

Abstract
Parkinson's disease (PD) is characterized pathologically by the formation of ubiquitin and α-synuclein (α-syn)-containing inclusions (Lewy bodies), dystrophic dopamine (DA) terminals, and degeneration of midbrain DA neurons. The precise molecular mechanisms underlying these pathological features remain elusive. Accumulating evidence has implicated dysfunctional autophagy, the cell self-digestion and neuroprotective pathway, as one of the pathogenic systems contributing to the development of idiopathic PD. Here we characterize autophagy-deficient mouse models and provide in vivo evidence for the potential role that impaired autophagy plays in pathogenesis associated with PD. Cell-specific deletion of essential autophagy gene Atg7 in midbrain DA neurons causes delayed neurodegeneration, accompanied by late-onset locomotor deficits. In contrast, Atg7-deficient DA neurons in the midbrain exhibit early dendritic and axonal dystrophy, reduced striatal dopamine content, and the formation of somatic and dendritic ubiquitinated inclusions in DA neurons. Furthermore, whole-brain-specific loss of Atg7 leads to presynaptic accumulation of α-syn and LRRK2 proteins, which are encoded by two autosomal dominantly inherited PD-related genes. Our results suggest that disrupted autophagy may be associated with enhanced levels of endogenous α-syn and LRRK2 proteins in vivo. Our findings implicate dysfunctional autophagy as one of the failing cellular mechanisms involved in the pathogenesis of idiopathic PD.
Introduction
Parkinson's disease (PD) is the most common neurodegenerative movement disorder and is characterized by the loss of nigrostriatal dopamine (DA) neurons, degeneration of striatal DAergic axons, and formation of intracellular Lewy bodies, which consist primarily of α-synuclein (α-syn) and ubiquitin. Both genetic and environmental factors contribute to PD pathogenesis, but the underlying molecular mechanisms remain elusive. The vast majority of PD occurs sporadically while inherited familial forms of the disease account for ∼5% of all cases (Dauer and Przedborski, 2003). The identification of genetic mutations has implicated several cellular pathways in PD etiology, with growing evidence suggesting a link between dysfunctional intracellular catabolism and PD pathogenesis.
The autophagy-lysosome system is a major catabolic pathway that degrades long-lived protein complexes/aggregates and cellular organelles. Macroautophagy (autophagy) involves the formation and trafficking of autophagosomes and their subsequent degradation by lysosomes (Mizushima et al., 2008). Several lines of evidence show that autophagy degrades α-syn, particularly aggregate-prone forms of PD-linked mutants (Webb et al., 2003; Spencer et al., 2009; Yu et al., 2009). Moreover, chaperone-mediated autophagy (CMA), another lysosomal pathway, was shown to degrade human wild-type, but not mutant forms, of α-syn (Cuervo et al., 2004). LRRK2, another PD-linked gene product, is occasionally found in α-syn-positive inclusions in postmortem PD brains (Zhu et al., 2006). Autosomal dominant mutations in LRRK2 are the most common genetic cause of familial PD and have been linked to sporadic forms of the disease. A recent study reports that LRRK2 overexpression causes impaired clearance of proteasomal substrates (Lichtenberg et al., 2011), and suggests a role for LRRK2 in the regulation of intracellular catabolism. Additionally, expression of PD-related LRRK2 mutations produces autophagic abnormalities (Alegre-Abarrategui et al., 2009; Ramonet et al., 2011), however it remains unclear whether altered autophagy can promote abnormal LRRK2 expression.
While numerous studies have shown that basal autophagy plays a vital neuroprotective role in CNS neurons (Hara et al., 2006; Komatsu et al., 2006), in vivo evidence linking dysfunctional autophagic degradation to nigrostriatal neurodegeneration is lacking. To understand the effects of autophagy impairment on midbrain DA neuron pathology, we characterized conditional knock-out mice with cell-specific deletion of the essential autophagy gene, Atg7, in DA neurons. Atg7 is an E1-like activating enzyme required for autophagosome formation (Kirisako et al., 2000). Genetic ablation of Atg7 in DA neurons unexpectedly causes a delayed and moderate loss of DAergic somata, which is preceded by the early and frequent appearance of enlarged DAergic axon and dendrite varicosities. In addition, Atg7 inactivation in the CNS promotes presynaptic accumulation of endogenous α-syn and LRRK2 proteins in the brain, indicating a possible link between impaired autophagy and the accumulation of two PD-related proteins. Our findings also suggest that autophagy deficiency in DA neurons leads to pathogenic events that partially resemble human disease progression. The present study, therefore, implicates dysfunctional autophagy as a potential contributing factor in idiopathic PD.
Materials and Methods
Animals.
Floxed Atg7 mice were characterized previously (Komatsu et al., 2006) and were crossed with either TH-IRES-Cre mice (gift from Dr. Ted Ebendal, Uppsala University, Uppsala, Sweden) to generate Atg7fl/fl;TH-IRES-Cre mice or with a Nestin-Cre driven line to produce Atg7fl/fl;Nestin-Cre mice. Both male and female mice were used for experimentation. Genomic DNA was purified from tail biopsy and mice were genotyped by PCR. All animal protocols were approved by the intramural Institutional Animal Care and Use Committee (Mount Sinai School of Medicine).
Immunohistological examination.
Mice were anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/ml) and transcardially perfused with 0.1 m PBS containing 4% paraformaldehyde. Tissues were postfixed for 12–16 h and cryoprotected in 30% sucrose/PBS. Frozen tissue was sectioned serially (30 μm) on a cryostat. Free-floating sections were blocked in PBS supplemented with 10% serum and 0.25% Triton X-100, and then incubated overnight with one or more of the following antibodies (where indicated): rabbit polyclonal Atg7 (1:200; gift from Dr. Takashi Ueno, Juntendo University, Tokyo, Japan), mouse monoclonal tyrosine hydroxylase (1:1000; Pel Freeze Biological), rabbit polyclonal TH (1:1000; Millipore), guinea pig polyclonal p62/SQSTM1 (1:1000; American Research Products), rabbit polyclonal ubiquitin (1:300; Dako), mouse monoclonal synaptophysin (1:100; American Research Products), mouse monoclonal calbindin D-28K (1:1000; Swant), rabbit polyclonal calbindin (1:500; Millipore). For mouse monoclonal Syn 303 (1:1000; gift from Dr. Virginia Lee, University of Pennsylvania, Philadelphia, Pennsylvania) and rabbit monoclonal LRRK2 C42–1 (1:250; Epitomics) antibodies, a heat-based antigen retrieval protocol was used (Jiao et al., 1999). Sections were incubated in species-appropriate biotinylated secondary antibodies and were visualized by either diaminobenzidine (DAB) for light microscopy or avidin-conjugated fluorophores and 4′,6-diamidino-2-phenylindole (DAPI) for confocal imaging.
Image analysis.
