIII and IV lack affinity for VDAC [22]. Sun et al., [23] showed that both HKI and HKII can bind VDAC and this binding can be facilitated by VDAC phosphorylation. Glucose and ATP both act as a positive catalytic substrate for HK-VDAC interaction [13]. Glutamic acid 73 of VDAC is important for HK-1 binding and closure of VDAC [24, 25].
2.3 Bcl-2 family members
Several bcl-2 family members have been reported to interact with VDAC. VDAC2 has been shown to bind BAK and keep BAK in an inactive conformation [26]. Bcl-2xl has also been reported to bind to VDAC and this interaction is suggested to reduce cell death, but there is disagreement as to whether bcl-2xl binding promotes the open or closed state of VDAC. Shimizu et al, [27], created liposomes containing VDAC and showed that Bcl-2 family proteins regulate the release of cytochrome c from the mitochondrial outer membrane. Pro-apoptotic Bcl-2 family protein, Bax and Bak were reported to facilitate VDAC opening, whereas the anti-apoptotic protein, Bcl-XL, closed VDAC [27]. This same group reported that the BH4 domain of antiapoptotic Bcl-2 family proteins, Bcl-2 and Bcl-XL, physically interacts with VDAC and inhibits VDAC activity (VDAC closure) [28]. In contrast other studies have suggested that Bcl-XL maintains VDAC in an open state which facilitates metabolic exchange between cytosol and mitochondria and by maintaining mitochondrial function opposes apoptosis [29, 30]. Consistent with a pro-apoptotic role for VDAC closure, in an in-vitro study using purified VDAC from rat liver mitochondria, the proapoptotic Bcl-2 family member Bid was shown to induce VDAC closure [31]. Thus, there seems to be agreement that bcl-2 family members can bind to VDAC, but the functional effect of this interaction is debated.
2.4 Post-translational modifications
Post-translational modifications have also been reported to occur on VDAC. A number of studies have reported phosphorylation of VDAC [15, 32-36]. Deng et al [34] performed a large scale proteomic analysis of mitochondrial phosphoproteins and identified phosphorylation of Ser 117 of VDAC1. Distler et al showed that VDAC1 was phosphorylated on Ser 12 and Ser 136 [36]. Ser 12 is a consensus PKC site, whereas Ser 136 is a site recognized by CAMKII/GSK. Both GSK-3β [15, 33] and PKCε [32] have been reported to phosphorylate VDAC. PKCε has been found to interact with VDAC and inhibit the mitochondrial permeability transition pore (mPTP) in cardiac mitochondria [32]. VDAC1 was shown to form a VDAC-PKCε complex in a phosphorylation dependent manner [32]. Baines et al. showed that PKCε is translocation to the mitochondria resulting in the phosphorylation of VDAC1. GSK has been reported to lead to phosphorylation of VDAC in both HeLa cells [15], and in heart [33] and both studies find that inhibition of GSK phosphorylation of VDAC reduces cell death. Pastorino et al [15] find that inhibition of GSK reduces VDAC phosphorylation which results in dissociation of HKII and enhanced cell death. Das et al find in rat heart that inhibition of GSK using catalytic inhibitors, like SB 216763 or SB 415286, results in dephosphorylation of VDAC2, along with reduced entry of ATP into the mitochondria under deenergized conditions. This inhibition of ATP entry into the mitochondria during anoxia would preserve the glycolytically generated ATP which would otherwise be consumed by reverse mode of the F1-F0-ATPase [33]. Schwertz et al., (2007), also reported that p38 MAPK can phosphorylate VDAC [35].
VDAC can undergo many other post-translational modifications. Jones and coworkers have reported that during preconditioning, VDAC undergoes an O-glc NAc modification and that this modification correlates with cardioprotection [37]. VDAC1, VDAC2, and VDAC3 all can undergo SNO modifications [38, 39]. SNO is a reversible protein modification that has the ability to alter the activity of the targeted protein. Using SNO-RAC proteomic analysis, Kohr et al has found that VDAC1 is S-nitrosylated on Cys 140 and 245, VDAC2 can be S-nitrosylated on Cys 48 and 211, and VDAC3 is phosphorylated on Cys 65 and 229 [38, 39]. The functional effects of S-nitrosylation of VDAC are under investigation; however Cheng-Q [40] found that VDAC was inhibited by addition of an NO donor. Interestingly, Keinan et al [41] have reported that apoptosis is associated with oligomerization of VDAC. S-nitrosylation has been shown to reduce oxidation of proteins. It will be interesting to determine if S-nitrosylation of VDAC reduces oligomerization of VDAC. Using top-down Fourier transform mass spectrometry, Ryan et al [42] have found that the VDAC inhibitor Ro 68-3400 modifies VDAC at Cys-232. VDAC can also be acetylated but the consequence of this modification needs to be determined.
