

Abstract
Atherosclerotic plaques develop in a non-random manner along the vasculature following a hemodynamically determined distribution profile. The pathogenesis of shear stress-induced inflammation and atherosclerotic lesion formation has led to discussions about personalized strategies in prevention and treatment. Recent discoveries involving the tyrosine kinase receptor, Tie1 in (1) mechanotransduction, (2) inflammation and (3) neovascularization has invigorated these efforts. In this review, we will present the current understanding on Tie1 and its role in these key components of atherogenesis.
Tie1: Overview
The Tie (Tyrosine kinase with Immunoglobulin-like and EGF homology) family of receptor tyrosine kinases, comprising of Tie1 and Tie2, was first reported in 1992 (Partanen et al. 1992). While significant attention has focused on the role of Tie2 in development and disease (Patan 1998), much less is known about Tie1. Identification of a true ligand for Tie1 has remained elusive. Tie1 can be activated by an Angiopoietin 1 (Ang1) chimeric protein (COMP-Ang1) as well as by native Ang1 and Ang4, but not by Ang2 or Ang3 (Saharinen et al. 2005). Notably, co-expression with Tie2 is required for robust Tie1 activation (Saharinen et al. 2005), and association of Tie1 with Tie2 has been reported to modulate Tie2 activation (Yuan et al. 2007). Tie1 is subject to extracellular proteolytic cleavage by various chemical stimuli (Yabkowitz et al. 1999) and physiological levels of shear stress (Chen-Konak et al. 2003)(see below). The truncated Tie1 product persists in the cytosol for several hours, and was also found to associate with the tyrosine phosphatase and adaptor protein Shp2 (Marron et al. 2000), thus providing an additional mechanism for ligand independent modulation of receptor tyrosine kinase signaling in endothelial cells (Singh et al. 2012).
To date, studies have shown that Tie1 is almost exclusively expressed only in endothelial cells and cells of hematopoietic lineage (Partanen et al. 1992, Puri et al. 2003). Tie1 is the last receptor tyrosine kinase expressed during fetal vascular development and ablation of Tie1 expression results in mid- to late-gestation embryonic lethality due to severe edema, hemorrhages and loss of microvessel integrity (Puri et al. 1995, Qu et al. 2010, Sato et al. 1995). Thus, Tie1 is not required for vasculogenesis, but it is required for the integrity (Patan 1998) and survival (Partanen et al. 1996) of vascular endothelial cells. The lymphatic vasculature appears to be particularly sensitive to the level of Tie1 expression as a hypomorphic allele with reduced expression resulted in normal formation of the lymphatic endothelium but failure of the lymphatic vasculature to remodel following initial formation of the vascular plexus (D'Amico et al. 2010, Qu et al. 2010).Tie1 is expressed in the heart, ki dneys, liver, lungs and brain between embryonic day 13.5 and birth. At 6 weeks after birth, Tie1 expression is attenuated throughout the vasculature but expression in the lungs increases up to 4-fold consistent with a role of Tie1 in formation and maturity of the pulmonary vascular bed (Taichman et al. 2003). Tie1 expression is not just increased in adult normal physiology but also in pathologic tumor angiogenesis and arthritis (Kaipainen et al. 1994, Lin et al. 1999, Shahrara et al. 2002).
The Role of Hemodynamic Shear Stress on the Endothelium and Atherosclerosis
Due to the nature of its viscosity and the pressures required for its delivery to the extremities, blood flow exerts a physical force on the vessel wall (Freund et al. 2012). This force can be resolved into primary vectors in three dimensions (Figure 1), namely shear, stretch and compression in each axis. Shear stress is the force in a plane parallel to the vessel wall representing the frictional force that blood flow exerts on the endothelial surface. Movement of fluid has two basic components, a directional scalar and a magnitude vector. Flow patterns in vivo have been broadly separated into laminar and non-laminar flow. The unidirectional movement of fluid and its component particles comprises laminar flow. Non-laminar flow (also commonly referred to as disturbed flow (Asakura et al. 1990, Gimbrone et al. 2000)) is a term for a group of flow profiles that include recirculatory and turbulent flow (Davies et al. 1999, Peacock 1990). Recirculatory flow is characteristic of vascular branch points such as the celiac, mesenteric and renal artery branches of the aorta, and the most studied of which are branches of the aortic arch (Suo et al. 2007). Laminar flow with physiological level of shear stress has been extensively studied and shown to have protective effects on the endothelium (Dimmeler et al. 1996, Levesque et al. 1990). In contrast, non-laminar flow with low shear stress promotes endothelial cell turnover and augments inflammatory responses.
Figure 1.