Fluorescent images were acquired on an LSM510 Meta confocal microscope (Zeiss). For quantitative analysis of dystrophic axon swellings and axon fiber density, confocal images were acquired with a 63×/1.4 N.A. oil-immersion objective and a zoom factor of two. Twenty-four images per animal (eight images/section through the coronal plane) were taken from the dorsolateral striatum in 3–5 animals per group. All images were acquired using the same microscope and camera settings. Puncta counts and density from deconvolved images were determined by MetaMorph software (Molecular Devices). Dystrophic axon terminals were defined as puncta ≥0.6 μm2. Brightfield images were acquired on an Axioplan 2IE microscope (Zeiss). For quantitative analysis of TH-labeled dendritic varicosities, 18 images per animal (six images/section) from the substantia nigra pars reticulata (SNpr) in three animals per group were taken with a 63×/1.4 N.A. oil-immersion lens. Punctate structures were quantified by ImageJ software (NIH). Quantitative analyses for dystrophic axon terminals, axon fiber density, and dendritic varicosities were performed by an observer who was not blinded to genotype and age.
Stereology.
Stereological analysis was performed as previously described (Li et al., 2010). Immunolabeling with anti-TH antibody and cresyl violet counterstain was performed on every fourth section through the substantia nigra pars compacta (SNpc). Unbiased stereological analysis was performed by a blinded observer using the optical fractionator method in Stereo Investigator 10 (Microbrightfield). Contours were drawn around the SNpc at 10× magnification. Stereological counting was performed at 63× magnification. Coefficient of error was set at p < 0.1.
Behavioral tests.
Locomotor behavior was assessed in mice at 4 months and 9 months of age. The automated open field (Accuscan) was used to monitor spontaneous locomotor activity. Mice were placed into a 16 × 16 inch chamber, which was equipped with photocell emitters projecting 32 infrared beams at ground level and 16 beams at 3 inches above the ground. Photocell analyzers recorded and tallied beam breaks to measure horizontal and vertical activity over 30 min. For the challenging beam traversal, mice were trained to cross a 100 cm long Plexiglas beam consisting of four 25 cm sections, which gradually narrowed in width (3.5, 2.5, 1.5, and 0.5 cm; Fleming et al., 2004). Animals received 2 d of training before testing. Training consisted of five trials across the beam from widest to narrowest section. On testing day, a mesh grid was placed on the beam to increase difficulty. Animals were videotaped as they traversed the beam over five trials. A blinded observer recorded the number of paw slips through the grid, which were counted as errors. The number of errors per step was reported.
HPLC analysis of DA.
The dorsal striatum from 1- and 4-month-old mice was dissected and quickly frozen on dry ice. Following homogenization with HPLC buffer (0.1 m trichloroacetic acid, 0.01 m sodium acetate, 0.1 μm EDTA, and 9% methanol, pH 3.9) samples were centrifuged at 13,000 rpm for 20 min. The supernatant was collected and analyzed by HPLC at the CMN/KC Neurochemistry Core Laboratory at Vanderbilt University in a blinded manner. Protein content was determined by homogenizing pellets in 0.1 m HCl and BCA method (Pierce) was used. Data were normalized to tissue mass.
Electron microscopy.
Tissue was prepared for electron microscopy as previously described (Komatsu et al., 2007a). Briefly, mice were perfused with 2% paraformaldehyde/2% glutaraldehyde. Thin sections (70 nm) from the substantia nigra were examined by transmission electron microscopy (Hitachi H7500). The cross-sectional area of aggregates were reported as a percentage of the cross sectional area of dendrites. Measurements from 18 micrographs were obtained using ImageJ software (NIH).
Cell culture and Western analysis.
Atg5−/− mouse embryonic fibroblasts (MEF; gift from Dr. Noboru Mizushima, Tokyo Medical and Dental University, Tokyo, Japan) and Atg7−/− MEF cells were maintained in DMEM supplemented with 10% FBS and 50 μg/ml penicillin/streptomycin. Cells were lysed (50 mm Tris, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, Protease inhibitor mixture, 1 μm Pepstatin, 1 mm PMSF) and centrifuged at 14,000 rpm for 10 min. Protein concentrations were determined by the BCA method following the manufacturer's protocol (Pierce). Lysates were electrophoresed on a 7% Tris acetate gel. Western blotting by rabbit monoclonal LRRK2 C41–2 (1:200; Epitomics) and guinea pig polyclonal p62/SQSTM1 (1:1000; American Research Products) was detected with the Odyssey Infrared Imaging System (LiCor).
Quantitative PCR.
Total RNA was extracted from cells with the RNeasy Extraction Kit (Qiagen). Complementary DNA (cDNA) was synthesized from 5 μg of purified total RNA derived from each sample using Sprint RT Complete-Double PrePrimed kit (Clontech). RT-qPCR of cDNA templates was performed in triplicate using FastStart SYBR Green Master Mix (Roche) and an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems). Levels of β-actin mRNA were used to normalize the amounts of total RNA between samples. Fold changes were calculated as described previously (Pfaffl, 2001).
Statistics.
All data are presented as mean ± SEM. Significance was determined using unpaired Student's t test and the p value for significance was set at p < 0.05. In all instances, Levene's test for homogeneity of variance was used for inclusion in parametric tests (p > 0.05 for Levene's tests). Statistical analyses and graphing were performed with GraphPad Prism v5.0 (GraphPad Software).
Results
Loss of Atg7 leads to development of intracellular p62/SQSTM1 and ubiquitinated inclusions in DA neurons
To determine the role of autophagy in catecholeminergic neurons, we generated mutant mice with tyrosine hydroxylase (TH) cell-specific deletion of essential autophagy gene Atg7. We crossed the previously characterized Atg7 floxed (Atg7fl/fl) mice (Komatsu et al., 2006) with a TH-IRES-Cre mouse line that harbors an IRES-Cre cassette at the 3′UTR of the TH gene (Lindeberg et al., 2004), to generate conditional knock-out (cKO) mice (Atg7fl/fl;TH-IRES-Cre or cKOTH; Fig. 1A). Inactivation of Atg7 in TH+ neurons of the substantia nigra pars compacta (SNpc) was examined by immunofluorescent labeling for Atg7 (Fig. 1B). The lack of Atg7 staining in TH+ neurons of cKOTH mice at postnatal day 10 (P10) compared with Atg7fl/fl control mice suggests that Atg7 expression is suppressed by P10. p62/SQSTM1 is a ubiquitin binding protein and a known autophagic substrate that rapidly accumulates when autophagy is suppressed (Wang et al., 2006; Komatsu et al., 2007b). To assure the functional inhibition of autophagy, we performed immunofluorescence labeling and found that Atg7-deficient TH neurons develop a large number of intracellular inclusions labeled for p62 and ubiquitin (Fig. 1C,D). To assess the efficiency of Cre-mediated recombination in our mouse model, we quantified the percentage of TH+ neurons containing two or more p62 inclusions at P30. Since p62 inclusions were not observed in TH+ cells of either Atg7fl/fl (0/797 cells, n = 3) or Atg7fl/wt;TH-IRES-Cre (0/810 cells, n = 3) control mice, and nearly all TH+ cells of cKOTH mice contained p62 inclusions (96.57 ± 0.80%, 1040/1079 cells, n = 4), we conclude that Cre-mediated deletion of Atg7 is nearly complete and that loss of Atg7 effectively inactivates autophagic activity in almost all midbrain DA neurons of cKOTH mice.