3.0 VDAC and cell death
3.1 VDAC as a component of mPTP
VDAC and ANT interact at contact sites between the inner and outer membranes, and it had been suggested that mPTP might be formed by such a complex between VDAC and ANT. However genetic depletion studies suggested that neither ANT nor VDAC were required for mPTP activity [43]. It would be expected that loss of VDAC would inhibit cell death if VDAC were an obligatory component of the mPTP, but Baines et al showed no inhibition of cell death with loss of either VDAC 1, 2 or 3 [43]. In agreement with other studies [26, 44], Baines et al found that VDAC-2 null MEFS exhibited enhanced cell death.
3.2 VDAC2 and BAK
Cheng et al [26] reported that VDAC2 binds BAK and prevents it from activating apoptosis. However a recent study [45] shows that VDAC2 is needed for t-BID induced apoptosis. Roy et al [45] showed that VDAC2 is required for truncated BID-induced mitochondrial apoptosis by recruiting BAK to the mitochondria. De Stefani et al [44] have recently reported that loss of VDAC2 enhances apoptosis, but that knock-down of VDAC1 reduces apoptosis.
3.3 VDAC1 and IP3R
In contrast to Baines et al [43], De Stefani et al [44] reported a reduction in H2O2 mediated apoptosis in HeLa cells in which VDAC1 was reduced with siRNA, suggesting that VDAC1 promotes cell death. Baines showed that VDAC 1/3 double null MEFs were not protected from H2O2 or ionomycin induced death, but at some doses showed increased death. In support of a detrimental role for VDAC1, Abu-Hamad found an increase in apoptosis when VDAC1 was overexpressed [46]. However, Cheng et al also found no difference in cell death in VDAC1-null MEFs treated with staurosporin [26]. A mechanism by which VDAC1 might promote death was provided by De Stefani et al, who report that VDAC1 forms a complex with the IP3R which results in the transfer of apoptotic Ca signals to the mitochondria. They find that VDAC1 immunoprecipitates with IP3R and it is suggested that this interaction facilitates low-amplitude Ca transfer to the mitochondria which enhances apoptosis [44]. Grp75 is reported to be involved in linking VDAC with IP3R. Thus, VDAC2 is involved in binding a number of bcl-2 family members and most reports suggest that loss of VDAC2 enhances cell death. There are somewhat conflicting data regarding whether the loss of VDAC is protective. Additional studies are needed to determine the different binding partners and post-translational modifications of different isoforms of VDAC and whether these modifications alter cell death signaling.
3.4 VDAC2 and Erastin
Erastin has been shown to bind to VDAC2 and increase cell death in tumors with RAS activating mutations [47]. Erastin mediated cell killing has been shown to be dependent on ERK1/2 phosphorylation; thus erastin is reported to selectively kill tumor cells that have oncogenic activation of RAS. Erastin decreased the metabolite permeability of VDAC as shown by a decreased NADH flux through VDAC [47]. Erastin mediated death in cells with oncogenic RAS was block with antioxidants such as a-tocopherol and BHT, suggesting that it acts by increasing ROS or that it involves a redox sensitive modification. Erastin was also shown to bind VDAC2 and the ability of erastin to increase cell death was lost in cell lacking VDAC2. It is interesting that the erastin-mediated increase in cell killing is enhanced in cells with activated RAS-MER-ERK1/2 signaling. This might suggest that the phosphorylation state of VDAC or some other component of the signaling pathway is important in the erastin stimulation of death. It will also be of interest to determine if erastin binding to VDAC2 releases BAK and whether this plays a role in the erastin mediated killing.
3.5 Does VDAC opening/closing regulate cell death
As discussed, anti-apototic bcl-2 family members have been reported to interact with VDAC and modulate cell death. Most groups find Bcl-xl/bcl-2 association with VDAC to reduce apoptosis, but they differ as to whether this is mediated by an opening or closing of VDAC.
Studies report that binding of Bcl-2 or Bcl-xl to VDAC maintains VDAC in an open state allowing metabolite entry that serves to maintain mitochondrial membrane potential which opposes mPTP opening [29]. Consistent with these studies Bcl-xl has been shown to promote VDAC1 opening in a lipid bilayer [30]. There are also reports that Bcl-xl can attenuate VDAC opening which has also been suggested to oppose mPTP [27]. The reasons for this discrepancy are unclear but there are several issues that should be considered. VDAC can undergo a number of post-translational modifications and can interact with a number of proteins. It is possible that the effect of any one of these “modifications/interactions” could be altered depending on what other modifications or protein interactions are present. For example HKII binding might lead to VDAC opening in the absence of a key post translational modification, but could close VDAC in the presence of these post translational modifications. A number of elegant studies have directly examined VDAC gating in lipid bilayers [10, 31]. These studies provide very direct measurements of VDAC activity, but one needs to be cautious that purified VDAC in a bilayer might respond differently than in situ VDAC which can be modulated by protein interactions and post translational modifications.
