

Abstract
Lung cancer is the most common cause of cancer-related mortality worldwide. Here, we report elevated expression of tribbles homolog 2 (TRIB2) in primary human lung tumors and in non-small cell lung cancer cells that express low levels of differentiation-inducing transcription factor CCAAT/enhancer-binding protein alpha (C/EBPα). In approximately 10–20% of cases, elevated TRIB2 expression resulted from gene amplification. TRIB2 knockdown was found to inhibit cell proliferation and in vivo tumor growth. In addition, TRIB2 knockdown led to morphological changes similar to C/EBPα overexpression and correlated with increased expression and activity of C/EBPα. TRIB2-mediated regulation of C/EBPα was found to occur through the association of TRIB2 with the E3 ligase TRIM21. Together, these data identify TRIB2 as a potential driver of lung tumorigenesis through a mechanism that involves downregulation of C/EBPα.
Keywords: tribbles, lung, C/EBPα, TRIM21
Introduction
Lung cancer is currently the leading cause of death from all cancers in the United States (Jemal et al., 2010). Despite the use of aggressive medical therapy, non-small cell lung cancers (NSCLCs), a major subset of lung cancers, have an overall 5-year survival rate of less than 15% (Jemal et al., 2007). Recently, the cancer stem cell hypothesis has been applied extensively to solid tumors, such as lung tumors, in addition to hematopoietic cancers (Al-Hajj et al., 2003; Hemmati et al., 2003; Singh et al., 2003; Galli et al., 2004; Bapat et al., 2005; Collins et al., 2005; Fang et al., 2005; Dalerba et al., 2007; O’Brien et al., 2007; Ricci-Vitiani et al., 2007; Eramo et al., 2008; Schatton et al., 2008). As such, much research has gone into uncovering the different populations of progenitor cells within the lung. These are considered to be a rare population of undifferentiated cells with enhanced ability to proliferate, self-renew, and eventually differentiate into phenotypically diverse and heterogeneous cell populations (Kim et al., 2005).
The CCAAT/enhancer-binding protein (C/EBP) family of transcription factors is known to affect different aspects of development and differentiation in the lung (Cassel and Nord 2003). In particular, expression of C/EBPα has been shown to be crucial for lung development, as knockout mice exhibit massive hyperproliferation of type II pneumocytes and do not survive after birth (Flodby et al., 1996). C/EBPα may act as a tumor suppressor in the lung, as its overexpression in lung tumor cells results in a dramatic decrease in cell proliferation (Halmos et al., 2002), and it is often downregulated in NSCLC (Halmos et al., 2002; Costa et al., 2007). Work done by Pear and colleagues showed that the human homolog of the Drosophila tribbles protein, TRIB2, associates with and inhibits the transcriptional activity of C/EBPα (Keeshan et al., 2006; Dedhia et al., 2010). Additionally, overexpression of TRIB2 induces acute myelogenous leukemia in mice through inactivation of C/EBPα (Keeshan et al., 2006), and downregulation of the tumor suppressor gene FOXO3a is mediated by TRIB2 in human melanoma cells (Zanella et al., 2010).
Based, in part, on these reports, we examined the roles of tumor-initiating cells and TRIB2 in lung tumor oncogenesis. Our results show that TRIB2 is overexpressed and often amplified in lung cancer. Upon short hairpin RNA (shRNA)-mediated knockdown of this putative oncogene, tumor cells lines ceased proliferation and failed to produce tumors in vivo when injected into non-obese diabetic/severe combined immunodeficient mice (NOD/SCID) mice. Finally, we show that TRIB2-mediated downregulation of C/EBPα requires the presence of the E3 ligase TRIM21.
Results
TRIB2 is overexpressed and amplified in a subset of human lung cancers
It has been established that growth of cell lines under non-adherent spheroid conditions can increase their tumor-initiating capabilities (for example Ricci-Vitiani et al., 2007; Todaro et al., 2007). To confirm that this was also true for lung cancer cell lines, NCI-H1650 cells that had been grown as monolayers or as spheroids were injected into NOD/SCID mice and tumor growth was measured over time. Cells grown as spheroids were more efficient at initiating tumorigenesis than the corresponding number of cells grown as monolayers (Supplementary Figure S1A). From this data and supporting literature, we reasoned that a gene upregulated in cancer cells grown as spheroids might be instrumental in lung tumorigenesis. To identify potential drivers of lung tumorigenesis, microarray expression analysis was performed comparing the transcriptional profile of eight NSCLC cell lines grown either as monolayers or as spheroids. From this transcriptional analysis, TRIB2 was identified as significantly overexpressed in the spheroid population in eight out of eight NSCLC cell lines examined (P<0.05; Supplementary Table S2). TRIB2 overexpression in the spheroid population ranged from 2 to 76-fold over that of the corresponding monolayer culture. Using quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) analysis, TRIB2 expression levels in the NSCLC cell lines propagated either as monolayers or non-adherent spheroids were confirmed. There was a significant increase in TRIB2 expression levels in all cell lines grown as spheroids relative to monolayer conditions (Figure 1a).
