

Abstract
After peripheral nerve injury, neurotrophins play a key role in the regeneration of damaged axons that can be augmented by exercise, although the distinct roles played by neurons and Schwann cells are unclear. In this study, we evaluated the requirement for the neurotrophin, brain-derived neurotrophic factor (BDNF), in neurons and Schwann cells for the regeneration of peripheral axons after injury. Common fibular or tibial nerves in thy-1-YFP-H mice were cut bilaterally and repaired using a graft of the same nerve from transgenic mice lacking BDNF in Schwann cells (BDNF−/−) or wild-type mice (WT). Two weeks postrepair, axonal regeneration into BDNF−/− grafts was markedly less than WT grafts, emphasizing the importance of Schwann cell BDNF. Nerve regeneration was enhanced by treadmill training posttransection, regardless of the BDNF content of the nerve graft. We further tested the hypothesis that training-induced increases in BDNF in neurons allow regenerating axons to overcome a lack of BDNF expression in cells in the pathway through which they regenerate. Nerves in mice lacking BDNF in YFP+ neurons (SLICK) were cut and repaired with BDNF−/− and WT nerves. SLICK axons lacking BDNF did not regenerate into grafts lacking Schwann cell BDNF. Treadmill training could not rescue the regeneration into BDNF−/− grafts if the neurons also lacked BDNF. Both Schwann cell- and neuron-derived BDNF are thus important for axon regeneration in cut peripheral nerves.
Introduction
Injured peripheral axons have the ability to regenerate, but the regenerative process is slow and functional recovery is generally very poor. One reason for decreased regeneration could be a decline in neurotrophic support in tissue surrounding the regenerating axons over time (for review, see Gordon, 2009). To facilitate regeneration, treatment approaches have included manipulation of neurotrophins. In particular, brain-derived neurotrophic factor (BDNF) has been shown to be important in the growth of regenerating axons into peripheral nerve grafts (Zhang et al., 2000; English et al., 2005).
After peripheral nerve injury, BDNF mRNA is upregulated in axotomized motoneurons as well as Schwann cells distal to the injury (Funakoshi et al., 1993). The upregulation of BDNF mRNA in axotomized motoneurons is transient and lasts only a few days (Gordon, 2009). The increase in BDNF in cells in the distal stump is much longer-lasting. Thus, much research has focused on the role of BDNF at the distal stump in facilitating axon regeneration after nerve injury. Because traditional genetic BDNF knock-out mice die shortly after birth (Jones et al., 1994; Ernfors et al., 1995; Conover and Yancopoulos, 1997), most research has focused on the pharmacologic manipulation of BDNF. Application of trophic factors such as BDNF and NT4/5 in pharmacologic doses at the site of injury has been shown to enhance the regeneration of axons into the distal stump (English et al., 2005). Reducing neurotrophin signaling through systemic infusion of BDNF antibodies (Zhang et al., 2000) significantly limits the regenerative elongation of axons.
Although the upregulation of BDNF mRNA in axotomized motoneurons is brief, it may provide a source of neurotrophic support to regenerating axons before the increase in BDNF in distal Schwann cells (Gordon, 2009). Increasing motoneuron-derived BDNF may enhance axon regeneration. Experiments examining the potential for motoneuron-derived BDNF to promote axon regeneration have been indirect. Treadmill training (Gómez-Pinilla et al., 2001) and electrical stimulation of axons in the proximal stump (Al-Majed et al., 2000) enhance BDNF mRNA expression in motoneurons after nerve cut. This increase in BDNF likely underlies the significant enhancement of axon elongation into a peripheral nerve graft after 2 weeks of treadmill training (Sabatier et al., 2008).
Until recently, the technology required to examine the differential contributions of Schwann cell-derived and motoneuron-derived BDNF has not been available. In the current study, we manipulated the expression of BDNF genetically in a cell type-specific manner, removing BDNF selectively from Schwann cells or motoneurons. We hypothesized that removing BDNF from the Schwann cells surrounding the regenerating axons would impair the growth of the regenerating axons. In addition, we propose a novel model in which motoneuron-derived BDNF regulates treadmill training-mediated elongation of axons after nerve cut. Preliminary reports of these findings have been made (Wilhelm et al., 2009).
Materials and Methods
Animals and surgical methods.
All procedures were approved by the Institutional Animal Care and Use Committee of Emory University and conformed to the Society for Neuroscience's Guidelines for the Use of Animals in Research. All mouse genotypes used in the following experiments were on a C57BL/6J background that were backcrossed for at least six generations. Experiments were conducted on adult (>2 months old) male and female mice weighing 18–48 g (mean, 22 g).