Consistent with a role for VDAC closure in cell death, Tikunov et al [48] found that closure of VDAC with G3139, an 18-mer phosphorothionate, enhanced oxidative stress and lead to opening of mPTP. Tan et al [49] showed that G3139 blocks rather than closes the VDAC pore, resulting in decreased permeability to anions and cations both, rather than the selective loss of anion flux with VDAC closure. Based on the kinetics they concluded that G3139 partially enters the pore and forms an unstable bond. G3139 is a phosphorothioate oligonucleotide, with a sulfur atom replacing a nonbridging oxygen. Because the phosphodiester congener of G3139 does not block VDAC it suggests a role for the sulfur atom. Tikunov et al [48] found that G3139 blocked superoxide anion transport from the intermembrane space to the cytosol, which they proposed enhanced mPTP opening.
HK binding to VDAC attenuates apoptotic cell death [50]. Majewski et al [20] suggest that HK binding promotes the open conformation of VDAC. Azoulay-Zohar et al [51] studied purified VDAC in a bilayer and reported the HKII binding to VDAC promoted VDAC closure. Additional studies will be required to resolve this issue.
If increased VDAC opening or closing can induce cell death, then why does loss of VDAC have so little effect on cell death? It is likely that if the cell is lacking all three VDAC isoforms then unless there is some compensatory change, there should be very reduced or altered metabolite, anion and cation exchange across the outer mitochondrial membrane. This loss of exchange across the outer mitochondria membrane would be expected to have large effects on cellular energetics, through loss of oxidative phosphorylation, and compensatory changes are likely. These changes could influence the response to cell death. For example if VDAC closure reduces cell death because it reduces ATP and metabolite entry into the mitochondria, allowing mitochondrial membrane potential to dissipate, and if there were some compensatory protein that took over this function with loss of VDAC (which would likely be necessary for viability of the cell) then loss of VDAC would not alter cell death because of the compensatory changes.
4.0 Does opening or closing VDAC promote cell death in cardiac ischemia/reperfusion?
Cardiac ischemia is defined as the lack of blood flow to the heart, which deprives the heart of oxygen and metabolic substrates. As reviewed in detail elsewhere [52], if ischemia is prolonged the heart undergoes irreversible injury. In heart, most of the ATP is generated by electron transport and oxidative phoshorylation in the mitochondria. During ischemia, with the lack of oxygen, electron transport is inhibited and the mitochondrial membrane potential becomes depolarized causing oxidative generation of ATP to stop. ATP and other high energy phosphate levels decline and anaerobic glycolysis is stimulated, leading to cellular acidosis which leads to a rise in intracellular Na via plasma membrane Na-H exchange. Na also enters the cell via non-inactivating Na channels [52]. The resultant rise in Na leads to an increase in cytosolic Ca via Na-Ca exchange. On reperfusion extracellular pH returns to normal and this stimulate additional Na-H which leads to further entry of Na and Ca into the cell. Oxygen is also restored with reperfusion leading to re-energization of the mitochondrial membrane potential which is the driving force for Ca uptake into the mitochondria. Elevated mitochondrial Ca can activate the mPTP. The return of oxygen and the reenergization of the mitochondrial membrane potential also lead to a burst of reactive oxygen species (ROS) which also activates mPTP.
There are a number of cardioprotective drugs and protocols that have been shown to reduce ischemia-reperfusion injury. Brief intermittent periods of ischemia and reperfusion, referred to as ischemic preconditioning (IPC), have been shown to reduce ischemia-reperfuion injury following a sustained period of ischemia. As reviewed elsewhere IPC treatment reduces ischemic acidosis and the rise in Na and Ca that occur during the sustained period ischemia [53]. Although IPC and many other cardioprotective drugs reduce ischemic acidosis the mechanism responsible is not clear.