Figure 1.

TRIB2 is overexpressed and amplified in a subset of human lung tumors. (a) Expression of TRIB2 in eight NSCLC cell lines grown as monolayers (white bars) or under low-adherence conditions as spheres (black bars). Expression levels in spheres were normalized to actin and expressed relative to the values in monolayer cultures (P<0.01). (b) Microarray expression analysis of TRIB2 in 68 primary lung tumor samples. Data were normalized to the corresponding value from matched non-tumor samples and expressed as log2. Changes greater than two-fold were considered significant, indicated by the dashed lines. (c) Histology of human lung tumors stained for TRIB2 and Ki67 expression (in brown). Human lung tumor sample LT4 (upper panels) and LT7 (lower panels) stained with TRIB2, hematoxylin and eosin (H&E) and Ki67 to identify dividing cells. Scale bar is 250 μm. (d) Quantitative PCR of TRIB2 gene copy number based on a custom Taqman probe to the second intron. Results are normalized to RNAseP and expressed relative to matched non-tumor samples. Black bars represent gene copy numbers in tumor samples, gray bars represent the matched control tissues from each patient. (e) Fluorescence in situ hybridization analysis from lung cancer cell lines LC-1/sq-SF and H2228, and dissociated cells from human lung tumor samples LT12 and LT13. Probes for TRIB2 gene (green) and centromere of chromosome 2 (red) were used. Yellow spots represent non-specific background.
Based upon these results, we hypothesized that TRIB2 may have a role in human lung tumorigenesis. The expression pattern of TRIB2 in 68 human primary lung tumor samples was subsequently examined using microarray-based mRNA expression analyses. mRNA expression levels were normalized to the corresponding control lung tissue before comparison of TRIB2 levels between tumors. TRIB2 was two- to six-fold overexpressed in 29.4% (20 of 68) of bulk, unsorted primary lung tumor samples (Figure 1b and Supplementary Table S1). The variation in TRIB2 expression levels between primary tumor samples could partially be attributed to the percentage of stem-like cells present within the tumor tissue. Immunohistochemical examination of the tumor samples revealed that for some samples, TRIB2 was overexpressed in a high percentage of cells throughout the tumor, whereas for other samples, high TRIB2 expression was localized to a subset of cells within the tumor tissue, and was relatively low in the surrounding tissues (Figure 1c and Supplementary Figure S1B). In the latter instances, overall TRIB2 expression appeared to be relatively low, but in fact, there were localized regions with intense staining. The subset of cells exhibiting high TRIB2 expression levels may represent tumor-initiating cell populations (Figure 1c and Supplementary Figure S1B).
The TRIB2 gene resides at 2p24, a chromosomal locus prone to frequent breaks and amplifications (Durkin and Glover, 2007). We examined TRIB2 gene copy number in primary human samples to determine whether increased gene copy number could provide an additional explanation for increased TRIB2 expression. Using single-nucleotide polymorphism analysis, gene amplification surrounding the TRIB2 locus was observed in 8 of the 68 samples (12%) previously analyzed (Supplementary Figure 1C). In addition, quantitative PCR analysis of an independent cohort of tumor samples revealed significant and specific gene amplification of 7- to 10-fold at the TRIB2 locus when normalized to non-cancerous counterparts for 3 of 14 (21%) tumor samples analyzed (LT4, LT6 and LT12; Figure 1d). Further, TRIB2 gene amplification was confirmed by fluorescence in situ hybridization analysis in both lung cancer cell lines and in LT12 (Figure 1e). Specific amplification of the TRIB2 locus was observed in LC-1/sq-SF lung cancer cells, whereas the elevated level of TRIB2 in H2228 cells was found to be due to polyploidy. The lung tumor samples examined were generally diploid, and TRIB2 gene copy numbers roughly matched the values obtained by quantitative PCR (Figure 1d). These results indicate that TRIB2 is overexpressed in a subset of lung tumors (around 30%), and that in approximately 10–20% of cases, the overexpression stems from gene amplification.
Depletion of TRIB2 gene expression inhibits proliferation, sphere formation and in vivo tumorigenicity
Based upon observations that overexpression of TRIB2 has a role in cellular transformation (Keeshan et al., 2006; Dedhia et al., 2010; Zanella et al., 2010), the effect of modulating TRIB2 levels using shRNA constructs specific for TRIB2 (sh1–4) or a non-targeting control (shNT) was examined. TRIB2 expression was efficiently depleted using independent shRNA constructs (Figure 2a and Supplementary Figure S2A) in multiple cell lines. Two shRNA constructs were chosen for further analysis, as they consistently exhibited the greatest level of TRIB2 knockdown (Figure 2a). TRIB2 sh1 is directed against the 3′ non-coding region of TRIB2, whereas TRIB2 sh2 is directed against the coding sequence of TRIB2. Both constructs consistently decreased mRNA levels by ~75% (Supplementary Figure S2A).