Under isofluorane anesthesia, the common fibular (CF) and tibial (TIB) nerves were exposed and cut ∼1 mm distal to their branching from the sciatic nerve. A segment of the CF or tibial nerve (8–10 mm long) was harvested from a wild-type (WT) or a BDNF-lacking donor mouse that did not contain a yellow fluorescent protein (YFP) transgene. The proximal end of the donor graft was apposed to the proximal segment of the cut nerve of the matching nerve of the host mouse on a small rectangle of SILASTIC film (Dow Corning) and secured using ∼5 μl of fibrin glue containing thrombin, fibrin, and fibronectin (de Vries et al., 2002; MacGillivray, 2003). In eight nerves, BDNF (2.0 μg) was added to the fibronectin portion of the fibrin glue at the time of the repair. The distal end of the donor graft was not secured to the host's distal stump to prevent any influence by potential-target-derived molecules. All surgeries were performed bilaterally, so that each limb contained one nerve repaired with a wild-type graft and one nerve repaired with a BDNF-lacking graft. The graft types were also balanced across nerves (TIB or CF) such that each mouse had one TIB repaired with a KO graft and the contralateral TIB repaired with a WT graft. Additionally, conditions were counterbalanced across animals so that equal numbers of mice had left limb TIB nerves repaired with grafts from WT and KO donors. No significant differences were found in the length of regenerating axon profiles based on the type of nerve grafted (CF vs TIB), the limb used (left vs right), or the sex of the host mouse. Therefore, data from all CF and TIB nerves were pooled together for analysis regardless of host sex or limb used.
Two different host models were used in the above transection paradigm to allow us to manipulate the BDNF expression in the regenerating axons (Table 1). The H strain of thy-1-YFP mice [thy-1-YFP-H (Feng et al., 2000)] was used in experiments in which hosts had wild-type BDNF expression levels. In these mice, a subset of motor and sensory axons in peripheral nerves is marked completely by YFP (Feng et al., 2000). Marked axons in these mice are assumed to represent a sample of all of the axons in the peripheral nerves (Groves et al., 2005). The second host model used lacked BDNF in YFP+ neurons. We used single-neuron labeling with inducible Cre-mediated knock-out (SLICK) mice (Young et al., 2008), in which YFP is expressed in neurons in a manner similar to that in the thy-1-YFP-H mice with a representative subset of axons containing YFP. In these mice, the YFP+ sensory and motor neurons also express a tamoxifen-inducible Cre-recombinase. When bred with transgenic mice expressing loxP-flanked BDNF genes, selected offspring (e.g., SLICK::BDNFf/f) were treated with tamoxifen (see Tamoxifen administration, below) and BDNF was knocked out in the YFP+ neurons. Host mice lacking either one (SLICK::BDNFf/w) or both (SLICK::BDNFf/f) BDNF alleles were used. The founders of the A strain of SLICK mice (B6.Cg-Tg(CAG-cre/Esr1*)5Amc/J; stock no. 004682) and of the thy-1-YFP-H mice (B6.Cg-Tg(Thy1-YFPH)2Jrs/J; stock no. 003782) were obtained from The Jackson Laboratory. The founders of our colony of floxed BDNF mice were a generous gift from Dr. Michael Sendtner (Universität Würzburg, Würzburg, Germany).
Table 1.
Transgenic mice were used to assess the roles of BDNF in axon regeneration
| Genotype | Treatment tamoxifen (Tx) or saline (UT) | Host mouse or nerve donor | BDNF alleles |
|---|---|---|---|
| WT, tomato | Tx or UT | Donor | Systemic BDNF +/+ |
| thy-1-YFP-H | UT | Host | Systemic BDNF +/+ |
| Tam-Cre::BDNFf/f | Tx | Donor | Systemic BDNF −/− |
| Tam-Cre::BDNFf/f | UT | Donor | Systemic BDNF +/+ |
| Tam-Cre::BDNFf/w | Tx | Donor | Systemic BDNF +/− |
| Tam-Cre::BDNFf/w | UT | Donor | Systemic BDNF +/+ |
| CNTF-Cre::BDNFf/f | UT | Donor | CNTF+ cells BDNF −/−, other cells BDNF+/+ |
| CNTF-Cre::BDNFf/w | UT | Donor | CNTF+ cells BDNF +/−, other cells BDNF+/+ |
| SLICK::BDNFf/f | Tx | Host | YFP+ neurons BDNF −/−, other cells BDNF +/+ |
| SLICK::BDNFf/w | Tx | Host | YFP+ neurons BDNF +/−, other cells BDNF +/+ |
Several different mouse models were used for the collection of graft tissues to allow us to manipulate the environment into which the axons could attempt to regenerate (Table 1). Graft tissue was collected from strains of non-YFP-expressing mice. Graft tissue containing BDNF was collected from a non-YFP-expressing littermate of the host mouse or a transgenic mouse (B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J; stock no. 007676; The Jackson Laboratory). When graft tissue from these transgenics, termed “tomato,” was used, a red background was observed because tomato mice express red fluorescence in all tissues unless Cre-recombinase is expressed (Muzumdar et al., 2007). Once Cre-recombinase is expressed in a cell, the red fluorescent protein gene is excised and green fluorescent protein is produced. Only non-Cre-expressing tomato mice were used as graft donors. We found no significant difference in the lengths of axons growing into grafts from wild-type littermates or tomato mice; therefore, data from these two groups were combined and labeled WT.