It might be useful to consider the functional consequence of VDAC opening and closing in the setting of ischemia and reperfusion, which would be different than in a more glycolytic cell in which many of the studies examining the effect of VDAC opening/closing on cell death have been done. There are several pieces of data consistent with a protective role for reducing ATP entry into the mitochondria. Murry et al showed in their initial report on ischemia preconditioning that IPC slowed the rate of decline in ATP [54]. Although there could be other mechanisms involved, reduced ATP entry via VDAC would account for this observation. Slowing the rate of ATP entry into the mitochondria during ischemia and its subsequent consumption by the F1 ATPase would allow better preservation of cell ATP [53]. The reduction in ATP breakdown would also reduce glycolysis and generation of lactate (which was also shown to occur with IPC) which would reduce ischemic acidosis and thereby reduce Na-H exchange, which otherwise would lead to an increase in intracellular Na which in turn results in reverse mode NCX that leads to an increase in Ca [52]. The ATP that is consumed by reverse mode of the F1ATPase is used to generate a mitochondria membrane potential. Although a higher membrane potential will oppose mPTP opening, a higher membrane potential is also associated with increased generation of ROS. In addition the membrane potential is the driving force for Ca entry into the matrix, and this matrix Ca can activate mPTP [53]. Consistent with this concept, there are data showing a more rapid decline in mitochondrial membrane potential during IPC [55]. Also consistent with this concept reducing mitochondrial membrane potential prior to ischemia, for example by over expression of uncoupling proteins or addition of uncoupler has been shown to be cardioprotective.
Data from Bcl-2 overexpressing heart also support of a role for reduced ATP consumption in cardioprotection. We [56] and others [57, 58], have found that overexpression of Bcl-2 reduced ischemia-reperfusion injury. Hearts from mice with cardiac specific overexpression of Bcl-2 showed improved post-ischemic recovery of left ventricular developed pressure, reduced infarct size, and improved post-ischemic recovery of phosphocreatine; we have also found that when Bcl-2 is overexpressed, hearts have a slower ATP hydrolysis rate during ischemia and reduced ischemic acidification [56]. The cardioprotection can be explained by the slower rate of ATP break-down. Imahashi et al also found VDAC association with Bcl-2 in these hearts and that this association increased with ischemia. They further found that binding of Bcl-2 to VDAC in isolated mitochondria reduced ATP consumption by reverse mode of the F1-F0ATPase under deenergized conditions. It should be noted that these data would be consistent with bcl-2 inhibition of ATP entry into the mitochondria (either by VDAC or ANT) or by a more direct inhibition of the F1-F0-ATPase.
A study examining the protective effect of GSK inhibition provided additional evidence for a role for VDAC. GSK is active under normal resting conditions when it is not phosphorylated, and phosphorylation at serine 9 leads to its inactivation. Studies have shown that GSK plays a role in ischemic preconditioning [59] and cardioprotection [60], and also targets the mPTP in cardiomyocytes [61, 62]. The mechanism by which GSK delays mPTP opening is unclear. Nishihara et al, [62] reported that inactive GSK-3β interacts with ANT at the inner mitochondrial membrane, which interferes with the binding between ANT and cyclophilin D, theoretically suppressing the opening of mPTP. Das et al [33] showed that GSK-3β inhibitors preserve ATP during ischemia by reducing adenine nucleotide transport by VDAC [33], as summarized in Figure 1. A role for VDAC, as opposed to ANT was determined by measuring the conversion of ADP to AMP by adenylate kinase located in the intermembrane space. This assay requires adenine nucleotide transport across the outer but not the inner mitochondrial membrane. GSK inhibitors also altered AMP production consistent with a reduction in ADP entry via VDAC.
Figure 1.
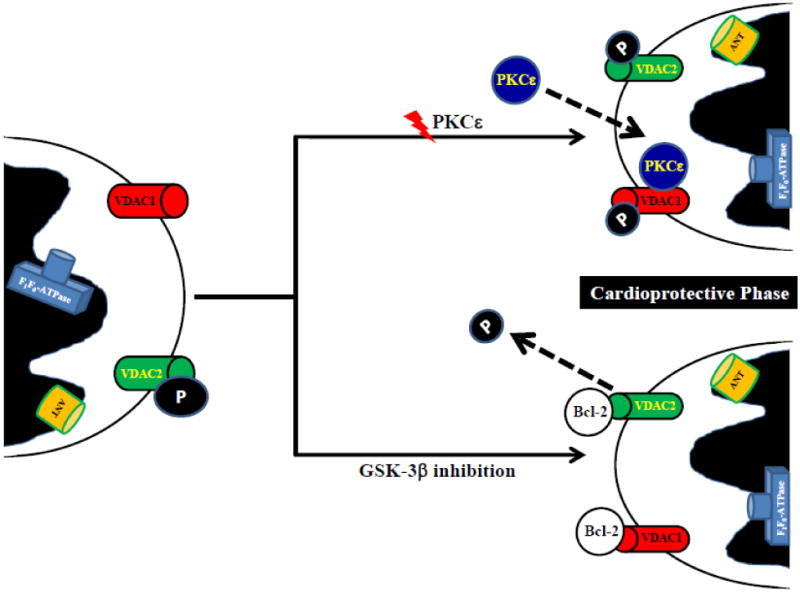
Proposed VDAC involvement to protect the heart by regulating adenine nucleotide transport, either by post-translational modifications or by interaction with other proteins.