Figure 2.

Knockdown of TRIB2 by shRNA inhibits cell proliferation and abrogates tumor growth. (a) Western blot of TRIB2 expression in U937 cells infected with sh1 or sh2. Actin is shown as a loading control. *Bands likely represent an alternate isoform of TRIB2. (b) Relative cell proliferation in H226 cells infected with sh1 or sh2. Values are expressed relative to cell number on day 0. *P<0.01 by Student’s t-test relative to non-targeting controls. (c) Tumor growth in NOD/SCID mice injected with 50 000 H226 cells infected (or mock infected) with the indicated shRNA construct directed against TRIB2. Animals were killed once tumor volume reached 1000mm3. Shown is a representative experiment (n=8). Error bars represent standard deviation.
Depletion of TRIB2 in two NSCLC lines, NCI-H226 and NCI-H1650 (H226 and H1650, respectively, hereafter), resulted in significant inhibition of cell proliferation (Figure 2b). This was accompanied by a modest increase in apoptosis (Supplementary Figure S2B). The effect of loss of TRIB2 expression on the ability of cells to grow under low-adherence conditions was also examined. Following knockdown of TRIB2 expression, the ability of cells to quickly re-form spheroids was slightly impaired, and there was marked inhibition of sphere formation during long-term culture (Supplementary Figure S2C). Importantly, the reduction in cell proliferation appears to be specific for transformed cells, as TRIB2 knockdown in normal lung fibroblast and epithelial cells did not inhibit proliferation (Supplementary Figure S2D–E). Together, these data suggest that TRIB2 specifically affects the proliferative capacity of lung cancer cells, and may have a role in cancer stem cell self-renewal.
To more closely mimic the tumor microenvironment, the tumorigenic capacity of cells in which TRIB2 expression had been depleted was assayed in vivo by injection into NOD/SCID mice. H226 or H1650 cells with confirmed knockdown phenotypes were embedded in collagen or matrigel, and injected subcutaneously. Mice injected with mock-infected cells quickly developed large tumors, as did cells expressing non-targeting shRNA after a lag period. In contrast, mice injected with cells in which TRIB2 had been knocked down remained tumor free for up to 240 days after transplant (Figure 2c), at which time the experiment was terminated.
To confirm that inhibition of cell proliferation was indeed due to loss of TRIB2 expression, we performed rescue experiments in which shRNA-resistant TRIB2 cDNAs were overexpressed. Constructs were generated for TRIB2 that either lack the 3′ non-coding region (Trib2s) and thus are not targeted for degradation by sh1, or are mutated such that they are no longer targeted for degradation by sh2 (Trib2m). The in vitro overexpression of Trib2m protein in lung cancer cell lines stably expressing sh2 was sufficient to rescue inhibition of cell proliferation (Figure 3a, Supplementary Figure S3). Similar results were obtained upon rescue of sh1-mediated TRIB2 knockdown with Trib2s (data not shown). We then examined whether introduction of Trib2s and Trib2m in vivo could overcome the antitumorigenic effect of TRIB2 knockdown. H226 or H1650 cells stably expressing sh1 or sh2 were infected with Trib2s or Trib2m, respectively, imbedded in collagen or matrigel and injected into NOD/SCID mice. Overexpression of Trib2m or Trib2s in the presence of shRNA directed against TRIB2 resulted in rapid tumorigenesis (Figure 3b and data not shown). Thus, both in vitro and in vivo inhibition of lung cancer tumor cell proliferation were specifically caused by inhibition of TRIB2 expression.
Figure 3.

Overexpression of TRIB2 rescues antitumorigenic phenotype of TRIB2 knockdown. (a) Relative cell proliferation of H226 cells infected (or mock infected) with shNT or sh2, followed by infection with Trib2m. (b) Tumor growth in NOD/SCID mice after injection of 100 000 H1650 cells infected (or mock infected) with shNT or sh2, followed by infection with Trib2m. All panels show representative experiments (n=6). Error bars represent standard deviation.
TRIB2 knockdown correlates with increased expression and activity of C/EBPα
Previously published work has suggested the existence of an interaction between C/EBPα and TRIB2 (Keeshan et al., 2006; Dedhia et al., 2010), and has shown that C/EBPα is expressed in lung cells during development (Flodby et al., 1996). When TRIB2 expression levels are decreased, several lung cancer cell lines exhibit dramatic phenotypic changes (Figure 4a). As TRIB2 has been shown to be responsible for both downregulation of C/EBPα and inhibition of cell differentiation (Keeshan et al., 2006; Naiki et al., 2007; Dedhia et al., 2010), we investigated whether the morphological changes observed upon TRIB2 knockdown in lung cancer cell lines could be due to increased levels of C/EBPα. Indeed, overexpression of C/EBPα in H226 cells resulted in morphological changes similar to those seen upon TRIB2 knockdown (Figure 4a). To further characterize these changes, immunofluorescence was performed using various lung lineage markers (Kim et al., 2005). Parental H226 and H226shNT cells retained multiple lineage markers indicative of a more primitive cell type, whereas cells that had been infected with sh1 or sh2 to knockdown TRIB2 expression became restricted to either the alveolar type I or II lineages (Supplementary Figure S4A). This result is consistent with the reported role of C/EBPα as a necessary factor for differentiation during development (Flodby et al., 1996), and suggests that upon TRIB2 knockdown, C/EPBα levels increase and drive lung cancer cell differentiation.