Figure 1.
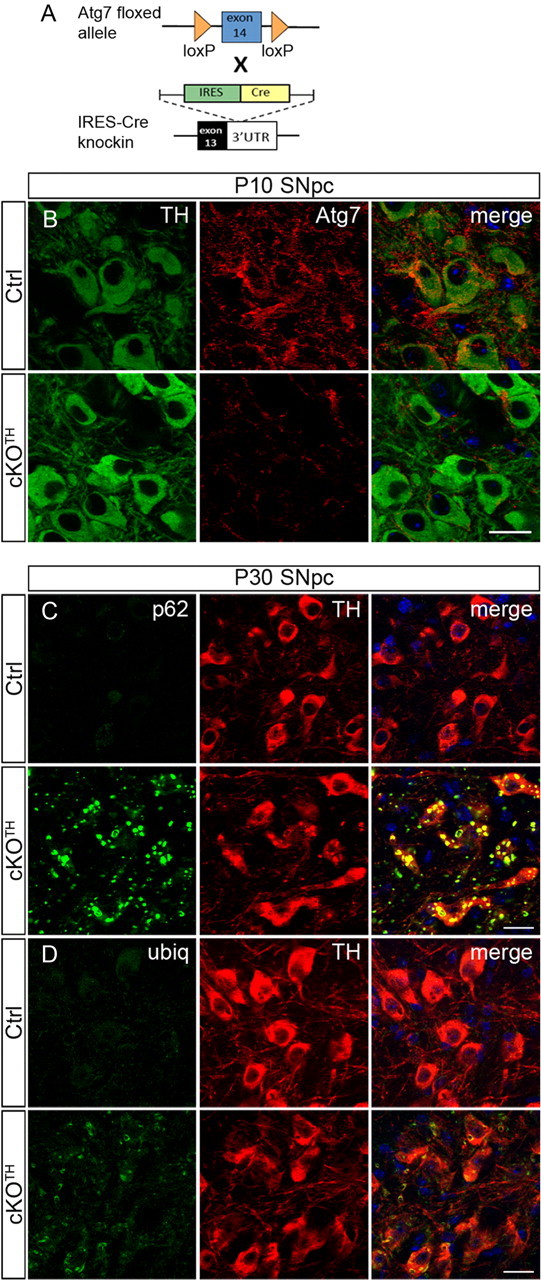
Generation of Atg7fl/fl;TH-IRES-Cre mice. A, Schematic representation of genetic cross between mice carrying the floxed Atg7 allele and knock-in mice carrying IRES-Cre inserted at the 3′UTR of the TH gene to generate Atg7fl/fl;TH-IRES-Cre (cKOTH) mice. B, Immunofluorescence labeling of TH (green) or Atg7 (red) in the SNpc at P10. Atg7 immunoreactivity is reduced in TH+ neurons by P10 in cKOTH mice. C, D, Numerous p62 and ubiquitin inclusions form in TH+ cell bodies at P30. Confocal images show TH (red) and (C) p62/SQSTM1 (green) or (D) ubiquitin (green) immunostaining in the SNpc. DAPI nuclear staining shown in blue. Scale bars, 20 μm.
Atg7 deletion causes delayed midbrain DA neuron degeneration and locomotor impairments
Previous studies show that Atg5 or Atg7 deletion in the CNS leads to varying degrees of cell loss in different neuron populations. The cerebellar Purkinje cells are among the most vulnerable neurons to undergo conspicuous degeneration, which has been demonstrated in mice with either whole brain or cell-specific deletion of Atg7 (Komatsu et al., 2006, 2007a). To examine the impact of impaired autophagy on DA neuronal survival, we performed an unbiased stereological analysis of TH+ neurons in the SNpc. While we observed no sign of TH+ cell loss in cKOTH mice at 4 months (n = 4 per group), we found a 39.8% decrease of TH-labeled cells (n = 4 per group, p = 0.003) in autophagy-deficient mice at 9 months of age (Fig. 2A,B). Since the loss of TH immunoreactivity is not a direct measure of cell death, we also assessed the total number of Nissl-positive neurons in the SNpc and report a 28.1% reduction in cKOTH mice (n = 4 per group, p = 0.03), indicating TH+ cell loss at 9 months.
Figure 2.

Delayed DA neuron cell death and locomotor impairment in cKOTH mice. The number of DA neurons in the SNpc was identified by TH immunoreactivity and cresyl violet stained nuclei at (A) 4 months (n = 4 per group) and (B) 9 months (n = 4 per group). Stereological analysis of TH-labeled neurons shown to the right. C, D, Locomotor activity of cKOTH mice at 4 and 9 months in an automated open-field. C, Horizontal and D, vertical activity was measured by the number of beam breaks over 30 min in 4-month-old (n = 7 per group) and 9-month-old mice (n = 7–8 per group). E, The number of errors per step was recorded as 4-month-old (n = 6 per group) and 9-month-old (n = 7–8 per group) mice cross the challenging beam. The data represent the average error rate over 5 trials. *p < 0.05, **p < 0.01, ***p < 0.001.
We next asked whether TH+ neuron degeneration correlates with impaired locomotor behavior. cKOTH and control mice were monitored for spontaneous movement in an automated open-field. While there was a trend for 4-month-old cKOTH mice to have decreased locomotor activity (n = 7 per group), cKOTH mice at 9 months showed significant reductions in horizontal activity (n = 7–8, p = 0.003) and vertical activity (n = 7–8, p < 0.0001) compared with control mice (Fig. 2C,D). The challenging beam traversal is a highly sensitive measure of motor performance and coordination, and has been used to detect subtle motor disturbances in mice with PD-like pathology (Fleming et al., 2004; Li et al., 2010). Consistent with the gradual decline in locomotor activity, 4-month-old cKOTH mice display few obvious abnormalities while crossing the challenging beam (n = 6 per group), whereas 9-month-old cKOTH mice make significantly more errors (n = 7–8, p = 0.02) than control mice while completing the beam task (Fig. 2E).
Early degeneration of DAergic axon terminals and reduced striatal DA levels
The above results suggest that DAergic cell loss is delayed in cKOTH mice, despite the early formation of many p62 and ubiquitinated aggregates upon Atg7 deletion (Fig. 1). We next examined the effects of impaired autophagy on nigrostriatal axons terminating in the dorsolateral striatum. TH immunolabeling at 1 month reveals enlarged TH-labeled puncta in cKOTH mice. Puncta ≥0.6 μm2 were defined as dystrophic DAergic axons. Furthermore, quantitative analysis of the overall TH-fiber density in the striatum of cKOTH mice at 1 month revealed a decrease compared with controls (34%, n = 3–5, p = 0.04; Fig. 3A). By 9 months, increased numbers of dystrophic swellings (n = 3–4, p = 0.03) and further reduction in TH+ fiber density was observed in cKOTH mice compared with controls (54%, n = 3–4, p = 0.01; Fig. 3B). Although a few dystrophic axons are observed as early as P10, TH+ fiber density in the striata of cKOTH mice is similar to control mice, suggesting that DAergic axons may develop normally and then degenerate over time (data not shown). Immunofluorescent labeling for synaptophysin and TH confirms that the enlarged TH puncta are indeed dystrophic DAergic axon terminals (Fig. 3C).