HKII has been shown to translocate to mitochondria with IPC [63]. Smeele et al [64] reported that perfusing a heart with a TAT-HK peptide that disrupts HKII association with the mitochondria increases ischemia-reperfusion injury and blocks IPC mediated protection. Although HK has been shown to bind to VDAC, it is not clear that VDAC is the target for HKII binding in these studies. The TAT-HK peptide that was used by Smeele et al has been shown to displace HKII from mitochondria and trigger apoptosis in cells lacking VDAC1 and 3 [65]. Thus although HKII displacement from mitochondria appears to block IPC, it is not clear that this effect is mediated by VDAC.
5.0 Summary and Future Directions
Although VDAC does not appear to be a required component of mPTP, there are data suggesting that VDAC can regulate cell death. VDAC2 has been shown in many studies to attenuate cell death, in part by binding and inactivating BAK. Other Bcl-2 family members bind to VDAC and the effect of this binding on VDAC and/or mitochondrial function is stilldebated. Different VDAC isoforms undergo a number of different post translational modifications and the precise role of these modifications and their effect on VDAC function is an important area for future study. There are multiple VDAC isoforms and their different roles and functions are just beginning to be understood. Clearly VDAC is important for communication between the mitochondria and the rest of the cell and understanding how it is regulated will be an important area for future study.
Highlights.
Discusses regulation of VDAC by protein-protein interactions and post-translational modifications
Discusses the role of the open versus the close state in VDAC and cell death
Discusses the role of VDAC in ischemia-reperfusion injury
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tan W, Colombini M. VDAC closure increases calcium ion flux. Biochim Biophys Acta. 2007;1768:2510–2515. doi: 10.1016/j.bbamem.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hodge T, Colombini M. Regulation of metabolite flux through voltage-gating of VDAC channels. J Membr Biol. 1997;157:271–279. doi: 10.1007/s002329900235. [DOI] [PubMed] [Google Scholar]
- 3.Craigen WJ, Graham BH. Genetic strategies for dissecting mammalian and Drosophila voltage-dependent anion channel functions. J Bioenerg Biomembr. 2008;40:207–212. doi: 10.1007/s10863-008-9146-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neumann D, Buckers J, Kastrup L, Hell SW, Jakobs S. Two-color STED microscopy reveals different degrees of colocalization between hexokinase-I and the three human VDAC isoforms. PMC Biophys. 2010;3:4. doi: 10.1186/1757-5036-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tornroth-Horsefield S, Neutze R. Opening and closing the metabolite gate. Proc Natl Acad Sci U S A. 2008;105:19565–19566. doi: 10.1073/pnas.0810654106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rapizzi E, Pinton P, Szabadkai G, Wieckowski MR, Vandecasteele G, Baird G, Tuft RA, Fogarty KE, Rizzuto R. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J Cell Biol. 2002;159:613–624. doi: 10.1083/jcb.200205091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bernier-Valentin F, Rousset B. Interaction of tubulin with rat liver mitochondria. J Biol Chem. 1982;257:7092–7099. [PubMed] [Google Scholar]
- 8.Carre M, Andre N, Carles G, Borghi H, Brichese L, Briand C, Braguer D. Tubulin is an inherent component of mitochondrial membranes that interacts with the voltage-dependent anion channel. J Biol Chem. 2002;277:33664–33669. doi: 10.1074/jbc.M203834200. [DOI] [PubMed] [Google Scholar]
- 9.Sackett DL, Bhattacharyya B, Wolff J. Tubulin subunit carboxyl termini determine polymerization efficiency. J Biol Chem. 1985;260:43–45. [PubMed] [Google Scholar]
- 10.Rostovtseva TK, Sheldon KL, Hassanzadeh E, Monge C, Saks V, Bezrukov SM, Sackett DL. Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc Natl Acad Sci U S A. 2008;105:18746–18751. doi: 10.1073/pnas.0806303105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Appaix F, Kuznetsov AV, Usson Y, Kay L, Andrienko T, Olivares J, Kaambre T, Sikk P, Margreiter R, Saks V. Possible role of cytoskeleton in intracellular arrangement and regulation of mitochondria. Exp Physiol. 2003;88:175–190. doi: 10.1113/eph8802511. [DOI] [PubMed] [Google Scholar]