Because traditional homozygous BDNF knock-out mice die shortly after birth (Jones et al., 1994; Ernfors et al., 1995; Conover and Yancopoulos, 1997), graft tissue lacking BDNF was collected from conditional BDNF knock-out mice created using Cre-lox technology. Cre-recombinase was expressed under one of two different promoters. Mice in which a ubiquitous tamoxifen-inducible Cre-recombinase was expressed (strain B6.Cg-Tg(Thy1-Cre/ESR1,-EYFP)AGfng/J; stock no. 007606; The Jackson Laboratory) (Hayashi and McMahon, 2002) were bred to floxed BDNF mice in which loxP sites were inserted around bdnf exon 8 to create inducible BDNF-null mice (Rauskolb et al., 2010). Offspring from these matings were treated with tamoxifen to induce ubiquitous Cre expression and excision of the bdnf gene in all tissues (tam-Cre::BDNFf/f Tx). After tamoxifen administration, tam-Cre::BDNFf/f Tx mice had an average weight of 38 g, whereas the average weight for all other mouse genotypes was 22 g. This weight gain may be due to a reduction in BDNF signaling in the hypothalamus (Unger et al., 2007). Nerve size did not appear to be altered by the elevated weight of these donor mice. Transgenic mice containing a construct that expresses Cre recombinase under the control of a exogenous promoter for ciliary neurotrophic factor (CNTF-Cre::BDNFf/f) were bred to the same floxed BDNF mice described above to produce offspring in which BDNF is knocked-out selectively in CNTF-expressing cells. Mice with only one floxed BDNF allele (tam-Cre:: BDNFf/w Tx and CNTF-Cre::BDNFf/w) were used to examine the effects of reduced BDNF. Transgenic mice not treated with tamoxifen (tam-Cre::BDNFf/f UT and tam-Cre::BDNFf/w UT) were used as additional controls. The lack of BDNF in tam-Cre::BDNFf/f Tx mice was confirmed using fluorescent in situ hybridization (FISH; Fig. 1C). To evaluate the expression of Cre in CNTF-Cre mice, we used a Cre-recombinase reporter mouse, the tomato mouse described above, as a measure of Cre-expression. The presence of GFP+ Schwann cells in the sciatic nerve of CNTF-Cre::tomato mice (Fig. 1D,E) confirms that Cre-recombinase is expressed in a cell-type-specific manner and, by analogy, that BDNF is knocked-out selectively in CNTF-Cre::BDNFf/f mice. RT-PCR was performed on the distal stumps of transected TIB and CF nerves from CNTF-Cre::BDNFf/f and WT mice 2 weeks posttransection to further assess the BDNF content of the nerves of these mice. In nerves from WT mice, a 25.0 ± 7.3 (SEM)-fold increase was found in BDNF mRNA, relative to that in nerves from CNTF-Cre::BDNFf/f mice. These RT-PCR results combined with the data from the reporter mice support our assumption that BDNF is highly reduced in CNTF-expressing Schwann cells in CF and TIB nerves of CNTF-Cre::BDNFf/f mice.
Figure 1.

BDNF mRNA is not present in cells of the transgenic knock-outs used as nerve graft donors. A, Based on results of FISH, BDNF mRNA is present in the cells in the ventral horn of the spinal cord of untreated wild-type mice. B, Two weeks of treadmill training (TT) resulted in increased labeling of BDNF mRNA-positive cells in wild-type mice. C, No BDNF mRNA was detected in tissues collected from tam-Cre::BDNFf/f mice, regardless of training condition. D, BDNF allele deletion for the CNTF-Cre::BDNFf/f mice was assessed using a Cre reporter mouse, termed tomato. All cells in the sciatic nerve of the tomato mouse are tomato fluorophore-positive. E, After expression of Cre-recombinase, Cre+ cells express green fluorescent protein. The presence of green fluorescence in presumptive Schwann cells indicates that Cre recombinase is active in cells of the sciatic nerve of CNTF-Cre-expressing mice. These data support the idea that Cre activity in CNTF+ cells in CNTF-Cre::BDNFf/f mice removes BDNF from CNTF+ cells.
Tamoxifen administration.
Powdered tamoxifen was dissolved in a solution consisting of 10% ethanol and 90% sunflower oil (all from Sigma-Aldrich) and heated to 37°C. The tamoxifen was stored at 4°C for 3 d. Adult tam-Cre::BDNF and SLICK mice were administered this tamoxifen solution orally, at a dose of 0.75 mg/20 g body weight per day for 3 d. The SLICK mice were administered a second round of tamoxifen 2 weeks after the original dosing because a second dose has been reported to be necessary to achieve complete knock-out in these animals (Young et al., 2008). All mice were used in experiments a minimum of 2 weeks after final tamoxifen administration.
Treadmill training.