Figure 3.
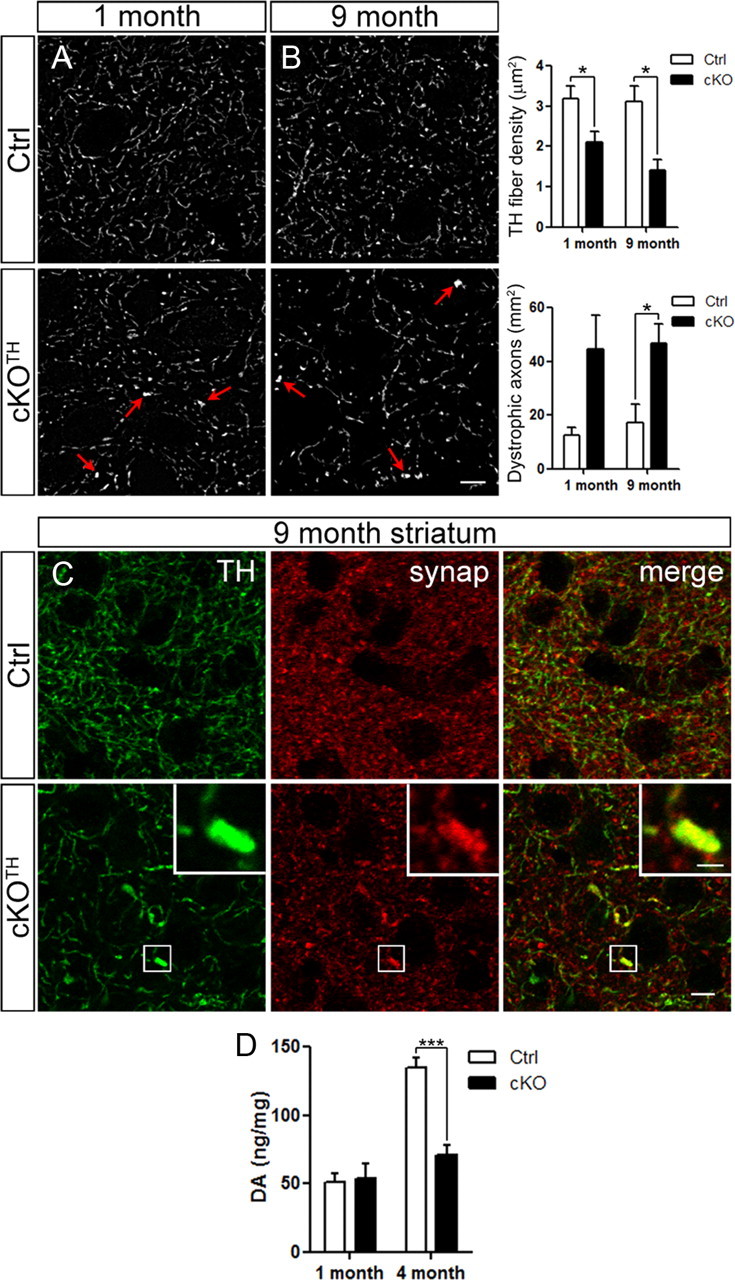
DAergic axon terminal degeneration and disrupted DA homeostasis. A, B, High resolution confocal images through the dorsolateral striatum show TH immunoreactivity. Images from (A) 1-month-old (n = 3–5 per group) and (B) 9-month-old mice (n = 3–4 per group) were deconvolved. Arrows indicate axon swellings in cKOTH striatum. Scale bar: (in B) A, B, 5 μm. TH+ fiber density and dystrophic axons, defined as puncta ≥0.6 μm2, were quantified. C, High resolution confocal images through the dorsolateral striatum show immunofluorescence labeling of TH (green) and synaptophysin (red), where yellow indicates DAergic axon terminals. High magnification of a dystrophic axon is shown in the inset. Scale bars: C, 10 μm; inset, 1.5 μm. D, HPLC analysis of DA levels in the dorsal striatum at 1 month (n = 6–7 per group) and 4 months (n = 5–6 per group). *p < 0.05, ***p < 0.001.
We next tested DA physiology in 1- and 4-month-old cKOTH mice by neurochemical analysis of dorsal striata. High performance liquid chromatography (HPLC) data indicate a significant reduction in striatal DA levels in 4-month-old cKOTH mice (53%, n = 5–6, p = 0.0002) compared with control mice, but no difference in DA content at 1 month (n = 6–7 per group; Fig. 3D). Although cKOTH mice do not display obvious behavioral abnormalities or significant loss of DA cell bodies at 4 months, the striatal DA content is severely affected and is likely associated with DAergic axon degeneration. Moreover, degeneration of DAergic terminals and the reduction of striatal DA levels may contribute to motor impairment observed at later time points.
Atg7-deficient TH+ neurons have dystrophic dendrites containing ubiquitinated inclusions
DAergic neurons in the SNpc have long dendrites that radiate into the pars reticulata (SNpr) and are vulnerable to the effects of neurotoxins, such as 6-OHDA and MPP+ (Bywood and Johnson, 2000). Immunohistochemical labeling with anti-TH antibody reveals that midbrain DAergic dendrites of 4-month-old cKOTH mice have numerous large swellings (or varicosities; 341.0 ± 37.66 swellings/mm2, n = 3) that are nearly undetectable in the corresponding region in control mice (9.801 ± 1.37 swellings/mm2, n = 3; Fig. 4A). Similarly, dendritic dystrophy was observed in the ventral tegmental area (data not shown) and TH+ noradrenergic neurons in the locus ceruleus (Fig. 4B). The aberrant dendritic beadings in the SNpr are filled with ubiquitinated structures or proteins, as indicated by immunofluorescence staining for ubiquitin and TH (Fig. 4C).
Figure 4.