- 12.Maldonado EN, Patnaik J, Mullins MR, Lemasters JJ. Free tubulin modulates mitochondrial membrane potential in cancer cells. Cancer Res. 2010;70:10192–10201. doi: 10.1158/0008-5472.CAN-10-2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zorov DB, Juhaszova M, Yaniv Y, Nuss HB, Wang S, Sollott SJ. Regulation and pharmacology of the mitochondrial permeability transition pore. Cardiovasc Res. 2009;83:213–225. doi: 10.1093/cvr/cvp151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perevoshchikova IV, Zorov SD, Kotova EA, Zorov DB, Antonenko YN. Hexokinase inhibits flux of fluorescently labeled ATP through mitochondrial outer membrane porin. FEBS Lett. 2010;584:2397–2402. doi: 10.1016/j.febslet.2010.04.033. [DOI] [PubMed] [Google Scholar]
- 15.Pastorino JG, Hoek JB, Shulga N. Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 2005;65:10545–10554. doi: 10.1158/0008-5472.CAN-05-1925. [DOI] [PubMed] [Google Scholar]
- 16.John S, Weiss JN, Ribalet B. Subcellular localization of hexokinases I and II directs the metabolic fate of glucose. PLoS One. 2011;6:e17674. doi: 10.1371/journal.pone.0017674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arzoine L, Zilberberg N, Ben-Romano R, Shoshan-Barmatz V. Voltage-dependent anion channel 1-based peptides interact with hexokinase to prevent its anti-apoptotic activity. J Biol Chem. 2009;284:3946–3955. doi: 10.1074/jbc.M803614200. [DOI] [PubMed] [Google Scholar]
- 18.Shoshan-Barmatz V, Zakar M, Rosenthal K, Abu-Hamad S. Key regions of VDAC1 functioning in apoptosis induction and regulation by hexokinase. Biochim Biophys Acta. 2009;1787:421–430. doi: 10.1016/j.bbabio.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 19.Shoshan-Barmatz V, Keinan N, Abu-Hamad S, Tyomkin D, Aram L. Apoptosis is regulated by the VDAC1 N-terminal region and by VDAC oligomerization: release of cytochrome c, AIF and Smac/Diablo. Biochim Biophys Acta. 2010;1797:1281–1291. doi: 10.1016/j.bbabio.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 20.Majewski N, Nogueira V, Bhaskar P, Coy PE, Skeen JE, Gottlob K, Chandel NS, Thompson CB, Robey RB, Hay N. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell. 2004;16:819–830. doi: 10.1016/j.molcel.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 21.Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem. 2002;277:7610–7618. doi: 10.1074/jbc.M109950200. [DOI] [PubMed] [Google Scholar]
- 22.Arora KK, Filburn CR, Pedersen PL. Structure/function relationships in hexokinase. Site-directed mutational analyses and characterization of overexpressed fragments implicate different functions for the N- and C-terminal halves of the enzyme. J Biol Chem. 1993;268:18259–18266. [PubMed] [Google Scholar]
- 23.Sun L, Shukair S, Naik TJ, Moazed F, Ardehali H. Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and II. Mol Cell Biol. 2008;28:1007–1017. doi: 10.1128/MCB.00224-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zaid H, Abu-Hamad S, Israelson A, Nathan I, Shoshan-Barmatz V. The voltage-dependent anion channel-1 modulates apoptotic cell death. Cell Death Differ. 2005;12:751–760. doi: 10.1038/sj.cdd.4401599. [DOI] [PubMed] [Google Scholar]
- 25.Villinger S, Briones R, Giller K, Zachariae U, Lange A, de Groot BL, Griesinger C, Becker S, Zweckstetter M. Functional dynamics in the voltage-dependent anion channel. Proc Natl Acad Sci U S A. 2010;107:22546–22551. doi: 10.1073/pnas.1012310108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science. 2003;301:513–517. doi: 10.1126/science.1083995. [DOI] [PubMed] [Google Scholar]
- 27.Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- 28.Shimizu S, Konishi A, Kodama T, Tsujimoto Y. BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc Natl Acad Sci U S A. 2000;97:3100–3105. doi: 10.1073/pnas.97.7.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vander Heiden MG, Chandel NS, Li XX, Schumacker PT, Colombini M, Thompson CB. Outer mitochondrial membrane permeability can regulate coupled respiration and cell survival. Proc Natl Acad Sci U S A. 2000;97:4666–4671. doi: 10.1073/pnas.090082297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vander Heiden MG, Li XX, Gottleib E, Hill RB, Thompson CB, Colombini M. Bcl-xL promotes the open configuration of the voltage-dependent anion channel and metabolite passage through the outer mitochondrial membrane. J Biol Chem. 2001;276:19414–19419. doi: 10.1074/jbc.M101590200. [DOI] [PubMed] [Google Scholar]