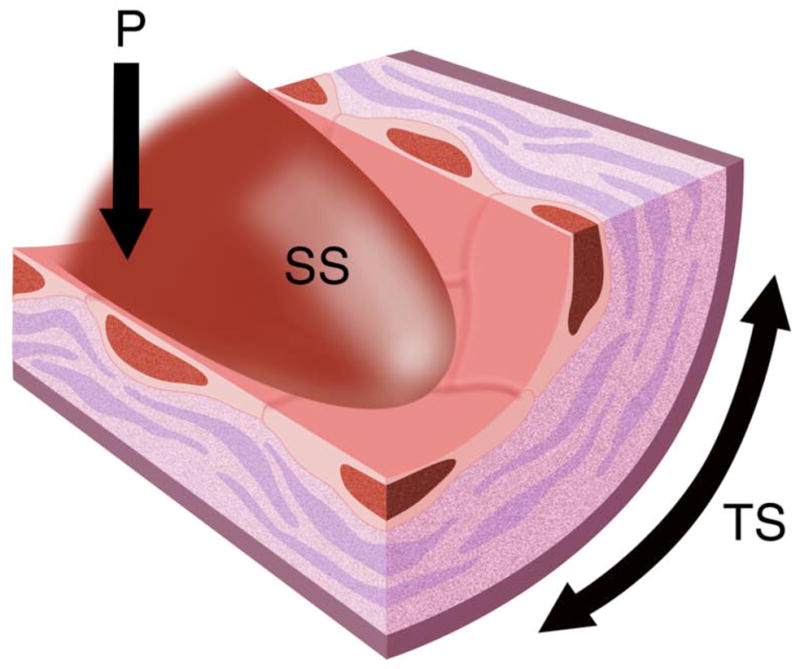
Illustration of pulsatile blood flow with exertion of shear stress (SS), pressure (P) and tensile stress (TS) forces on the vessel wall.
The evolution of shear stress-induced responses in the endothelium is essential to the homeostasis of the vascular trees. Mechanosensors of shear stress include integrins, G protein-coupled receptors, ion channels, the cytoskeleton, PECAM-1, the membrane lipid layer and receptor tyrosine kinases including vascular endothelial growth factor receptor-2 (VEGFR2) (Li et al. 2005), (Figure 2A). Downstream of mechanosensors, the signal is propagated by increases in activation of several signaling cascades including activation of the Rho family of GTPases, mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase (PI3K), extracellular related kinase (ERK1/2), tumor necrosis factor-alpha (TNF-α) and nuclear factor-kappaB (NF-κB) (Li et al. 2005). In addition, endothelial nitric oxide synthase (eNOS) is essential for the synthesis and release of NO, a potent vasodilator, antioxidant and anti-inflammatory molecule. eNOS expression is increased with laminar flow and attenuated with non-laminar flow (Gimbrone et al. 2000). Impaired expression of eNOS is a dominant component of endothelial dysfunction (Moncada 2006), thus eNOS is an important risk factor in atherosclerosis.
Figure 2.




(A) Cell surface shear stress response molecules, vascular endothelial growth factor receptor (VEGFR1/FLT, VEGFR2/KDR), G protein-coupled receptors (GPCR), tyrosine kinase with immunoglobulin-like and epidermal growth factor homology (Tie1, Tie2), integrins, primary cilia, platelet derived growth factor receptor (PDGFR) and ion channels. Endothelial nitric oxide synthase (eNOS) is also depicted. (B) Non-laminar flow preferentially occurs at vessel branch points. (C) Vessel branch points subjected to non-laminar flow are prone to inflammation and atherosclerotic plaque formation. (D) Reduction of endothelial Tie1 expression attenuates atherosclerotic plaque progression and neovascularization.
Atherosclerotic lesions are preferentially localized to the outer walls of arterial branches and to the inner curvatures of tortuous vessels, where flow is disturbed causing increased susceptibility of the vessel wall to endothelial inflammation (Figure 2B) (Asakura et al. 1990, Caro et al. 1971). Immunohistochemistry has shown a predilection of monocyte chemotactic protein-1 (MCP-1) expression at aortic branch points (Malinauskas et al. 1995) and, intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) at the inner curvature of the aortic arch (Suo et al. 2007). The sparing of high net flow regions to lesion formation, and the susceptibility of regions with disturbed flow suggest that laminar flow with high shear stress protects against atherosclerosis, whereas non-laminar flow with low shear stress act as detrimental mechanical stimuli contributing to atherogenesis (Figure 2C) (Gimbrone et al. 2000).