Beginning the third day after nerve transection and repair, mice were treadmill-trained for 5 d a week for 2 weeks using either an interval or continuous paradigm (Sabatier et al., 2008). We have previously reported a sex difference in the elongation of axons in response to treadmill training (Wood et al., 2011). In these experiments, for each sex, we used the training paradigm that has been shown to enhance axon regeneration. Thus, male mice were trained continuously for 1 h at a slow speed of 10 m/min (continuous training) and female mice performed four repetitions at a faster speed of 20 m/min for 2 min with a 5 min rest (interval training). There was no statistically significant difference in the values obtained from males and females; thus, the data from both sexes were combined.
Tissue collection.
Animals were deeply anesthetized with pentobarbital (150 mg/kg, i.p.) and transcardially perfused with 0.9% saline and 4% paraformaldehyde. Sciatic nerves and spinal cord segments L3–L5 were harvested and cryoprotected for 3 d in 20% sucrose. Both CF and TIB nerves were mounted onto slides using VectaShield (Vector Labs) and stored at 4°C. Fixed spinal cord segments (L3–L5) were collected to analyze the amount of BDNF in the tissue. The spinal cord segments were cut (transverse sections; 20 μm thick) on a cryostat (Reichert-Jung) and stored at −50°C.
Axon length analysis.
The confocal imaging and measurements of regenerating axon profiles were performed as previously described (English et al., 2005; Groves et al., 2005). Briefly, the nerves were imaged at a low magnification using a Zeis LSM510 confocal microscope. Stacks of serial optical sections (10 μm thick) were pasted together using Adobe Photoshop to reconstruct the entire length of the repaired nerve and graft. The lengths of YFP+ axon profiles were measured using Image Pro Plus or ImageJ software.
FISH.
The expression of BDNF mRNA was examined using FISH as previously described with modifications (Muddashetty et al., 2007). BDNF riboprobes of 745 bp corresponding to nucleotides 485–1229 of the mouse BDNF (GenBank accession number EF125685) were generated from linearized pcDNA3-BDNF plasmid by in vitro transcription with DIG RNA Labeling Mix (Roche). T7 and SP6 RNA polymerases were used to obtain both antisense- and sense-labeled probes, respectively. Riboprobes were alkaline-hydrolyzed to reduce the size. The spinal sections were prehybridized in hybridization buffer (4× SSC, 50% deionized formamide, 1× Denhardt's solution, 10% dextran sulfate, 0.5 mg/ml salmon sperm DNA, 0.25 mg/ml yeast t-RNA) at 55°C for 1 h before they were incubated with DIG-labeled antisense and sense riboprobes at 55°C overnight. The hybridized DIG-probe-labeled sections were detected using a Cy3-conjugated mouse anti-DIG antibody (1:200; Jackson ImmunoResearch). Images were acquired on a Zeis LSM510 confocal microscope using a 20× objective.
RT-PCR.
The dissected sciatic nerves were subjected to RNA extraction using TRIzol reagent (Invitrogen). Reverse transcription was done with random hexamer primers by Superscript III First-Strand Synthesis System (Invitrogen). Real-time PCR for BDNF mRNA from the sciatic nerves was performed as previously described (Donnelly et al., 2011; Willis et al., 2011). In brief, the reverse-transcribed RNA was amplified using Prism7000 Sequence Detection System (Applied Biosystems) with QuantiTect SYBR Green PCR Kits (Qiagen). The following primers were used for real-time PCR amplification: mouse BDNF (forward: 5′-CGGCGCCCATGAAAGAAGTA-3′; reverse: 5′-AGACCTCTCGAACCTGCCCT-3′) and mouse 18S rRNA (forward: 5′-TTAACGAGGATCCATTGGAG-3′; reverse: 5′-GGCCTGCTTTGAACACTCTA-3′). Relative cycle threshold quantification (ΔΔCt) was performed using 18S rRNA as endogenous control to obtain ΔCt and then ΔCt from the knock-out nerve tissue being normalized to the one from wild-type to obtain ΔΔCt. Fold change was expressed by 2−ΔΔCt.
Statistical analysis.
Data are expressed as the average ± SEM. Differences in the number of axons as well as the median length of axon profiles were assessed using a one-way ANOVA with Fisher's least significant differences post hoc analysis (α < 0.05) where appropriate. A Student's t test was used to assess differences based on the sex of the host, the nerve cut and repaired, and the limb used. Because axon profile lengths are not normally distributed (English et al., 2005), the differences between axon profile length populations were examined using the Kolmogorov–Smirnov two-sample test. Using this nonparametric method, the probability that two distributions measured could be samples drawn from the same population is determined. Its use requires no prior assumptions about the distributions of the data.