Figure 4.

TRIB2 inhibits C/EBPα function. (a) Phase-contrast images of H226 cells infected with shNT, sh1, sh2 or overexpressing C/EBPα. (b) Expression levels of TRIB2 and C/EBPα in the Origene lung cancer panel as measured using qRT-PCR. Values are normalized to actin and expressed relative to the average of the normal samples. Pearson correlation coefficient −0.38, P<0.05. (c) 293T cells were cotransfected with the indicated constructs as well as a reporter vector carrying a 4 × C/EBPα-binding site. Top panel: luciferase activity of the transfected cells. Activity was measured at 24 h after transfection. Shown is a representative experiment (n=6). Error bars are standard error of the mean. *, P<0.05, relative to reporter + CEBPA. Lower panel: western blot of TRIB2 and C/EBPα levels at 24 h after transfection with the indicated constructs. (d) Relative mRNA expression of TRIB2 and C/EBPα target genes normalized to actin. U937 cells were infected with sh1or shNT on day 0. The next day, a subset of cells was collected (day 1 expression levels), and all-trans retinoic acid (ATRA, for granulocyte colony-stimulating factor receptor) or granulocyte colonystimulating factor (for neutrophil elastase) was added to the culture media. Cells surviving puromycin selection were collected on day 4. Error bars represent standard deviation.
As the phenotypic changes observed upon TRIB2 knockdown were similar to the phenotypic changes induced by C/EBPα overexpression in lung cancer cell lines (Figure 4a), C/EBPα levels were also examined in primary lung tumor samples. Pearson product-moment correlation analysis of microarray data of the same 68 primary samples detailed in Figure 1 indicates a negative correlation between TRIB2 and C/EBPα expression (−0.306, P<0.02; Supplementary Figure S4B). To confirm this correlation, qRT-PCR was performed on an independent cohort of primary lung tumor cDNAs, and a significant inverse correlation between TRIB2 and C/EBPα expression levels was again observed (Figure 4b). This indicates that, similar to mouse acute myelogenous leukemia studies (Keeshan et al., 2006; Dedhia et al., 2010), TRIB2 expression might affect the level of C/EBPα expression in human lung cancer.
To determine whether TRIB2 also affects the transactivation capabilities of C/EBPα, a transient transfection system in which forced expression of C/EBPα leads to luciferase reporter activation was employed. Specifically, a stable 293T cell line that expresses a luciferase reporter behind a 4 × C/EBP-binding site was generated, and transient transfections of TRIB2, C/EBPα or both species simultaneously were performed. Transfection of the reporter with C/EBPα alone led to reporter activation approximately 10-fold greater than background levels (Figure 4c). In contrast, cotransfection of both C/EBPα and TRIB2 reproducibly resulted in a three-fold decrease in reporter activation (Figure 4c). Although there was a reduction in reporter activity, C/EBPα protein levels were not affected (Figure 4c). Thus, although coexpression of TRIB2 and C/EBPα leads to a decrease in C/EBPα transactivation activity, it is not due to degradation of C/EBPα.
To further validate the observation that TRIB2 expression affects C/EBPα function, the expression levels of C/EBPα target genes were examined after modulation of TRIB2 levels. After knockdown of expression of TRIB2 using shRNA constructs was confirmed using qRT-PCR and upregulation of C/EBPα was confirmed by western blotting (Figure 4d and Supplementary Figure S4C, respectively), expression of the C/EBPα target genes GCSFR (CSF3; Smith et al., 1996) and NE2 (Oelgeschlager et al., 1996) were examined (Figure 4d). The expression levels of both C/EBPα target genes were increased as much as 5- to 10-fold when TRIB2 expression levels were reduced. This finding further supports the hypothesis that expression of TRIB2 inhibits C/EBPα activity.