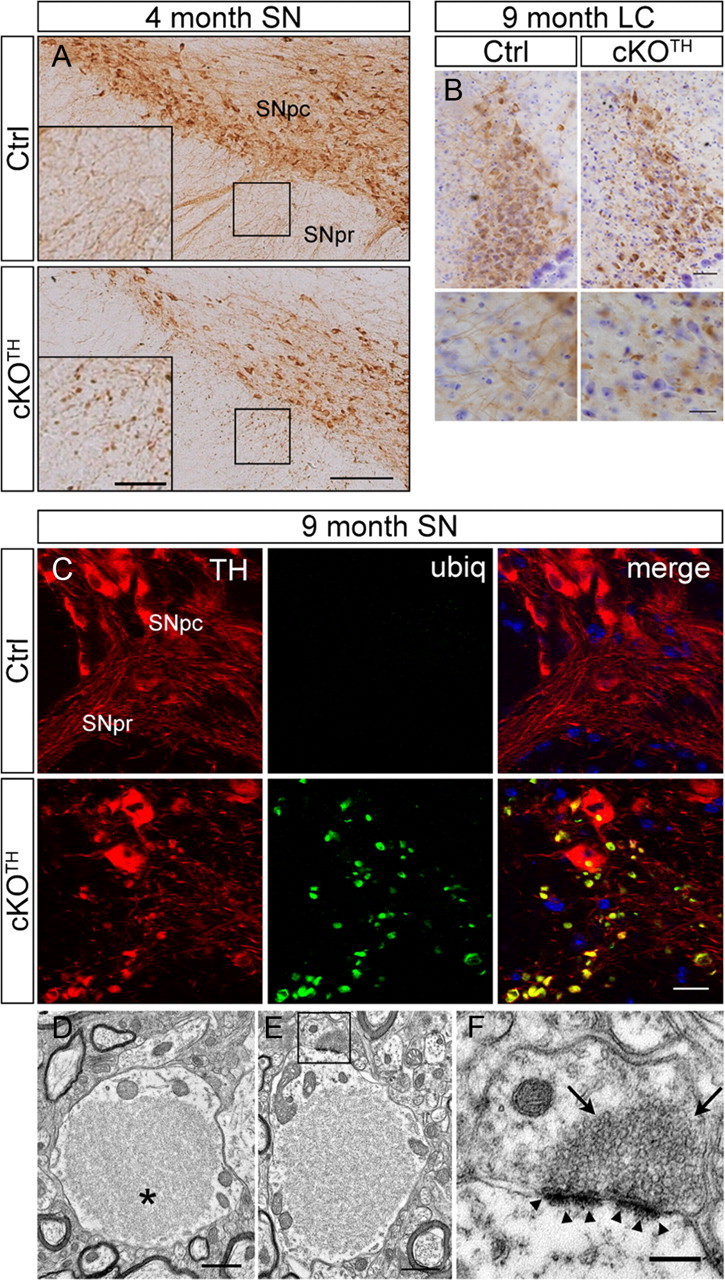
Dendritic dystrophy associated with ubiquitinated aggregate accumulation. A, Immunostaining with TH in midbrain DA neurons of cKOTH mice at 4 months. High-magnification images are shown in inset. SNpc, substantia nigra pars compacta, SNpr, substantia nigra pars reticulata. Scale bars: A, 150 μm; inset, 50 μm. B, Immunohistochemical staining with TH in noradrenergic neurons of the locus ceruleus in cKOTH mice at 9 months reveals dendritic swellings and degeneration. Cresyl violet staining labels nuclei. High-magnification images of noradrenergic dendrites are below. Scale bars: B, 50 μm; magnified image, 20 μm. C, High resolution confocal images through the midbrain at 9 months with ubiquitin (green) and TH (red). Scale bar: 20 μm. D–F, Representative electron micrographs of dendrites in the substantia nigra of cKOTH mice at 9 months. D, Large filamentous inclusion, as indicated by asterisk, within a swollen dendrite. E, Synapses adjacent to large inclusions remain morphologically intact (boxed area). F, High-magnification image of synapse (boxed area in E). Arrowheads indicate normal postsynaptic densities and arrows point to normal presynaptic vesicle pool. Scale bars: D, E, 1 μm; F, 250 nm.
Ultrastructural studies were conducted to visualize the enlarged dendritic profiles in the midbrain of cKOTH mice. The dendritic swellings observed in these studies contained large inclusions, 1.3–7 μm in longest cross-sectional diameter (Fig. 4D). The inclusions did not have limiting membranes and appeared to be comprised primarily of a feltwork of disorganized filaments, as well as some scattered microtubules. The inclusions occupied ∼72% of the cross-sectional area of the dendritic swelling (71.5 ± 2.8%). Such inclusions were observed in the cell bodies of TH+ neurons of cKOTH (Fig. 1) and have also been previously reported in the cell bodies of neurons deficient in autophagy (Hara et al., 2006; Komatsu et al., 2006, 2007b), suggesting that a single autophagy-mediated clearance mechanism is shared by the somatic and dendritic compartments of the cell. The mitochondria within these dendritic swellings were usually restricted to the cytoplasmic rim between the large filamentous inclusion and the plasma membrane (Fig. 4D). However, despite these large abnormal inclusions and the associated dendritic swellings, synapses involving the distorted dendrites appeared morphologically intact; the synaptic clefts, postsynaptic densities, and presynaptic vesicle populations appeared largely normal (Fig. 4E,F). The results suggest that the accumulation of large filamentous inclusions contributes directly to the swelling and dystrophy of DAergic dendrites and likely results in the obstruction of the exchange of molecules between the distal dendrites and the soma of affected neurons.
Accumulation of pathogenic α-syn protein in presynaptic terminals of Atg7-deficient brains
Since multiple studies have linked autophagy to the clearance of α-syn (Webb et al., 2003; Rideout et al., 2004), we asked whether TH+ neurons of cKOTH mice develop aberrant α-syn levels. Surprisingly, immunolabeling with Syn303 antibody, which recognizes a pathogenic form of α-syn (Duda et al., 2002), did not reveal abnormal accumulation in midbrain TH+ cells bodies, even as late as 20 months old (Fig. 5A). Nevertheless, α-syn aggregates were detected in striatal axonal swellings of 20-month-old cKOTH mice (Fig. 5B). Although somatic α-syn inclusions in DA neurons were absent, these results indicate that the loss of autophagy may contribute to the buildup of α-syn in presynaptic DAergic terminals in aged brains. To test whether other neuron populations are susceptible to augmented levels of endogenous α-syn when autophagic clearance is compromised, we examined Atg7fl/fl;Nestin-Cre (cKONes) mice where Atg7 is depleted in all cell types of the CNS, as has been described previously (Komatsu et al., 2006). Since the survival rate of cKONes mice declines considerably by 4 weeks after birth, all studies with cKONes mice were conducted between 1 and 2 months of age. Immunofluorescence staining with Syn303 reveals substantial accumulations of α-syn within the deep cerebellar nuclei (DCN) of cKONes mice at P35. The DCN receive inhibitory input from Purkinje cells, which are specifically labeled by antibodies against calbindin. Syn303 aggregates appear in severely dystrophic calbindin-positive Purkinje cell axons terminating in the DCN (Fig. 5C). The large, punctate calbindin staining pattern was similarly observed in mice with Purkinje cell-specific deletion of Atg5 or Atg7 (Komatsu et al., 2007a; Nishiyama et al., 2007). Together, these data suggest that impaired autophagic clearance leads to the accumulation of α-syn in axons.
Figure 5.