Results
Axon regeneration is reduced significantly in grafts lacking BDNF
The CF and TIB nerves of thy-1-YFP-H mice were cut and repaired with 8–10-mm-long grafts harvested from the corresponding nerve in a mouse lacking both BDNF alleles in all cells (tam-Cre::BDNFf/f Tx), a mouse missing one BDNF allele in all cells (tam-Cre::BDNFf/w Tx), or a mouse with both BDNF alleles in all cells (WT, tam-Cre::BDNFf/f UT, or tam-Cre::BDNFf/w UT). Two weeks postrepair, host mice were killed, and the regeneration of YFP+ axon profiles was analyzed using confocal microscopy. We found that the lengths of axon profiles from grafts lacking BDNF (tam-Cre:: BDNFf/f Tx) were significantly decreased, as demonstrated by a significant shift in the distribution to the left of data from thy-1-YFP-H mice whose cut axons regenerated into grafts from WT mice (p ≤ 0.003; Fig. 2A). Regenerating axons grow readily into (BDNF-containing) grafts from WT donors, but most axons growing into grafts from BDNF−/− donors stop just distal to the surgical repair site. We also analyzed the median lengths of axon profiles growing into each graft type (Fig. 2B). The average median length of axon profiles from nerves repaired with grafts from tam-Cre::BDNFf/f Tx mice was significantly shorter than that in nerves repaired with grafts from WT, tam-Cre::BDNFf/f UT, or tam-Cre::BDNFf/w UT mice (p ≤ 0.0006) or from tam-Cre::BDNFf/w Tx mice (p ≤ 0.0001). No significant difference was found in the distributions of axon profile lengths or in the median lengths of axon profiles growing into WT, tam-Cre::BDNFf/f UT, tam-Cre::BDNFf/w UT, or tam-Cre:: BDNFf/w Tx grafts. The lack of difference in axon growth into WT and BDNFf/w environments is not surprising because this effect has been reported in previous papers where traditional BDNF heterozygotes were used as graft donors (English et al., 2005). Thus, we feel that these data are strong evidence that BDNF in the environment surrounding regenerating axons (the graft) is necessary for elongation of axon profiles after nerve cut and repair.
Figure 2.

Profiles of axons growing into grafts lacking BDNF (tam-Cre::BDNFf/f treated and CNTF-Cre::BDNFf/f) are significantly shorter than controls. A, C, The cumulative frequency histogram of profile lengths of axons growing into tam-Cre::BDNFf/f-treated (A) and CNTF-Cre::BDNFf/f (C) grafts are significantly shifted to the left compared with controls, indicating an overall decrease in profile lengths. For all cumulative histograms, the size of each symbol is proportional to the SEM of that data point and the dashed line indicates the 50th percentile. B, The median length of axon profiles growing into tam-Cre::BDNFf/f-treated grafts are significantly reduced compared to those growing into WT and untreated grafts (**p ≤ 0.0006) and tam-Cre::BDNFf/w Tx grafts (*p ≤ 0.0001). D, The median length of axon profiles from CNTF-Cre::BDNFf/f-treated grafts are significantly reduced compared with the lengths of regenerating axon profiles growing into grafts containing BDNF (**p ≤ 0.0001 compared to WT, *p ≤ 0.002 compared to CNTF-Cre::BDNFf/w). For all bar graphs, circles represent individual data points and error bars represent ±SEM.
To isolate the cellular source of BDNF important for axon regeneration into a graft, we generated transgenic mice that lack BDNF only in CNTF-Cre-recombinase-expressing cells. Grafts from CNTF-Cre::BDNFf/f mice lack BDNF in CNTF+ Schwann cells. Similar to the results with the tamoxifen-inducible BDNFf/f mice, the distribution of axon profile lengths growing into CNTF-Cre::BDNFf/f grafts is significantly shifted to the left (Fig. 2C) compared with WT (p ≤ 0.004) or heterozygous CNTF-Cre::BDNFf/w grafts (p ≤ 0.01). No difference was found in the median length of axon profiles or in the population distributions of axons growing into WT or CNTF-Cre::BDNFf/w grafts. The median length of axon profiles growing into CNTF-Cre:: BDNFf/f grafts was significantly shorter than those growing into WT (p ≤ 0.0001) or CNTF-Cre::BDNFf/w grafts (p ≤ 0.002). Axons did not grow very far into the grafts lacking BDNF in Schwann cells (Fig. 3E). When considered together with the tam-Cre::BDNFf/f data presented above, we conclude that Schwann cell-derived BDNF is critical for the elongation of axons into nerve grafts after nerve injury.
Figure 3.