TRIB2 interacts with TRIM21 and leads to degradation of C/EBPα
The ability of TRIB2 to affect C/EBPα activity could occur through at least two mechanisms. TRIB2 might bind C/EBPα directly and prevent its transactivation capabilities. Alternatively, the ability of TRIB2 to modulate expression of C/EBPα has been hypothesized to occur via targeting of the transcription factor for proteasomal degradation (Keeshan et al., 2006; Dedhia et al., 2010), although the exact mechanism remains unknown. To distinguish between these two possibilities and identify protein interactors of TRIB2, immunoprecipitation– mass spectrometry was performed. H1650 cells were retrovirally transduced with a C-terminal FLAG-6xHis dual-affinity-tagged TRIB2 or with a TRIB2 construct devoid of the C-terminal dual-affinity tag as a negative control. Clarified lysates were subjected to dual-affinity purification, and bound proteins were identified by mass spectrometry. The E3 ubiquitin ligase TRIM21 (also called Ro52; (Wada and Kamitani 2006; Wada et al., 2009)) was clearly identified in the affinity-tagged construct sample but not observed in the negative control experiment (Supplementary Table S3). Coprecipitation of TRIB2 and TRIM2 in TRIB2-overexpressing H226 cells confirmed the TRIB2/TRIM21 protein interaction (Figure 5a). This, in combination with our results and previously published results detailing the regulation of C/EBPα by TRIB2 (Keeshan et al., 2006; Dedhia et al., 2010), suggests that TRIM21 might be an E3 ubiquitin ligase responsible for targeting C/EBPα for degradation in lung cancer, and that interaction between TRIM21 and TRIB2 is important to this process.
Figure 5.
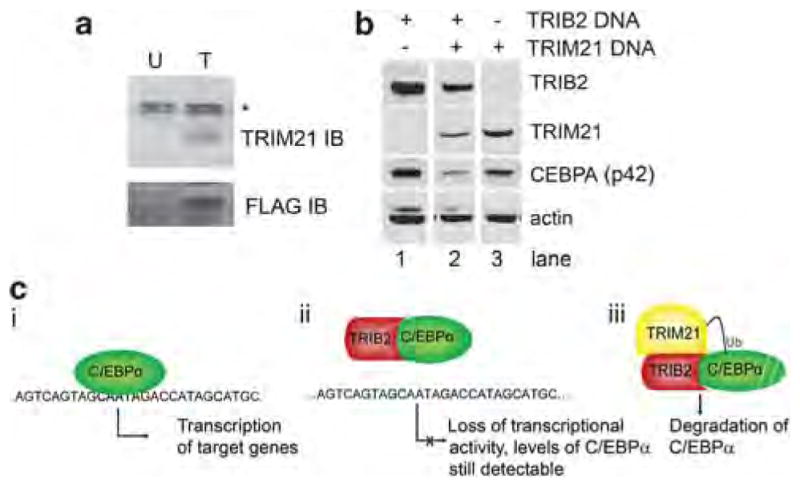
TRIB2 interacts with TRIM21 and regulates C/EPBα. (a) H226 cells infected with a C-terminal FLAG-6xHis dual-affinity- tagged TRIB2 (T) or with a TRIB2 construct devoid of the C-terminal dual-affinity tag (U) as a negative control were subjected to nickel resin precipitation and immunoblotted (IB) with the indicated antibodies. *Non-specific band. (b) 293T cells were transiently transfected with CEBPA in the presence or absence of TRIB2 and TRIM21 transfection, then immunoblotted with the indicated antibodies. All lanes were derived from the same blot with the same exposure, but were non-adjacent in the original image. (c) Depiction of hypothesized protein interactions of TRIB2 and observed consequences. (i) C/EBPα acts as a transcription factor in the absence of TRIB2 and is able to activate transcription. (ii) In the presence of TRIB2, C/EBPα is no longer able to activate transcription, most likely because of an inability to bind DNA. (iii) Upon recruitment of TRIM21 to the TRIB2/C/EBPα complex, degradation of C/EBPα occurs, possibly through ubiquitylation and targeting to the proteasome.
To test the hypothesis that TRIM21 has a role in regulation of C/EBPα levels, a 293T transient transfection system was utilized. Cotransfection of C/EBPα and TRIB2 in the absence of TRIM21 resulted in very little or no degradation of C/EBPα (Figure 5b, lane 1). Likewise, cotransfection of TRIM21 and C/EBPα resulted in little or no change in C/EBPα levels (Figure 5b, lane 3). However, cotransfection of all three species led to a significant decrease in C/EBPα levels (Figure 5b, lane 2). The apparent reduction in TRIM21 expression may be due to TRIM21 autoubiquitination and subsequent degradation (Sabile et al., 2006). Taken together, these results suggest that both TRIB2 and TRIM21 are necessary for modulation of C/EBPα levels.
Discussion
The transcription factor C/EBPα has been shown to be a major player in lung differentiation, and recent reports have suggested that loss of C/EBPα expression may be instrumental in driving the formation of lung adenomas (Koschmieder et al., 2009). Although loss of the chromosomal region surrounding C/EBPα is a common feature of up to 50% of NSCLCs, sequencing studies by several groups failed to identify mutations within the coding region of the gene itself, suggesting an alternative mechanism for C/EBPα downregulation (Koschmieder et al., 2009). One possible mechanism may involve proteasomal degradation of the transcription factor driven by expression of TRIB2 (Grosshans and Wieschaus 2000; Mata et al., 2000; Seher and Leptin 2000; Naiki et al., 2007; Dedhia et al., 2010; Keeshan et al., 2006, 2010). The results presented here support this hypothesis, and further suggest that TRIM21 may be the E3 ubiquitin ligase necessary for C/EBPα degradation. Additionally, the juxtaposition of the reporter data (Figure 4c) and the C/EBPα degradation data (Figure 5b) suggest that although TRIB2 alone is sufficient to block the transcriptional activity of C/EBPα, TRIM21 expression is necessary for downregulation of C/EBPα levels.