A Role for Tie1 in Mechanotransduction
The most recent additions to the list of shear stress mediators are the Tie family of receptor tyrosine kinases, Tie1 (Chen-Konak et al. 2003) and Tie2 (Lee et al. 2003). Levels of Tie1 decrease with laminar flow in vitro (Woo et al. 2011) and its response alters with acute changes in shear stress magnitudes (Chen-Konak et al. 2003). Conversely, non-laminar flow conditions in vitro upregulates Tie1 promoter activity (Porat et al. 2004). However, the role of Tie1 in shear stress induced vascular diseases has not been extensively studied. As noted above, Tie1 cleavage and generation of an endodomain support the possibility of a shear stress mediated, ligand independent activation of Tie1.
Association of cell surface Tie1 with Tie2 inhibited Ang-2 mediated Tie2 activation (Kim et al. 2006). Additionally, proteolytic processing of Tie1 reduces the accumulation of phosphorylated Tie1 at the cell surface and enhances the responsiveness of Tie2 to Ang1 induced activation (Marron et al. 2007). Interestingly, both Tie1 holoreceptor and endodomain bind with Tie2 (Marron et al. 2000, Tsiamis et al. 2002). These studies together suggest that either the Tie1 holoreceptor or endodomain may play a role in regulating ligand activated Tie2 signaling through the formation of heterodimeric complexes.
Tie1 expression is increased in areas of proatherogenic shear stress, specifically at bifurcations of aortic branches (Woo et al. 2011) and microvasculature (Porat et al. 2004). Notably, expression persists at the aortic arch and its branch vessels, and bifurcations of the vertebral, celiac, mesenteric and renal arteries, established locales subjected to atherogenic disturbed flow. Tie1 expression was also observed at the aortic sinus and the aortic valves, areas characterized by turbulent flow. Tie1 is also pervasive in the aorta of young mice consistent with aortic immaturity; in contrast, expression decreases in the descending aorta characterized by atheroprotective laminar flow with high shear stress. In vivo experiments increasing the shear stress magnitude acutely reduced Tie1 promoter activity (Woo et al. 2011) while increasing eNOS (Cheng et al. 2005). Notably, Tie1 promoter activity was observed in areas of non-laminar flow with wide variations in shear stress, and in areas experiencing low shear stress. In vitro experiments mirrored these findings showing that laminar flow (Woo et al. 2011) downregulates Tie1 expression. Thus, atherogenic non-laminar flow augments Tie1 expression while protective laminar flow suppresses its promoter activity. Whether this is a negative effect of promoter suppression by atheroprotective laminar shear or activation by atherogenic shear is not known. An octameric negative shear stress response element (nSSRE) has been previously reported downregulating Tie1 expression (Chen-Konak et al. 2003). Shear stress response elements are targets of NF-κB transcription factor, which in turn can be regulated by shear stress (Gimbrone et al. 1999). In light of the fact that SSREs have been found in MCP-1, VCAM-1, platelet-derived growth factor (PDGF) and transforming growth factor-β (TGF-β), Tie1 expression may also be regulated by NF-κB. Hence, the shear stress-induced Tie1 cleavage product suggests a potential role for Tie1 in mechanotransduction. Tie1 may also only play a role in response to shear stress by upregulating its own expression and modulating the early atherogenic activity.
A Role for Tie1 in Inflammation
Tie1 expression is increased at the inner curvature of the aortic arch (Woo et al. 2011) and persistent at shoulders of atherosclerotic lesions (Porat et al. 2004). Both are areas of local inflammation that experience non-laminar flow and harbor increased VCAM-1 and ICAM-1 expression. Tie1 is commonly found associated with Tie2 (Marron et al. 2000, Saharinen et al. 2005, Seegar et al. 2010, Yuan et al. 2007). Ang1 induced Tie2 activation promotes endothelial integrity (Papapetropoulos et al. 2000). Stimulation with Ang1 induces association of Tie1 with Tie2 (Saharinen et al. 2005) with the effect of modulating angiopoietin activated Tie2 signaling (Kim et al. 2006, Marron et al. 2000). Thus, Tie1 likely plays a role in endothelial activation by directly inhibiting the ability of Tie2 to maintain endothelial quiescence.