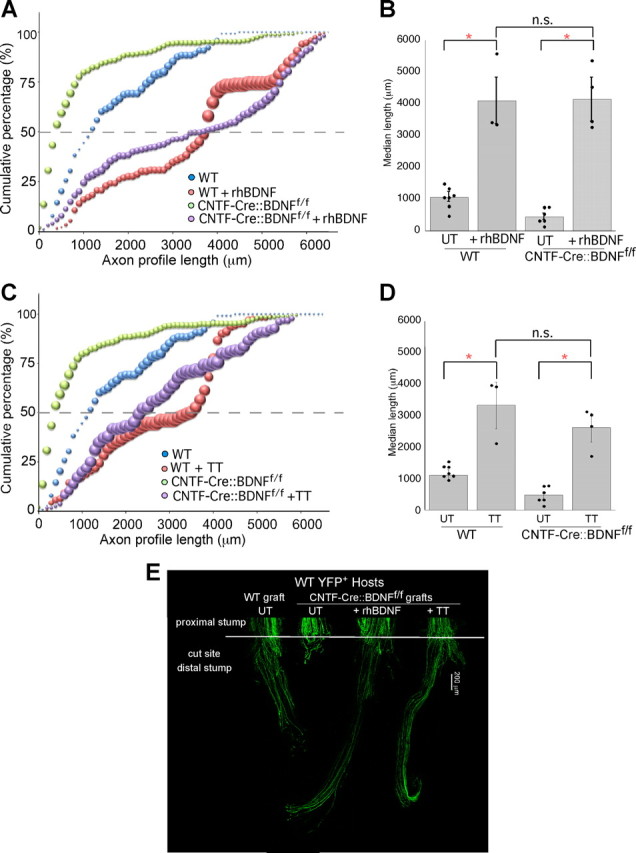
Two weeks of treadmill training (TT) or the administration of BDNF to the surgical repair site significantly enhances axon profile lengths. A, The cumulative histogram of profile lengths of axons growing into wild-type or CNTF-Cre::BDNFf/f grafts treated with rhBDNF is shifted significantly to the right of those from mice whose nerves were repaired with either untreated wild-type or CNTF-Cre::BDNFf/f grafts, suggesting that their axons are significantly longer. B, The median axon profile lengths are significantly longer into BDNF-treated wild-type and CNTF-Cre::BDNFf/f grafts compared with controls (*p ≤ 0.0001). C, The cumulative histograms of the lengths of axon profiles in wild-type and CNTF-Cre::BDNFf/f mice are shifted significantly to the right of those of control mice after treadmill training, indicating a significant increase in the lengths of all regenerating axon profiles. D, The median axon profile length of wild-type and CNTF-Cre::BDNFf/f is significantly increased after treadmill training (*p ≤ 0.0001). E, Low-magnification images of representative nerves from thy-1-YFPH hosts 2 weeks after nerve repair. The white bar across the nerve indicates the site of surgical repair.
Exogenous BDNF enhances axon regeneration
To examine whether the lack of growth into BDNF-lacking grafts could be reversed by the exogenous administration of BDNF, we performed a series of experiments in which recombinant human BDNF was added at the time of surgery to the fibrin glue used to secure the repaired nerve. We hypothesized that, if BDNF is important for the elongation of axons into a graft after nerve injury, then axons would grow into BDNF-lacking grafts if the neurotrophin was added to the repair site. We found that adding a presumed pharmacologic dose of BDNF to the fibrin glue at the time of repair enhanced the growth of axons, shifting the distribution of WT axon profile lengths significantly to the right compared with data from animals repaired similarly but without BDNF treatment (p ≤ 0.04; Fig. 3A). This shift to the right was similar to that found when BDNF was added to grafts from transgenic mice lacking BDNF (p ≤ 0.0001). This trend toward longer axons was also reflected in a very large increases in the median axon profile lengths of axons growing into BDNF-treated WT grafts (p ≤ 0.0001; Fig. 3B) and BDNF-treated CNTF-Cre::BDNFf/f grafts (p ≤ 0.0001). There is no significant difference in the lengths of axon profiles growing into WT grafts treated with BDNF and CNTF-Cre::BDNFf/f grafts treated with BDNF. The dramatic elongation of axons after the addition of BDNF is similar to data reported previously (English et al., 2005) regarding the addition of BDNF to wild-type grafts. In both the current and previously published experiments, the addition of BDNF produces axons that grow much longer than similarly repaired nerves without treatment. Thus, the addition of BDNF at the time of nerve cut results in elongation of axon profiles even in the absence of Schwann cell-derived BDNF.
Treadmill training enhances axon regeneration
Previous studies have shown that BDNF is upregulated after exercise (Gómez-Pinilla et al., 2002). To determine whether the source of the increased BDNF is Schwann cell-derived, we performed a set of experiments in which we treadmill-trained thy-1-YFP-H mice with cut CF and TIB nerves repaired with CNTF-Cre::BDNFf/f or WT grafts. All host mice ran 5 d a week for 2 weeks on a level-incline treadmill beginning 2 d postnerve transection. The cumulative population of axon profile lengths for trained WT (p ≤ 0.03) and CNTF-Cre::BDNFf/f (p ≤ 0.0002) mice was significantly shifted to the right, indicating a larger number of long axons compared with untrained mice (Fig. 3C). No difference was found in the length of axon profiles growing into either graft type in trained hosts. The median axon profile lengths in trained WT (p ≤ 0.0001) and trained CNTF-Cre::BDNFf/f (p ≤ 0.0001) mice are significantly longer than untrained controls (Fig. 3D). Thus, treadmill training results in an enhancement of axon regeneration despite the lack of Schwann cell-derived BDNF from the graft that was shown earlier to stimulate axon regeneration. The enhanced growth of axons into grafts lacking BDNF found in trained mice could be due to the upregulation of factors other than BDNF in the graft or an upregulation of BDNF in the regenerating neurons themselves.