Formation of the TRIB2/TRIM21 complex and subsequent downregulation of C/EBPα suggest that TRIB2 may function as a molecular adapter, facilitating assembly of a TRIM21 E3 ligase complex and aiding in the subsequent ubiquitylation and degradation of its substrates, such as C/EBPα. A similar property for the protein kinase dual specificity tyrosine phosphorylation regulated kinase 2 (DYRK2) has been identified, in which DYRK2 functions as a scaffold that mediates assembly of the EDVP E3 ligase complex (Maddika and Chen, 2009). Depletion of DYRK2 by small interfering RNA disrupts formation of the EDVP complex. Additionally, the kinase activity of DYRK2 is necessary for substrate ubiquitylation and degradation by the EDVP complex. TRIB2 may perform a scaffolding function similar to DYRK2, although it may not possess the phosphorylation capabilities, as TRIB2 has been proposed to be a pseudokinase (Hegedus et al., 2007).
An interaction between TRIB2 and the E3 ligase COP1 has previously been reported (Dedhia et al., 2010; Keeshan et al., 2010). The apparent ability of TRIB2 to bind to multiple E3 ligase proteins also supports the hypothesis that TRIB2 functions as a molecular scaffold, and is involved in many processes including C/EBPα degradation via the proteasome. Using an unbiased immunoprecipitation–mass spectrometry approach, we did not observe interaction between TRIB2 and COP1 in lung cancer cell lines, even though COP1 was expressed in those cell lines (data not shown). Perhaps the interaction of TRIB2 with different E3 ligases is context dependent, with the TRIB2/COP1 interaction involved in hematopoietic tumor generation, whereas TRIB2/TRIM21 is involved in solid tumor formation such as lung tumors.
Based on our results and those of others (Keeshan et al., 2006; Dedhia et al., 2010), we propose the following hypothesis for TRIB2 protein interactions and subsequent activities: C/EBPα functions as a transcription factor in the absence of TRIB2 (Figure 5ci). When TRIB2 is present and bound to C/EBPα, transcriptional activity is lost, possibly because of its inability to bind DNA (Figure 5cii). Upon recruitment of TRIM21 to the TRIB2:C/EBPα complex, C/EBPα is degraded in a proteasome-dependent manner (Figure 5ciii). Thus, when TRIM21 is present, increased TRIB2 expression will result in a decrease in CEBPα levels. Moreover, as C/EBPα has been shown to undergo autoregulation (Timchenko et al., 1995), decreased levels of the protein will lead to continued downregulation at the mRNA level.
As TRIM21 appears to mediate the effects of TRIB2, modulation of TRIM21 should phenocopy TRIB2 modulation. However, it has recently been shown that loss of TRIM21 results in the induction of a massive cytokine response, leading to cell death (Yoshimi et al., 2009). Thus, we were unable to confirm the expected phenocopy (data not shown).
Here, we described the role of TRIB2 as a potential oncogene in human lung cancers. We found that the gene is enriched in cells grown as spheroids and, importantly, is overexpressed in a significant percentage of primary lung tumors. Further, we found that this overexpression could be due to genomic amplification of the gene. This finding could enable both better diagnostics and screening for lung cancer as well as improved predictions of lung cancer outcomes. Additionally, inhibition of overexpressed and/or amplified TRIB2 in lung cancers could provide a possible means of treatment, as our results indicate that reducing levels of TRIB2 can efficiently inhibit tumor growth. Together, our findings indicate that overexpression of TRIB2 inhibits the expression of C/EBPα through multiple mechanisms, including transcriptional inactivation and protein degradation, and could consequently lock cells in an undifferentiated, tumor-initiating cell state.
Materials and methods
Primary lung tumor samples, cell proliferation and tumorigenicity
The human primary tumor samples utilized in this study consisted of matched pairs including a bulk tumor sample and adjacent non-tumorous lung tissue from the same patient. They were obtained from Asterand (Detroit, MI, USA; Supplementary Table S1), with approval from the appropriate institutional review boards. Total RNA, complementary DNA (cDNA) or genomic DNA were either obtained directly from Asterand (Figure 1b, Supplementary Figure S1C and S4B) and Origene (Rockville, MD, USA; Figure 4b) or were extracted from bulk tissue samples and used to generate DNA (Qiagen, Valencia, CA, USA; Figure 1d). To assess cell proliferation, cells were seeded at 5000 cells/well in 96-well tissue culture plates and analyzed using CellTiterGlo (Promega, Madison, WI, USA). Tumor growth studies in NOD/SCID mice were performed according to the National Institutes of Health guidelines with protocols approved by the institutional review board of the Genomics Institute of the Novartis Research Foundation.