- 31.Rostovtseva TK, Antonsson B, Suzuki M, Youle RJ, Colombini M, Bezrukov SM. Bid, but not Bax, regulates VDAC channels. J Biol Chem. 2004;279:13575–13583. doi: 10.1074/jbc.M310593200. [DOI] [PubMed] [Google Scholar]
- 32.Baines CP, Song CX, Zheng YT, Wang GW, Zhang J, Wang OL, Guo Y, Bolli R, Cardwell EM, Ping P. Protein kinase Cepsilon interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ Res. 2003;92:873–880. doi: 10.1161/01.RES.0000069215.36389.8D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Das S, Wong R, Rajapakse N, Murphy E, Steenbergen C. Glycogen synthase kinase 3 inhibition slows mitochondrial adenine nucleotide transport and regulates voltage-dependent anion channel phosphorylation. Circ Res. 2008;103:983–991. doi: 10.1161/CIRCRESAHA.108.178970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deng N, Zhang J, Zong C, Wang Y, Lu H, Yang P, Wang W, Young GW, Korge P, Lotz C, Doran P, Liem DA, Apweiler R, Weiss JN, Duan H, Ping P. Phosphoproteome analysis reveals regulatory sites in major pathways of cardiac mitochondria. Mol Cell Proteomics. 2011;10:M110 000117. doi: 10.1074/mcp.M110.000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwertz H, Carter JM, Abdudureheman M, Russ M, Buerke U, Schlitt A, Muller-Werdan U, Prondzinsky R, Werdan K, Buerke M. Myocardial ischemia/reperfusion causes VDAC phosphorylation which is reduced by cardioprotection with a p38 MAP kinase inhibitor. Proteomics. 2007;7:4579–4588. doi: 10.1002/pmic.200700734. [DOI] [PubMed] [Google Scholar]
- 36.Distler AM, Kerner J, Peterman SM, Hoppel CL. A targeted proteomic approach for the analysis of rat liver mitochondrial outer membrane proteins with extensive sequence coverage. Anal Biochem. 2006;356:18–29. doi: 10.1016/j.ab.2006.03.053. [DOI] [PubMed] [Google Scholar]
- 37.Ngoh GA, Watson LJ, Facundo HT, Dillmann W, Jones SP. Non-canonical glycosyltransferase modulates post-hypoxic cardiac myocyte death and mitochondrial permeability transition. J Mol Cell Cardiol. 2008;45:313–325. doi: 10.1016/j.yjmcc.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kohr MJ, Sun J, Aponte A, Wang G, Gucek M, Murphy E, Steenbergen C. Simultaneous measurement of protein oxidation and S-nitrosylation during preconditioning and ischemia/reperfusion injury with resin-assisted capture. Circ Res. 2011;108:418–426. doi: 10.1161/CIRCRESAHA.110.232173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kohr MJ, Aponte AM, Sun J, Wang G, Murphy E, Gucek M, Steenbergen C. Characterization of potential S-nitrosylation sites in the myocardium. Am J Physiol Heart Circ Physiol. 2011;300:H1327–1335. doi: 10.1152/ajpheart.00997.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng Q, Sedlic F, Pravdic D, Bosnjak ZJ, Kwok WM. Biphasic effect of nitric oxide on the cardiac voltage-dependent anion channel. FEBS Lett. 2011;585:328–334. doi: 10.1016/j.febslet.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Keinan N, Tyomkin D, Shoshan-Barmatz V. Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol Cell Biol. 2010;30:5698–5709. doi: 10.1128/MCB.00165-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ryan CM, Souda P, Bassilian S, Ujwal R, Zhang J, Abramson J, Ping P, Durazo A, Bowie JU, Hasan SS, Baniulis D, Cramer WA, Faull KF, Whitelegge JP. Post-translational modifications of integral membrane proteins resolved by top-down Fourier transform mass spectrometry with collisionally activated dissociation. Mol Cell Proteomics. 2010;9:791–803. doi: 10.1074/mcp.M900516-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–555. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Stefani D, Bononi A, Romagnoli A, Messina A, De Pinto V, Pinton P, Rizzuto R. VDAC1 selectively transfers apoptotic Ca(2+) signals to mitochondria. Cell Death Differ. 2011 doi: 10.1038/cdd.2011.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roy SS, Ehrlich AM, Craigen WJ, Hajnoczky G. VDAC2 is required for truncated BID-induced mitochondrial apoptosis by recruiting BAK to the mitochondria. EMBO Rep. 2009;10:1341–1347. doi: 10.1038/embor.2009.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abu-Hamad S, Zaid H, Israelson A, Nahon E, Shoshan-Barmatz V. Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: mapping the site of binding. J Biol Chem. 2008;283:13482–13490. doi: 10.1074/jbc.M708216200. [DOI] [PubMed] [Google Scholar]