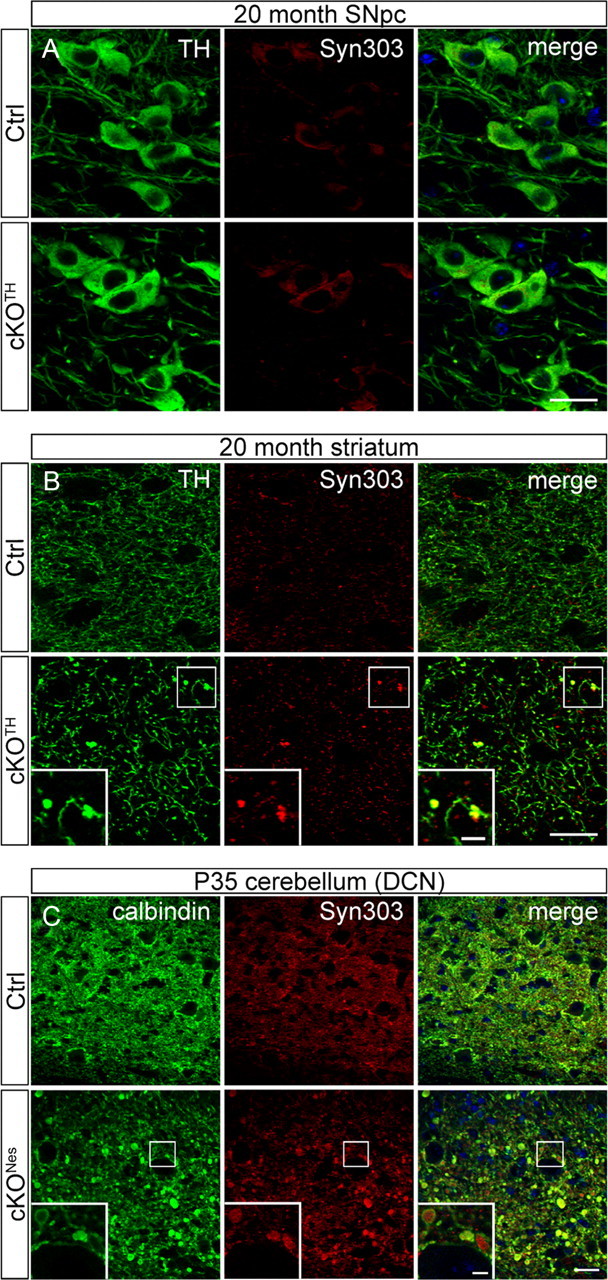
Abnormal presynaptic accumulation of α-syn protein in cKOTH and cKONes mice brain. A, Immunofluorescence labeling with TH (green) and Syn303 (red) in cKOTH mice at 20 months shows no difference in α-syn expression in DAergic cell bodies of the SNpc. B, Confocal images through the dorsal striatum of 20-month-old cKOTH mice with TH (green) and Syn303 (red). High magnification of swollen TH+ axon terminals containing Syn303 deposits shown in inset. Scale bars: A, B, 20 μm; B, inset, 5 μm. C, Immunofluorescence labeling of Syn303 (red) and calbindin (green) in Purkinje cell axons in the DCN of cKONes mice at P35. DAPI nuclear staining shown in blue. Scale bars: C, 25 μm; inset, 5 μm.
Loss of Atg7 contributes to enhanced levels of LRRK2 in neurons and cell culture
We next asked whether autophagy inhibition would alter endogenous LRRK2 protein levels. We did not detect any changes in LRRK2 immunostaining in midbrain TH+ cell bodies or striatal TH+ axons in 20 month-old cKOTH mice compared with corresponding regions in control mice (data not shown). However, like α-syn, LRRK2 protein is markedly accumulated in the DCN of cKONes mice, and is in part, colocalized in calbindin-positive Purkinje cell axonal swellings (Fig. 6A). Interestingly, LRRK2 inclusions also appear within large dendritic varicosities of Purkinje cells in cKONes mice (Fig. 6B). The formation of LRRK2-containing inclusions within dystrophic Purkinje dendrites resembles the morphology of the ubiquitinated structures observed in midbrain DAergic dendrites of cKOTH mice (Fig. 4). To confirm this novel in vivo finding, we further analyzed LRRK2 protein levels in MEF cell lines deficient in Atg7 or another essential autophagy gene, Atg5 (Atg5−/−; Mizushima et al., 2001). Consistent with our immunofluorescence staining results in cKONes mice, Atg7−/− MEFs contained significantly higher LRRK2 protein levels than control Atg7+/− MEFs (n = 5, p = 0.02; Fig. 6C). We also observed increased LRRK2 protein in Atg5−/− MEFs compared with control Atg5+/+ MEFs (n = 5, p < 0.0001; Fig. 6D). As expected, p62 levels are enhanced in both autophagy-deficient cell lines. We next performed quantitative PCR (qPCR) to evaluate LRRK2 mRNA levels and found a fourfold increase in LRRK2 mRNA in Atg5−/− MEFs over Atg5+/+control MEFs (n = 6, p = 0.009). These results suggest that elevated LRRK2 protein levels are attributable to heightened LRRK2 transcription caused by impaired autophagy.
Figure 6.
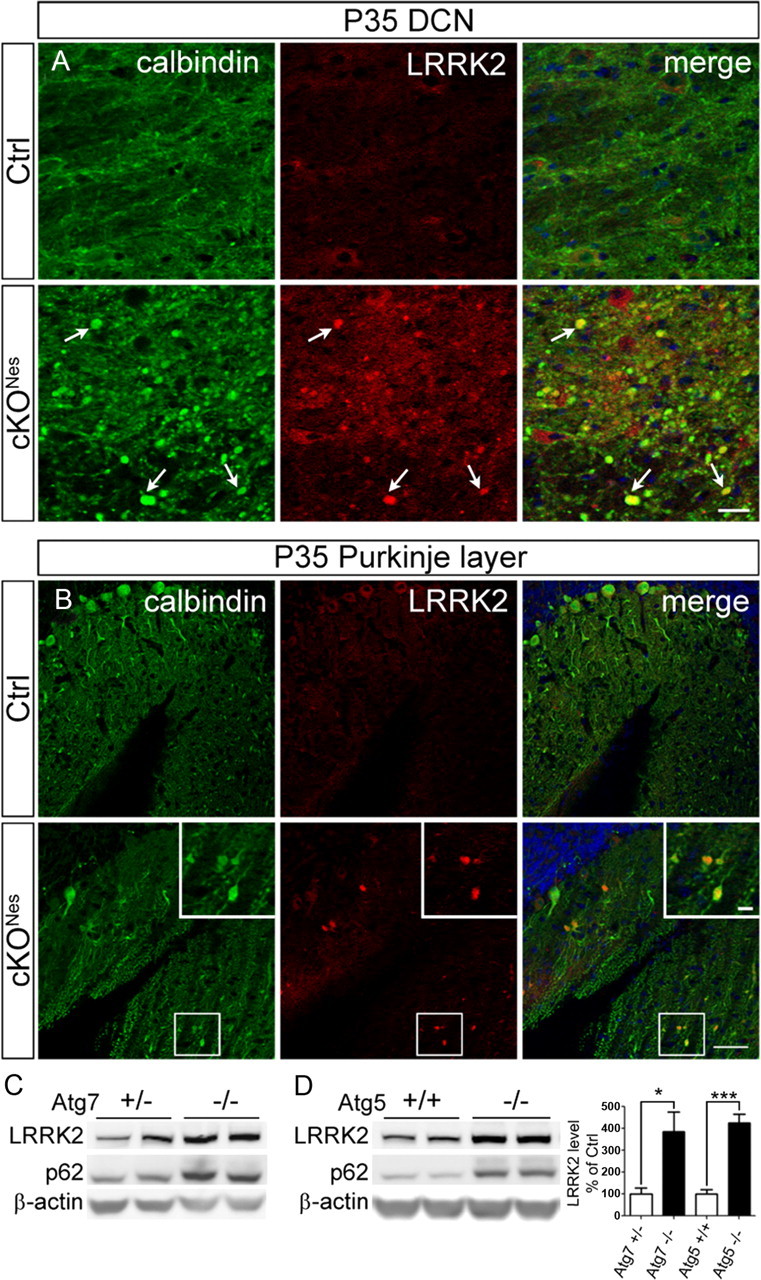
Elevated LRRK2 levels in autophagy deficient neurons and cell lines. Immunofluorescence staining of LRRK2 (red) and calbindin (green) in (A) Purkinje cell axons and (B) Purkinje cell dendrites in cKONes mice at P35. Inset shows magnified image of boxed areas. Scale bars: A, B, 50 μm; B, inset, 10 μm. C, Representative Western blot of endogenous LRRK2 in Atg7 heterozygous (+/−) and Atg7 KO (−/−) or (D) Atg5 wild-type (+/+) and Atg5 KO (−/−) MEF cells. Densitometric quantification shown on the right (n = 5 per group). *p < 0.05, ***p < 0.001.