Constructs and lentiviruses
A 1032-bp fragment encoding the entire human TRIB2 cDNA was subcloned into pcDNA3.1/V5/HIS (Invitrogen, Carlsbad, CA, USA) and pHAGE/CMV/MCS/IRES/ZSgreen vectors (Pan et al., 2008). To generate the Trib2m mutant, silent mutations of the TRIB2 sh2-binding region were introduced by site-directed mutagenesis. Lentiviral vectors expressing shRNA sequences directed against human TRIB2 (sh1–sh4) as well as a non-targeting construct (shNT) were obtained from Sigma-Aldrich (St Louis, MO, USA). The C/EBPα luciferase reporter vector has previously been described (Behre et al., 2002).
Other experimental methods
Additional details and methodologies regarding cell culture, antibodies, microarray procedures, constructs, lentiviruses, immunohistochemistry, immunofluorescence, expression analysis, immunoprecipitations, in vivo tumorigenicity, gene copy number analysis and mass spectrometry can be found in the Supplementary Methods.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This work was supported by grants from the NIH (HL56745 and CA90578) to DGT.
Footnotes
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
Conflict of interest
The authors declare no conflict of interest.
References
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bapat SA, Mali AM, Koppikar CB, Kurrey NK. Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res. 2005;65:3025–3029. doi: 10.1158/0008-5472.CAN-04-3931. [DOI] [PubMed] [Google Scholar]
- Behre G, Singh SM, Liu H, Bortolin LT, Christopeit M, Radomska HS, et al. Ras signaling enhances the activity of C/EBP alpha to induce granulocytic differentiation by phosphorylation of serine 248. J Biol Chem. 2002;277:26293–26299. doi: 10.1074/jbc.M202301200. [DOI] [PubMed] [Google Scholar]
- Cassel TN, Nord M. C/EBP transcription factors in the lung epithelium. Am J Physiol Lung Cell Mol Physiol. 2003;285:L773–L781. doi: 10.1152/ajplung.00023.2003. [DOI] [PubMed] [Google Scholar]
- Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65:10946–10951. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- Costa DB, Li S, Kocher O, Feins RH, Keller SM, Schiller JH, et al. Immunohistochemical analysis of C/EBPalpha in non-small cell lung cancer reveals frequent down-regulation in stage II and IIIA tumors: a correlative study of E3590. Lung Cancer. 2007;56:97–103. doi: 10.1016/j.lungcan.2006.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalerba P, Dylla SJ, Park IK, Liu R, Wang X, Cho RW, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci USA. 2007;104:10158–10163. doi: 10.1073/pnas.0703478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedhia PH, Keeshan K, Uljon S, Xu L, Vega ME, Shestova O, et al. Differential ability of Tribbles family members to promote degradation of C/EBPalpha and induce acute myelogenous leukemia. Blood. 2010;116:1321–1328. doi: 10.1182/blood-2009-07-229450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durkin SG, Glover TW. Chromosome fragile sites. Annu Rev Genet. 2007;41:169–192. doi: 10.1146/annurev.genet.41.042007.165900. [DOI] [PubMed] [Google Scholar]
- Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A, et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008;15:504–514. doi: 10.1038/sj.cdd.4402283. [DOI] [PubMed] [Google Scholar]
- Fang D, Nguyen TK, Leishear K, Finko R, Kulp AN, Hotz S, et al. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005;65:9328–9337. doi: 10.1158/0008-5472.CAN-05-1343. [DOI] [PubMed] [Google Scholar]
- Flodby P, Barlow C, Kylefjord H, Ahrlund-Richter L, Xanthopoulos KG. Increased hepatic cell proliferation and lung abnormalities in mice deficient in CCAAT/enhancer binding protein alpha. J Biol Chem. 1996;271:24753–24760. doi: 10.1074/jbc.271.40.24753. [DOI] [PubMed] [Google Scholar]
- Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- Grosshans J, Wieschaus E. A genetic link between morphogenesis and cell division during formation of the ventral furrow in Drosophila. Cell. 2000;101:523–531. doi: 10.1016/s0092-8674(00)80862-4. [DOI] [PubMed] [Google Scholar]
- Halmos B, Huettner CS, Kocher O, Ferenczi K, Karp DD, Tenen DG. Down-regulation and antiproliferative role of C/EBPalpha in lung cancer. Cancer Res. 2002;62:528–534. [PubMed] [Google Scholar]