Tie1 is expressed in inflammatory tissue of rheumatoid arthritis and osteoarthritis patients (Shahrara et al. 2002). Overexpression of Tie1 in vitro augments inflammatory markers VCAM-1, ICAM-1 and endothelial (E)-selectin potentially via a PI3K-mediated pathway (Chan et al. 2008). Conversely, analyses of Tie1 siRNA transfected HUVECs decreased inflammatory marker expression (Chan et al. 2009). In vitro experiments in murine aortic endothelial cells showed that 24 hours of atheroprotective laminar flow suppressed Tie1 expression while increasing eNOS (Woo et al. 2011). Tie1 promoter activity in HUVECs was found augmented by pro-inflammatory non-laminar flow conditions (Porat et al. 2004). Loss of Tie1 in murine aortic endothelial cells augmented the anti-inflammatory effect of laminar flow by increasing eNOS response, inhibitor of kappa B (I-kB) activity and decreasing ICAM-1 expression (Woo et al. 2011). Additionally, in vivo analyses of Tie1 deleted mice revealed attenuated Rho-associated protein kinase (ROCK) signaling leading to increased eNOS response. Therefore, Tie1 plays a role in endothelial cell activation associated with inflammation.
A Role for Tie1 in Neovascularization
Development of atherosclerosis begins in childhood with the development of fatty streaks. As the diffusion capacity of oxygen declines with plaque thickening, increasing angiogenic factors promote vessel formation from the luminal surface and vasa vasorum. Advanced lesions often contain a necrotic lipid-rich core and are self-sustaining via neovascularization (Moreno et al. 2004). Intraplaque vessels deliver oxygen and inflammatory factors essential for the survival and progression of lesions. These advanced plaques are often at risk for focal rupture of the thin fibrous cap, leading to formation of thrombi or release of microemboli into the bloodstream, and further enlarging the plaque by inward hemorrhage. Superimposed thrombus formation on the surface of the ruptured plaques greatly increases the risk of local arterial stenosis and worsens the prognosis. Recent studies have documented that plaque microvessel formation not only supplies nutrients but is a major entry pathway essential for leukocyte invasion and progression of disease (Eriksson 2011). Inhibition of angiogenesis in vivo attenuated neovascularization events and decreased atherosclerosis burden (Moulton et al. 2003). Previous documentation of intraplaque Tie1 expression (Porat et al. 2004), combined with its known role in capillary and small vessel viability raises the possibility of a third role for Tie1 in atherosclerosis progression via it’s role in maintaining vessel stability, though this has yet to be validated.
A Role for Tie1 in Atherosclerosis
Given its role in endothelial mechanotransduction, endothelial response to inflammation, and endothelial stability, it is not surprising that Tie1 plays a central role in atherosclerotic disease. Our laboratory documented that moderate reductions in Tie1 elicited 35% and 38% reductions in atherosclerotic lesions of 24 and 49 week-old mice respectively. Remarkably, further reduction of Tie1 by up to 80% resulted in 68% and 70% reduction in lesions of 12 and 24 week-old mice, respectively (Figure 3). This suggests a unique dose dependent amelioration of atherosclerosis progression conferred by attenuation of Tie1 induced inflammation (Figure 2D). Additionally, as discussed above, the reduction of lesion burden may also be due in part to reduction of intraplaque angiogenic events.
Figure 3.

Endothelial Specific Tie1 Deletion Reduces Atherosclerosis Burden. (A) Representative image of H&E stained aortic valve showing endothelial specific LacZ expression (arrowheads) from tamoxifen treated SCL-ERT-Cre:Rosa26R-LacZ mouse (100× magnification). (B) RT-PCR analysis of aortic Tie1 levels from tamoxifen treated Tie1−/flox:SCL-ERT-Cre mice (65% reduction, p<0.05) (C) Representative images of sudan IV stained distal aorta from Tie1−/flox: SCL-ERT-Cre:ApoE−/− and Tie1flox/flox:ApoE−/− mice depicting lesion reductions in Tie1 deleted mice. Graphical comparisons of mean aortic lesion area in tamoxifen treated Tie1−/flox: SCL-ERT-Cre:ApoE−/− and Tie1flox/flox:ApoE−/− mice analyzed at (D) 12 weeks (0.765 ± 0.114% vs. 0.244 ± 0.046%, p<0.0005) and (E) 24 weeks (3.644 ± 0.865% vs. 1.108 ± 0.207%, p<0.006). Data points denote individual animals, and horizontal bars indicate group average.