- 47.Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM, Boniface JJ, Smith R, Lessnick SL, Sahasrabudhe S, Stockwell BR. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447:864–868. doi: 10.1038/nature05859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tikunov A, Johnson CB, Pediaditakis P, Markevich N, Macdonald JM, Lemasters JJ, Holmuhamedov E. Closure of VDAC causes oxidative stress and accelerates the Ca(2+)-induced mitochondrial permeability transition in rat liver mitochondria. Arch Biochem Biophys. 2010;495:174–181. doi: 10.1016/j.abb.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tan W, Loke YH, Stein CA, Miller P, Colombini M. Phosphorothioate oligonucleotides block the VDAC channel. Biophys J. 2007;93:1184–1191. doi: 10.1529/biophysj.107.105379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pastorino JG, Hoek JB. Regulation of hexokinase binding to VDAC. J Bioenerg Biomembr. 2008;40:171–182. doi: 10.1007/s10863-008-9148-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Azoulay-Zohar H, Israelson A, Abu-Hamad S, Shoshan-Barmatz V. In self-defence: hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem J. 2004;377:347–355. doi: 10.1042/BJ20031465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murphy E, Steenbergen C. Ion transport and energetics during cell death and protection. Physiology (Bethesda) 2008;23:115–123. doi: 10.1152/physiol.00044.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 55.Ylitalo KV, Ala-Rami A, Liimatta EV, Peuhkurinen KJ, Hassinen IE. Intracellular free calcium and mitochondrial membrane potential in ischemia/reperfusion and preconditioning. J Mol Cell Cardiol. 2000;32:1223–1238. doi: 10.1006/jmcc.2000.1157. [DOI] [PubMed] [Google Scholar]
- 56.Imahashi K, Schneider MD, Steenbergen C, Murphy E. Transgenic expression of Bcl-2 modulates energy metabolism, prevents cytosolic acidification during ischemia, and reduces ischemia/reperfusion injury. Circ Res. 2004;95:734–741. doi: 10.1161/01.RES.0000143898.67182.4c. [DOI] [PubMed] [Google Scholar]
- 57.Chen Z, Chua CC, Ho YS, Hamdy RC, Chua BH. Overexpression of Bcl-2 attenuates apoptosis and protects against myocardial I/R injury in transgenic mice. Am J Physiol Heart Circ Physiol. 2001;280:H2313–2320. doi: 10.1152/ajpheart.2001.280.5.H2313. [DOI] [PubMed] [Google Scholar]
- 58.Kirshenbaum LA, de Moissac D. The bcl-2 gene product prevents programmed cell death of ventricular myocytes. Circulation. 1997;96:1580–1585. doi: 10.1161/01.cir.96.5.1580. [DOI] [PubMed] [Google Scholar]
- 59.Tong H, Imahashi K, Steenbergen C, Murphy E. Phosphorylation of glycogen synthase kinase-3beta during preconditioning through a phosphatidylinositol-3-kinase--dependent pathway is cardioprotective. Circ Res. 2002;90:377–379. doi: 10.1161/01.res.0000012567.95445.55. [DOI] [PubMed] [Google Scholar]
- 60.Gross ER, Hsu AK, Gross GJ. Opioid-induced cardioprotection occurs via glycogen synthase kinase beta inhibition during reperfusion in intact rat hearts. Circ Res. 2004;94:960–966. doi: 10.1161/01.RES.0000122392.33172.09. [DOI] [PubMed] [Google Scholar]
- 61.Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113:1535–1549. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nishihara M, Miura T, Miki T, Tanno M, Yano T, Naitoh K, Ohori K, Hotta H, Terashima Y, Shimamoto K. Modulation of the mitochondrial permeability transition pore complex in GSK-3beta-mediated myocardial protection. J Mol Cell Cardiol. 2007;43:564–570. doi: 10.1016/j.yjmcc.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 63.Zuurbier CJ, Eerbeek O, Meijer AJ. Ischemic preconditioning, insulin, and morphine all cause hexokinase redistribution. Am J Physiol Heart Circ Physiol. 2005;289:H496–499. doi: 10.1152/ajpheart.01182.2004. [DOI] [PubMed] [Google Scholar]
- 64.Smeele KM, Southworth R, Wu R, Xie C, Nederlof R, Warley A, Nelson JK, van Horssen P, van den Wijngaard JP, Heikkinen S, Laakso M, Koeman A, Siebes M, Eerbeek O, Akar FG, Ardehali H, Hollmann MW, Zuurbier CJ. Disruption of hexokinase II-mitochondrial binding blocks ischemic preconditioning and causes rapid cardiac necrosis. Circ Res. 2011;108:1165–1169. doi: 10.1161/CIRCRESAHA.111.244962. [DOI] [PubMed] [Google Scholar]
- 65.Chiara F, Castellaro D, Marin O, Petronilli V, Brusilow WS, Juhaszova M, Sollott SJ, Forte M, Bernardi P, Rasola A. Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage-dependent anion channels. PLoS One. 2008;3:e1852. doi: 10.1371/journal.pone.0001852. [DOI] [PMC free article] [PubMed] [Google Scholar]