- Hegedus Z, Czibula A, Kiss-Toth E. Tribbles: a family of kinase-like proteins with potent signalling regulatory function. Cell Signal. 2007;19:238–250. doi: 10.1016/j.cellsig.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci USA. 2003;100:15178–15183. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- Keeshan K, He Y, Wouters BJ, Shestova O, Xu L, Sai H, et al. Tribbles homolog 2 inactivates C/EBPalpha and causes acute myelogenous leukemia. Cancer Cell. 2006;10:401–411. doi: 10.1016/j.ccr.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeshan K, Bailis W, Dedhia PH, Vega ME, Shestova O, Xu L, et al. Transformation by Tribbles homologue 2 (Trib2) requires both the Trib2 kinase domain and COP1 binding. Blood. 2010;116:4948–4957. doi: 10.1182/blood-2009-10-247361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121:823–835. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- Koschmieder S, Halmos B, Levantini E, Tenen DG. Dysregulation of the C/EBPalpha differentiation pathway in human cancer. J Clin Oncol. 2009;27:619–628. doi: 10.1200/JCO.2008.17.9812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddika S, Chen J. Protein kinase DYRK2 is a scaffold that facilitates assembly of an E3 ligase. Nat Cell Biol. 2009;11:409–419. doi: 10.1038/ncb1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata J, Curado S, Ephrussi A, Rorth P. Tribbles coordinates mitosis and morphogenesis in Drosophila by regulating string/CDC25 proteolysis. Cell. 2000;101:511–522. doi: 10.1016/s0092-8674(00)80861-2. [DOI] [PubMed] [Google Scholar]
- Naiki T, Saijou E, Miyaoka Y, Sekine K, Miyajima A. TRB2, a mouse Tribbles ortholog, suppresses adipocyte differentiation by inhibiting AKT and C/EBPbeta. J Biol Chem. 2007;282:24075–24082. doi: 10.1074/jbc.M701409200. [DOI] [PubMed] [Google Scholar]
- O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- Oelgeschlager M, Nuchprayoon I, Luscher B, Friedman AD. C/EBP, c-Myb, and PU.1 cooperate to regulate the neutrophil elastase promoter. Mol Cell Biol. 1996;16:4717–4725. doi: 10.1128/mcb.16.9.4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H, Mostoslavsky G, Eruslanov E, Kotton DN, Kramnik I. Dual-promoter lentiviral system allows inducible expression of noxious proteins in macrophages. J Immunol Methods. 2008;329:31–44. doi: 10.1016/j.jim.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- Sabile A, Meyer AM, Wirbelauer C, Hess D, Kogel U, Scheffner M, et al. Regulation of p27 degradation and S-phase progression by Ro52 RING finger protein. Mol Cell Biol. 2006;26:5994–6004. doi: 10.1128/MCB.01630-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatton T, Murphy GF, Frank NY, Yamaura K, Waaga-Gasser AM, Gasser M, et al. Identification of cells initiating human melanomas. Nature. 2008;451:345–349. doi: 10.1038/nature06489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seher TC, Leptin M. Tribbles, a cell-cycle brake that coordinates proliferation and morphogenesis during Drosophila gastrulation. Curr Biol. 2000;10:623–629. doi: 10.1016/s0960-9822(00)00502-9. [DOI] [PubMed] [Google Scholar]
- Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- Smith LT, Hohaus S, Gonzalez DA, Dziennis SE, Tenen DG. PU.1 (Spi-1) and C/EBP alpha regulate the granulocyte colony-stimulating factor receptor promoter in myeloid cells. Blood. 1996;88:1234–1247. [PubMed] [Google Scholar]
- Timchenko N, Wilson DR, Taylor LR, Abdelsayed S, Wilde M, Sawadogo M, et al. Autoregulation of the human C/EBP alpha gene by stimulation of upstream stimulatory factor binding. Mol Cell Biol. 1995;15:1192–1202. doi: 10.1128/mcb.15.3.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todaro M, Alea MP, Di Stefano AB, Cammareri P, Vermeulen L, Iovino F, et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell. 2007;1:389–402. doi: 10.1016/j.stem.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Wada K, Kamitani T. Autoantigen Ro52 is an E3 ubiquitin ligase. Biochem Biophys Res Commun. 2006;339:415–421. doi: 10.1016/j.bbrc.2005.11.029. [DOI] [PubMed] [Google Scholar]
- Wada K, Niida M, Tanaka M, Kamitani T. Ro52-mediated monoubiquitination of IKK\{b. J Biochem. 2009;146:821–832. doi: 10.1093/jb/mvp127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimi R, Chang TH, Wang H, Atsumi T, Morse HC, III, Ozato K. Gene disruption study reveals a nonredundant role for TRIM21/Ro52 in NF-kappaB-dependent cytokine expression in fibroblasts. J Immunol. 2009;182:7527–7538. doi: 10.4049/jimmunol.0804121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanella F, Renner O, Garcia B, Callejas S, Dopazo A, Peregrina S, et al. Human TRIB2 is a repressor of FOXO that contributes to the malignant phenotype of melanoma cells. Oncogene. 2010;29:2973–2982. doi: 10.1038/onc.2010.58. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.