(modified with permission from (Woo et al. 2011))
Interestingly, attenuation of Tie1 did not significantly alter the extent of atherosclerotic lesions in the aortic valve region. Instead, Tie1 attenuation elicited a trend of increased lesions at the aortic sinus, an effect opposite to that observed in the distal aorta. Two recent studies highlighted regional differences in atherosclerotic plaque reduction of PECAM-1 knockout mice. PECAM-1 deletion in mice with reduced LDL receptor (Goel et al. 2008) or apolipoprotein E (ApoE) expression (Harry et al. 2008) significantly increased lesion formation in the aorta, however lesions at the lesser curvature of the aortic arch were decreased. The aortic valve experiences a unique and complex shear stress profile not evidenced elsewhere in the vasculature. Peacock previously described the flow profile in the aortic sinus as turbulent, comprising spinning vortices superimposed with random motion of fluid particles (Peacock 1990). In contrast, the flow profile at aortic bifurcations distal to the aortic sinus has been described as recirculatory and is characterized by bidirectional blood flow with the resultant effect of a low time-averaged shear stress. Notably, Tie1 expression was augmented on the fibrosa side of the aortic valve leaflet (non-laminar flow) but was absent from the ventricularis surface (laminar flow) (Woo et al. 2011). Hence, Tie1 may regulate atherosclerosis progression in a novel shear stress-defined, location-specific manner.
Conclusion and Future Directions
While Tie1 appears to play a role in development and progression of atherosclerosis, we are just beginning to unravel its role in this complicated process. Low shear stress is a key factor in the transition of immature plaques to high risk, rupture-prone thin cap fibroatheromas (Koskinas et al. 2009). Tie1 expression is maintained in similar regions of intrinsic lower shear stress, and expression can be enhanced at induced lower shear stress regions using in vivo shear stress modifying casts (Woo et al. 2011). Since Tie1 plays a role in local endothelial inflammation, a process that is critical for the evolution of early plaques into rupture-prone atheromas, we opine that Tie1 signaling may mediate the development of these high-risk plaques. The effect of Tie1 attenuation on the transformation of thin cap fibroatheromas is currently under investigation.
While Tie1 responds to shear stress by altering expression and modulating downstream signaling activity, it is unclear whether the holoreceptor is mechanically altered by forces it experiences. Specifically, does the receptor conformation change with the flow profile and/or the magnitude of force? Alteration of extracellular receptor conformation may facilitate ligand association or dimerization, potentially with Tie2, as inhibition of Tie2 protects against atherosclerosis (Ahmed et al. 2009, Hauer et al. 2009) and our experiments indicate that Tie2 activation is enhanced in the face of Tie1 deletion in vitro (Woo et al. 2011).While Tie1 activity is clearly modulated by hemodynamics, it is not clear whether it is a true mechanosensor, i.e. activation of Tie1 by shear stress results in a direct effect on signal transduction, or whether Tie1 expression may merely be upregulated in response to shear stress-induced signals.
As discussed above and illustrated in Figure 1,the biomechanical force imposed by blood flow in vivo comprises three components: shear, stretch and compression, it is likely that these components in concert effect Tie1 mediated signal transduction. Cyclic stretch increases Tie1 expression in vitro in coronary endothelial cells (Zheng et al. 2004), and in vivo at the aortic valve leaflet (Woo et al. 2011). Arterial segments exposed to cyclic flexure are subjected to increased apoptosis and macromolecular permeability at the endothelium (Van Epps et al. 2009). Thus, it will be beneficial to elucidate the response of Tie1 to the combined effects of stretch and shear stress as seen in aortic valve development and disease. It will also be important to define the effect of more complicated turbulent flow profiles characteristic of the aortic sinuses.