Treadmill training upregulates BDNF mRNA in motoneurons
To test the hypothesis that treadmill training upregulates BDNF expression in motoneurons, we used FISH to detect BDNF mRNA in lamina IX cells of the spinal cord. FISH was performed on tissue from spinal segments L3–L5 from trained and untrained WT and tam-Cre::BDNFf/f Tx mice whose CF and TIB nerves were cut and repaired. Significantly more BDNF mRNA signal was found in intact and trained WT mice (Fig. 1B) compared with intact and untrained WT mice (Fig. 1A). The distribution of the cells labeled as BDNF mRNA positive also changed with training. More small cells surrounding lamina IX were labeled after training in WT mice. No fluorescence was found in either trained or untrained tam-Cre::BDNFf/f Tx mice (Fig. 1C), consistent with the idea that the transgenic mice are BDNF-null in all tissues. The FISH data, considered together with the finding that axons elongate into BDNF-lacking grafts when the hosts are treadmill trained, are evidence that the training paradigm used may be adequate to upregulate motoneuron BDNF to levels sufficient to overcome any lack of BDNF in Schwann cells in the pathway taken by the axons.
Treadmill training-induced axon regeneration is dependent on neuron-derived BDNF
To test the hypothesis that neuronal BDNF is sufficient to produce training-mediated axon elongation, we created a transgenic mouse in which BDNF was null in a subset of axons that also contained YFP. Using these SLICK::BDNFf/f mice as hosts, we were able to measure the length of BDNF-lacking, YFP+ axon profiles growing into BDNF-rich (WT) or BDNF-null (tam-Cre::BDNFf/f Tx) grafts (Fig. 4). Marked axons of SLICK::BDNFf/f hosts grow into WT grafts in which Schwann cells produce BDNF. Axons from SLICK::BDNFf/f mice marked by YFP do not grow very far into grafts lacking Schwann cell BDNF. The entire population of YFP+ SLICK::BDNFf/f axons growing into grafts from tam-Cre::BDNFf/f Tx mice is shifted significantly to the left of the population of YFP+ SLICK::BDNFf/f axons growing into WT grafts (p ≤ 0.04; Fig. 4A). Thus, a lack of BDNF in both the neurons and the graft leads to an increase in short versus long axons growing into the graft. Hosts that lacked only one BDNF allele (SLICK::BDNFf/w) were used as controls and grew in patterns similar to the BDNF-rich, thy-1-YFP-H hosts. Similar to the thy-1 YFP-H hosts, untrained SLICK::BDNFf/w hosts did not grow as far into BDNF-lacking grafts (BDNFf/f) compared with WT grafts (p ≤ 0.04), again demonstrating the importance of Schwann cell-derived BDNF in axon regeneration. However, axons in treadmill-trained SLICK::BDNFf/w hosts did successfully grow into BDNF-lacking grafts compared with untrained SLICK::BDNFf/w hosts (p ≤ 0.0005; Fig. 4D). Axons in treadmill-trained SLICK::BDNFf/f hosts grew longer into WT grafts than did axons from untrained SLICK::BDNFf/f mice (p ≤ 0.0001), with a larger overall population of long axons (p ≤ 0.05; Fig. 4C). These findings are similar to those of axons in thy-1-YFP-H hosts growing into wild-type and BDNF-null grafts. However, unlike axons from treadmill-trained thy-1-YFP-H host mice, axons in treadmill-trained SLICK::BDNFf/f hosts do not grow into grafts lacking Schwann cell BDNF. The significant shift left in the cumulative histogram for these axons confirms a larger percentage of very short regenerating axons (p ≤ 0.01; Fig. 4C). There is no significant difference among treadmill-trained SLICK::BDNFf/f hosts growing into WT grafts and treadmill-trained SLICK::BDNFf/w hosts growing into WT or BDNFf/f Tx grafts. Thus, in the absence of Schwann cell-derived BDNF in the pathway taken by regenerating axons, increased expression of neuron-derived BDNF is necessary to drive optimal regeneration. We conclude that both axonal and Schwann cell sources of BDNF are necessary for axon regeneration.
Figure 4.