Discussion
While recent breakthroughs have shed light on the cellular mechanisms involved in familial PD, little is understood about the causes underlying idiopathic PD pathogenesis. In the current study, we attempt to uncover how the loss of an essential catabolic pathway contributes to the pathological features associated with PD by examining the effects of impaired autophagy in DA neurons and its role in maintaining the levels of endogenous PD-related proteins in the brain. We show that the targeted deletion of Atg7 in TH+ neurons results in early axonal dystrophy and degeneration, striatal DA depletion, enlarged dendritic swellings, and delayed cell loss and locomotor dysfunction in mutant mice. Furthermore, we demonstrate a potential link between autophagy deficiency and the accumulation of α-syn and LRRK2 proteins in neurons.
Our results indicate that disrupted autophagy has distinct consequences in DA neurons compared with other neuron populations. While inactivation of Atg7 in Purkinje cells causes rapid cell death at 8 weeks (Komatsu et al., 2007a), Atg7 deletion in DA neurons leads to a delayed and moderate loss of cell bodies (∼30%) as late as 9 months. Our stereological analysis suggests that the majority of TH-labeled cell bodies are surprisingly resistant to dysfunctional autophagy even in mice at 20 months (Fig. 4A). On the other hand, Atg7-deficient DAergic processes undergo substantial degeneration, which is likely associated with the reduced striatal DA levels observed at 4 months. Despite the severity of these early pathogenic events, locomotor function does not become impaired until 9 months of age, when there is significant DA neuron cell death in cKOTH mice. Whether the onset of behavioral abnormalities is directly correlated to significant cell loss or reflects the point at which DA depletion or TH+ axon degeneration exceeds a certain threshold remains unclear. However, the gradual decline in locomotor activity observed in our mouse model suggests the latter.
α-Syn is the primary constituent of Lewy bodies and has a significant pathological role in both familial and idiopathic PD. A recent study showed that overexpression of wild-type α-syn impairs autophagic activity (Winslow et al., 2010), and implies a functional relationship between α-syn and autophagic degradation. Conversely, growing evidence also indicates a role for autophagy in controlling α-syn protein levels (Spencer et al., 2009; Yu et al., 2009) and suggests that dysfunctional autophagic clearance may contribute to the development of α-syn inclusions in idiopathic PD. The extent to which autophagy regulates α-syn levels is still unclear, since other catabolic pathways have been implicated (Webb et al., 2003; Cuervo et al., 2004; Rideout et al., 2004). A recent report suggests that although α-syn is mostly degraded by the ubiquitin-proteasome system (UPS) under normal conditions in vivo, autophagy is recruited as the primary clearance system in transgenic mice expressing elevated levels of oligomeric α-syn (Ebrahimi-Fakhari et al., 2011). In our study, DAergic axons from cKOTH mice develop α-syn accumulation at 20 months, well after other neuropathology is observed and in contrast to the early and pervasive appearance of p62 and ubiquitinated inclusions. However, even at this late stage, α-syn deposits are not present in all affected DAergic axons. We detect similar α-syn localization in cerebellar Purkinje axons in cKONes mice, but at a much younger age (P35) and higher frequency. Purkinje cells are particularly vulnerable to the loss of autophagy at earlier time points (Komatsu et al., 2007a; Nishiyama et al., 2007), which may be one of the factors contributing to the early appearance of α-syn aggregates. Presumably, additional neuron populations would display α-syn pathology if cKONes mice lived beyond several months, therefore future studies should examine autophagy impairment in other cell-specific conditional models. In both cases, endogenous α-syn accumulates in Atg7-deficient axons, but not within cell bodies. These findings suggest that autophagy is involved in axonal α-syn protein homeostasis in vivo. While other degradation pathways, such as UPS or chaperone-mediated autophagy serve as the primary mechanisms for α-syn turnover in the soma, autophagy may play a more prominent role in clearing presynaptic α-syn, particularly when the cell is stressed.
We also asked whether autophagy deficiency would increase LRRK2 protein levels, as previous examination of postmortem PD brains revealed that LRRK2 is occasionally found in α-syn-containing Lewy bodies (Zhu et al., 2006) and a recent study shows that LRRK2 exacerbates α-syn-mediated neuropathology (Lin et al., 2009). We show for the first time that impaired autophagy leads to LRRK2 accumulation in certain brain regions (e.g., cerebellum), as well as in autophagy-deficient cell lines. Interestingly, our qPCR data in cell culture indicates that LRRK2 accumulation is not the consequence of impaired LRRK2 protein turnover per se, but rather may be related to the marked increase in LRRK2 mRNA. These intriguing results suggest that LRRK2 levels are upregulated in certain cell types in response to the loss of autophagy. Several reports have shown that autophagic flux is impeded by familial LRRK2 mutations and indicates a role for LRRK2 in autophagy regulation (Plowey et al., 2008; Alegre-Abarrategui et al., 2009; Ramonet et al., 2011). In light of these findings, LRRK2 upregulation in our cell model may reflect a compensatory response to decreased autophagic activity. Alternatively, enhanced LRRK2 transcription may serve as a response to cellular stress, which suggests that LRRK2 accumulation is an indirect consequence of dysfunctional autophagy. Further studies should examine the mechanisms underlying increased LRRK2 levels induced by impaired autophagy and how it may, in turn, affect α-syn accumulation.
The current study suggests that autophagy-deficient DA neurons in aged animals are susceptible to presynaptic accumulation of α-syn, but not LRRK2. In addition, cerebellar Purkinje cells undergo early accumulation of both α-syn and LRRK2 in mice with brain-specific deletion of Atg7, which reflects the unique vulnerability of Purkinje cells with impaired autophagy, as shown previously (Hara et al., 2006; Komatsu et al., 2006, 2007a; Nishiyama et al., 2007). Although the cerebellum is unaffected in PD, these findings highlight the potential consequences on PD-related protein levels when neuronal autophagy is severely compromised. While our mouse models do not recapitulate all of the pathogenic features in human PD, our study supports the notion that autophagy is one of several cellular systems that may deteriorate with age and contributes to PD pathogenesis. We propose that insufficient autophagy in CNS neurons, particularly midbrain DA neurons, represents a risk for the development of the disease, even in the absence of PD-related gene mutations. Consequently, the manipulation of autophagic activity should be explored as a potential therapeutic strategy for the treatment of PD.