In conclusion, Tie1 is involved in key atherogenic elements including (1) mechanotransduction of hemodynamic forces, (2) regulating inflammation and (3) modulating angiogenic events in advanced lesions. The prophylactic alleviation of atherosclerosis burden by the reduction of Tie1 expression and signaling suggests that Tie1 might prove to be a strong and attractive candidate for a targeted therapeutic approach, but we are just beginning to understand its role in this important disease process.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmed A, Fujisawa T, Niu XL, et al. Angiopoietin-2 confers Atheroprotection in apoE−/− mice by inhibiting LDL oxidation via nitric oxide. Circ Res. 2009;104:1333–1336. doi: 10.1161/CIRCRESAHA.109.196154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asakura T, Karino T. Flow patterns and spatial distribution of atherosclerotic lesions in human coronary arteries. Circ Res. 1990;66:1045–1066. doi: 10.1161/01.res.66.4.1045. [DOI] [PubMed] [Google Scholar]
- Caro CG, Fitz-Gerald JM, Schroter RC. Atheroma and arterial wall shear. Observation, correlation and proposal of a shear dependent mass transfer mechanism for atherogenesis. Proc R Soc Lond B Biol Sci. 1971;177:109–159. doi: 10.1098/rspb.1971.0019. [DOI] [PubMed] [Google Scholar]
- Chan B, Yuan HT, Ananth Karumanchi S, Sukhatme VP. Receptor tyrosine kinase Tie-1 overexpression in endothelial cells upregulates adhesion molecules. Biochem Biophys Res Commun. 2008;371:475–479. doi: 10.1016/j.bbrc.2008.04.091. [DOI] [PubMed] [Google Scholar]
- Chan B, Sukhatme VP. Suppression of Tie-1 in endothelial cells in vitro induces a change in the genome-wide expression profile reflecting an inflammatory function. FEBS Lett. 2009;583:1023–1028. doi: 10.1016/j.febslet.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Konak L, Guetta-Shubin Y, Yahav H, et al. Transcriptional and post-translation regulation of the Tie1 receptor by fluid shear stress changes in vascular endothelial cells. Faseb J. 2003;17:2121–2123. doi: 10.1096/fj.02-1151fje. [DOI] [PubMed] [Google Scholar]
- Cheng C, van Haperen R, de Waard M, et al. Shear stress affects the intracellular distribution of eNOS: direct demonstration by a novel in vivo technique. Blood. 2005;106:3691–3698. doi: 10.1182/blood-2005-06-2326. [DOI] [PubMed] [Google Scholar]
- D'Amico G, Korhonen EA, Waltari M, Saharinen P, Laakkonen P, Alitalo K. Loss of endothelial Tie1 receptor impairs lymphatic vessel development-brief report. Arterioscler Thromb Vasc Biol. 2010;30:207–209. doi: 10.1161/ATVBAHA.109.196618. [DOI] [PubMed] [Google Scholar]
- Davies PF, Polacek DC, Handen JS, Helmke BP, DePaola N. A spatial approach to transcriptional profiling: mechanotransduction and the focal origin of atherosclerosis. Trends Biotechnol. 1999;17:347–351. doi: 10.1016/s0167-7799(99)01348-7. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Haendeler J, Rippmann V, Nehls M, Zeiher AM. Shear stress inhibits apoptosis of human endothelial cells. FEBS Lett. 1996;399:71–74. doi: 10.1016/s0014-5793(96)01289-6. [DOI] [PubMed] [Google Scholar]
- Eriksson EE. Intravital microscopy on atherosclerosis in apolipoprotein e-deficient mice establishes microvessels as major entry pathways for leukocytes to advanced lesions. Circulation. 2011;124:2129–2138. doi: 10.1161/CIRCULATIONAHA.111.030627. [DOI] [PubMed] [Google Scholar]
- Freund JB, Goetz JG, Hill KL, Vermot J. Fluid flows and forces in development: functions, features and biophysical principles. Development. 2012;139:1229–1245. doi: 10.1242/dev.073593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimbrone MA, Jr, Anderson KR, Topper JN, et al. Special communicationthe critical role of mechanical forces in blood vessel development, physiology and pathology. J Vasc Surg. 1999;29:1104–1151. doi: 10.1016/s0741-5214(99)70252-1. [DOI] [PubMed] [Google Scholar]
- Gimbrone MA, Jr, Topper JN, Nagel T, Anderson KR, Garcia-Cardena G. Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann N Y Acad Sci. 2000;902:230–239. doi: 10.1111/j.1749-6632.2000.tb06318.x. discussion 239–240. [DOI] [PubMed] [Google Scholar]
- Goel R, Schrank BR, Arora S, et al. Site-specific effects of PECAM-1 on atherosclerosis in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2008;28:1996–2002. doi: 10.1161/ATVBAHA.108.172270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harry BL, Sanders JM, Feaver RE, et al. Endothelial cell PECAM-1 promotes atherosclerotic lesions in areas of disturbed flow in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2008;28:2003–2008. doi: 10.1161/ATVBAHA.108.164707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauer AD, Habets KL, van Wanrooij EJ, et al. Vaccination against TIE2 reduces atherosclerosis. Atherosclerosis. 2009;204:365–371. doi: 10.1016/j.atherosclerosis.2008.09.039. [DOI] [PubMed] [Google Scholar]
- Kaipainen A, Vlaykova T, Hatva E, et al. Enhanced expression of the tie receptor tyrosine kinase mesenger RNA in the vascular endothelium of metastatic melanomas. Cancer Res. 1994;54:6571–6577. [PubMed] [Google Scholar]
- Kim KL, Shin IS, Kim JM, et al. Interaction between Tie receptors modulates angiogenic activity of angiopoietin2 in endothelial progenitor cells. Cardiovasc Res. 2006;72:394–402. doi: 10.1016/j.cardiores.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Koskinas KC, Chatzizisis YS, Baker AB, Edelman ER, Stone PH, Feldman CL. The role of low endothelial shear stress in the conversion of atherosclerotic lesions from stable to unstable plaque. Curr Opin Cardiol. 2009 doi: 10.1097/HCO.0b013e328331630b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Koh GY. Shear stress activates Tie2 receptor tyrosine kinase in human endothelial cells. Biochem Biophys Res Commun. 2003;304:399–404. doi: 10.1016/s0006-291x(03)00592-8. [DOI] [PubMed] [Google Scholar]
- Levesque MJ, Nerem RM, Sprague EA. Vascular endothelial cell proliferation in culture and the influence of flow. Biomaterials. 1990;11:702–707. doi: 10.1016/0142-9612(90)90031-k. [DOI] [PubMed] [Google Scholar]
- Li YS, Haga JH, Chien S. Molecular basis of the effects of shear stress on vascular endothelial cells. J Biomech. 2005;38:1949–1971. doi: 10.1016/j.jbiomech.2004.09.030. [DOI] [PubMed] [Google Scholar]
- Lin WC, Li AF, Chi CW, et al. tie-1 protein tyrosine kinase: a novel independent prognostic marker for gastric cancer. Clin Cancer Res. 1999;5:1745–1751. [PubMed] [Google Scholar]
- Malinauskas RA, Herrmann RA, Truskey GA. The distribution of intimal white blood cells in the normal rabbit aorta. Atherosclerosis. 1995;115:147–163. doi: 10.1016/0021-9150(94)05497-7. [DOI] [PubMed] [Google Scholar]
- Marron MB, Hughes DP, Edge MD, Forder CL, Brindle NP. Evidence for heterotypic interaction between the receptor tyrosine kinases TIE-1 and TIE-2. J Biol Chem. 2000;275:39741–39746. doi: 10.1074/jbc.M007189200. [DOI] [PubMed] [Google Scholar]
- Marron MB, Hughes DP, McCarthy MJ, Beaumont ER, Brindle NP. Tie-1 receptor tyrosine kinase endodomain interaction with SHP2: potential signalling mechanisms and roles in angiogenesis. Adv Exp Med Biol. 2000;476:35–46. doi: 10.1007/978-1-4615-4221-6_3. [DOI] [PubMed] [Google Scholar]
- Marron MB, Singh H, Tahir TA, et al. Regulated proteolytic processing of Tie1 modulates ligand responsiveness of the receptor-tyrosine kinase Tie2. J Biol Chem. 2007;282:30509–30517. doi: 10.1074/jbc.M702535200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncada S. Adventures in vascular biology: a tale of two mediators. Philos Trans R Soc Lond B Biol Sci. 2006;361:735–759. doi: 10.1098/rstb.2005.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno PR, Purushothaman KR, Fuster V, et al. Plaque neovascularization is increased in ruptured atherosclerotic lesions of human aorta: implications for plaque vulnerability. Circulation. 2004;110:2032–2038. doi: 10.1161/01.CIR.0000143233.87854.23. [DOI] [PubMed] [Google Scholar]
- Moulton KS, Vakili K, Zurakowski D, et al. Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc Natl Acad Sci U S A. 2003;100:4736–4741. doi: 10.1073/pnas.0730843100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetropoulos A, Fulton D, Mahboubi K, et al. Angiopoietin-1 inhibits endothelial cell apoptosis via the Akt/survivin pathway. J Biol Chem. 2000;275:9102–9105. doi: 10.1074/jbc.275.13.9102. [DOI] [PubMed] [Google Scholar]
- Partanen J, Armstrong E, Makela TP, et al. A novel endothelial cell surface receptor tyrosine kinase with extracellular epidermal growth factor homology domains. Mol Cell Biol. 1992;12:1698–1707. doi: 10.1128/mcb.12.4.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partanen J, Puri MC, Schwartz L, Fischer KD, Bernstein A, Rossant J. Cell autonomous functions of the receptor tyrosine kinase TIE in a late phase of angiogenic capillary growth and endothelial cell survival during murine development. Development. 1996;122:3013–3021. doi: 10.1242/dev.122.10.3013. [DOI] [PubMed] [Google Scholar]
- Patan S. TIE1 and TIE2 receptor tyrosine kinases inversely regulate embryonic angiogenesis by the mechanism of intussusceptive microvascular growth. Microvasc Res. 1998;56:1–21. doi: 10.1006/mvre.1998.2081. [DOI] [PubMed] [Google Scholar]