Footnotes
This work was supported by the National Institutes of Health (NIH)–National Institute of Neurological Disorders and Stroke Grants R01NS060123, NS060809, and RNS055683A; the Michael J. Fox Foundation; the Bachmann-Strauss Dystonia and Parkinson Foundation (to Z.Y.); the Parkinson Alliance (to G.R.H.), and the Deutsche Forschungsgemeinschaft (to M.L.L.). Confocal laser scanning microscopy was performed at the Mount Sinai School of Medicine–Microscopy Shared Resource Facility, supported with funding from NIH–National Cancer Institute shared resources Grant 5R24 CA095823-04, National Science Foundation Major Research Instrumentation Grant DBI-9724504, and NIH shared instrumentation Grant 1 S10 RR0 9145-01. We thank Dr. Nina Pan and Dr. Ewa Kukielka for excellent technical assistance.
The authors declare no competing financial interests.
References
- Alegre-Abarrategui J, Christian H, Lufino MM, Mutihac R, Venda LL, Ansorge O, Wade-Martins R. LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum Mol Genet. 2009;18:4022–4034. doi: 10.1093/hmg/ddp346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bywood PT, Johnson SM. Dendrite loss is a characteristic early indicator of toxin-induced neurodegeneration in rat midbrain slices. Exp Neurol. 2000;161:306–316. doi: 10.1006/exnr.1999.7259. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Duda JE, Giasson BI, Mabon ME, Lee VM, Trojanowski JQ. Novel antibodies to synuclein show abundant striatal pathology in Lewy body diseases. Ann Neurol. 2002;52:205–210. doi: 10.1002/ana.10279. [DOI] [PubMed] [Google Scholar]
- Ebrahimi-Fakhari D, Cantuti-Castelvetri I, Fan Z, Rockenstein E, Masliah E, Hyman BT, McLean PJ, Unni VK. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of α-synuclein. J Neurosci. 2011;31:14508–14520. doi: 10.1523/JNEUROSCI.1560-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming SM, Salcedo J, Fernagut PO, Rockenstein E, Masliah E, Levine MS, Chesselet MF. Early and progressive sensorimotor anomalies in mice overexpressing wild-type human alpha-synuclein. J Neurosci. 2004;24:9434–9440. doi: 10.1523/JNEUROSCI.3080-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Sun Z, Lee T, Fusco FR, Kimble TD, Meade CA, Cuthbertson S, Reiner A. A simple and sensitive antigen retrieval method for free-floating and slide-mounted tissue sections. J Neurosci Methods. 1999;93:149–162. doi: 10.1016/s0165-0270(99)00142-9. [DOI] [PubMed] [Google Scholar]
- Kirisako T, Ichimura Y, Okada H, Kabeya Y, Mizushima N, Yoshimori T, Ohsumi M, Takao T, Noda T, Ohsumi Y. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J Cell Biol. 2000;151:263–276. doi: 10.1083/jcb.151.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Wang QJ, Holstein GR, Friedrich VL, Jr, Iwata J, Kominami E, Chait BT, Tanaka K, Yue Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A. 2007a;104:14489–14494. doi: 10.1073/pnas.0701311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, Natsume T, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007b;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- Li X, Patel JC, Wang J, Avshalumov MV, Nicholson C, Buxbaum JD, Elder GA, Rice ME, Yue Z. Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson's disease mutation G2019S. J Neurosci. 2010;30:1788–1797. doi: 10.1523/JNEUROSCI.5604-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenberg M, Mansilla A, Zecchini VR, Fleming A, Rubinsztein DC. The Parkinson's disease protein LRRK2 impairs proteasome substrate clearance without affecting proteasome catalytic activity. Cell Death Dis. 2011;2:e196. doi: 10.1038/cddis.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Parisiadou L, Gu XL, Wang L, Shim H, Sun L, Xie C, Long CX, Yang WJ, Ding J, Chen ZZ, Gallant PE, Tao-Cheng JH, Rudow G, Troncoso JC, Liu Z, Li Z, Cai H. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson's-disease-related mutant alpha-synuclein. Neuron. 2009;64:807–827. doi: 10.1016/j.neuron.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeberg J, Usoskin D, Bengtsson H, Gustafsson A, Kylberg A, Söderström S, Ebendal T. Transgenic expression of Cre recombinase from the tyrosine hydroxylase locus. Genesis. 2004;40:67–73. doi: 10.1002/gene.20065. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152:657–668. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama J, Miura E, Mizushima N, Watanabe M, Yuzaki M. Aberrant membranes and double-membrane structures accumulate in the axons of Atg5-null Purkinje cells before neuronal death. Autophagy. 2007;3:591–596. doi: 10.4161/auto.4964. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plowey ED, Cherra SJ, 3rd, Liu YJ, Chu CT. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem. 2008;105:1048–1056. doi: 10.1111/j.1471-4159.2008.05217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramonet D, Daher JP, Lin BM, Stafa K, Kim J, Banerjee R, Westerlund M, Pletnikova O, Glauser L, Yang L, Liu Y, Swing DA, Beal MF, Troncoso JC, McCaffery JM, Jenkins NA, Copeland NG, Galter D, Thomas B, Lee MK, et al. Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS One. 2011;6:e18568. doi: 10.1371/journal.pone.0018568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rideout HJ, Lang-Rollin I, Stefanis L. Involvement of macroautophagy in the dissolution of neuronal inclusions. Int J Biochem Cell Biol. 2004;36:2551–2562. doi: 10.1016/j.biocel.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, Adame A, Wyss-Coray T, Masliah E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson's and Lewy body diseases. J Neurosci. 2009;29:13578–13588. doi: 10.1523/JNEUROSCI.4390-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QJ, Ding Y, Kohtz DS, Kohtz S, Mizushima N, Cristea IM, Rout MP, Chait BT, Zhong Y, Heintz N, Yue Z. Induction of autophagy in axonal dystrophy and degeneration. J Neurosci. 2006;26:8057–8068. doi: 10.1523/JNEUROSCI.2261-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S, Brown S, O'Kane CJ, Rubinsztein DC. α-synuclein impairs macroautophagy: implications for Parkinson's disease. J Cell Biol. 2010;190:1023–1037. doi: 10.1083/jcb.201003122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu WH, Dorado B, Figueroa HY, Wang L, Planel E, Cookson MR, Clark LN, Duff KE. Metabolic activity determines efficacy of macroautophagic clearance of pathological oligomeric alpha-synuclein. Am J Pathol. 2009;175:736–747. doi: 10.2353/ajpath.2009.080928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Siedlak SL, Smith MA, Perry G, Chen SG. LRRK2 protein is a component of Lewy bodies. Ann Neurol. 2006;60:617–618. doi: 10.1002/ana.20928. author reply 618–619. [DOI] [PubMed] [Google Scholar]