- Peacock JA. An in vitro study of the onset of turbulence in the sinus of Valsalva. Circ Res. 1990;67:448–460. doi: 10.1161/01.res.67.2.448. [DOI] [PubMed] [Google Scholar]
- Porat RM, Grunewald M, Globerman A, et al. Specific induction of tie1 promoter by disturbed flow in atherosclerosis-prone vascular niches and flow-obstructing pathologies. Circ Res. 2004;94:394–401. doi: 10.1161/01.RES.0000111803.92923.D6. [DOI] [PubMed] [Google Scholar]
- Puri MC, Rossant J, Alitalo K, Bernstein A, Partanen J. The receptor tyrosine kinase TIE is required for integrity and survival of vascular endothelial cells. Embo J. 1995;14:5884–5891. doi: 10.1002/j.1460-2075.1995.tb00276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri MC, Bernstein A. Requirement for the TIE family of receptor tyrosine kinases in adult but not fetal hematopoiesis. Proc Natl Acad Sci U S A. 2003;100:12753–12758. doi: 10.1073/pnas.2133552100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Tompkins K, Batts L, Puri MC, Baldwin HS. Abnormal embryonic lymphatic vessel development in Tie1 hypomorphic mice. 2010 doi: 10.1242/dev.043380. paper in review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saharinen P, Kerkela K, Ekman N, et al. Multiple angiopoietin recombinant proteins activate the Tie1 receptor tyrosine kinase and promote its interaction with Tie2. J Cell Biol. 2005;169:239–243. doi: 10.1083/jcb.200411105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato TN, Tozawa Y, Deutsch U, et al. Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature. 1995;376:70–74. doi: 10.1038/376070a0. [DOI] [PubMed] [Google Scholar]
- Seegar TC, Eller B, Tzvetkova-Robev D, et al. Tie1-Tie2 interactions mediate functional differences between angiopoietin ligands. Mol Cell. 2010;37:643–655. doi: 10.1016/j.molcel.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahrara S, Volin MV, Connors MA, Haines GK, Koch AE. Differential expression of the angiogenic Tie receptor family in arthritic and normal synovial tissue. Arthritis Res. 2002;4:201–208. doi: 10.1186/ar407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh H, Hansen TM, Patel N, Brindle NP. The Molecular Balance between Receptor Tyrosine Kinases Tie1 and Tie2 Is Dynamically Controlled by VEGF and TNFalpha and Regulates Angiopoietin Signalling. PLoS One. 2012;7:e29319. doi: 10.1371/journal.pone.0029319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suo J, Ferrara DE, Sorescu D, Guldberg RE, Taylor WR, Giddens DP. Hemodynamic shear stresses in mouse aortas: implications for atherogenesis. Arterioscler Thromb Vasc Biol. 2007;27:346–351. doi: 10.1161/01.ATV.0000253492.45717.46. [DOI] [PubMed] [Google Scholar]
- Taichman DB, Schachtner SK, Li Y, Puri MC, Bernstein A, Scott Baldwin H. A unique pattern of Tie1 expression in the developing murine lung. Exp Lung Res. 2003;29:113–122. doi: 10.1080/01902140303767. [DOI] [PubMed] [Google Scholar]
- Tsiamis AC, Morris PN, Marron MB, Brindle NP. Vascular endothelial growth factor modulates the Tie-2:Tie-1 receptor complex. Microvasc Res. 2002;63:149–158. doi: 10.1006/mvre.2001.2377. [DOI] [PubMed] [Google Scholar]
- Van Epps JS, Chew DW, Vorp DA. Effects of cyclic flexure on endothelial permeability and apoptosis in arterial segments perfused ex vivo. J Biomech Eng. 2009;131:101005. doi: 10.1115/1.3192143. [DOI] [PubMed] [Google Scholar]
- Woo KV, Qu X, Babaev VR, et al. Tie1 attenuation reduces murine atherosclerosis in a dose-dependent and shear stress-specific manner. J Clin Invest. 2011;121:1624–1635. doi: 10.1172/JCI42040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabkowitz R, Meyer S, Black T, Elliott G, Merewether LA, Yamane HK. Inflammatory cytokines and vascular endothelial growth factor stimulate the release of soluble tie receptor from human endothelial cells via metalloprotease activation. Blood. 1999;93:1969–1979. [PubMed] [Google Scholar]
- Yuan HT, Venkatesha S, Chan B, et al. Activation of the orphan endothelial receptor Tie1 modifies Tie2-mediated intracellular signaling and cell survival. Faseb J. 2007;21:3171–3183. doi: 10.1096/fj.07-8487com. [DOI] [PubMed] [Google Scholar]
- Zheng W, Christensen LP, Tomanek RJ. Stretch induces upregulation of key tyrosine kinase receptors in microvascular endothelial cells. Am J Physiol Heart Circ Physiol. 2004;287:H2739–2745. doi: 10.1152/ajpheart.00410.2004. [DOI] [PubMed] [Google Scholar]