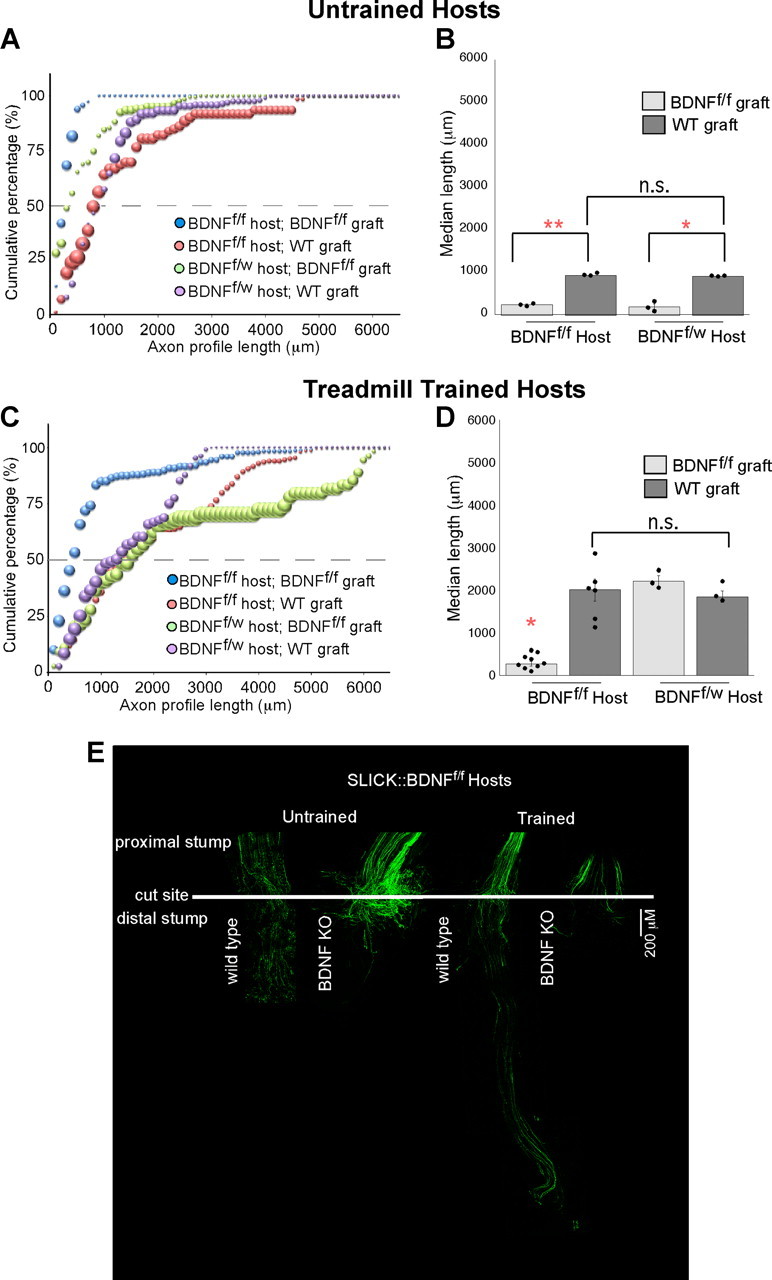
Axons lacking BDNF do not significantly regenerate into BDNF-lacking grafts regardless of the training. A, The cumulative histograms of axon profile lengths of SLICK::BDNFf/f and SLICK::BDNFf/w axons growing into tam-Cre::BDNFf/f Tx grafts are shifted significantly to the left of those of controls, suggesting that these axons are significantly shorter than those growing in WT grafts (p ≤ 0.04). B, The median axon profile lengths are significantly shorter into BDNFf/f grafts compared with WT (*p ≤ 0.009; **p ≤ 0.0001). C, The cumulative histogram of the lengths of axon profiles of YFP+ SLICK::BDNFf/f axons is significantly shifted to the left when nerves are repaired with BDNFf/f grafts and hosts are treadmill-trained for 2 weeks postrepair (p ≤ 0.01). D, The median axon profile length of SLICK::BDNFf/f axons growing into BDNFf/f grafts remains significantly decreased after treadmill training compared with SLICK::BDNFf/f axons growing into a WT graft (p ≤ 0.0001). E, Low-magnification images of representative nerves from trained and untrained SLICK::BDNFf/f hosts 2 weeks after nerve repair. The white bar across the nerve indicates the site of surgical repair.
BDNF content in the regenerating axons themselves or in the cells of the distal stump does not affect regenerative sprouting
In addition to the elongation of axons, we investigated whether the lack of BDNF in the graft or axons affects regenerative sprouting. We assessed the number of YFP+ axons whose lengths were measured in the grafts as a ratio of the total number of YFP+ axons in the proximal stump. This sprouting index was used as a global estimate of the number of branches per axon (Sabatier et al., 2008). We found no significant difference in the sprouting index regardless of the graft type or host model used (data not shown). Thus, the lack of BDNF production by cells in the segment of nerve distal to the transection or by the axons themselves did not affect the number of regenerative sprouts forming from the segment of the nerve proximal to the transection.
Discussion
In this study, we found that BDNF derived from either Schwann cells or motoneurons can facilitate axon regeneration. Two weeks following nerve transection and repair, the lengths of profiles of regenerating axons growing into grafts lacking BDNF in Schwann cells were strikingly shorter than those growing into wild-type grafts. The addition of recombinant human BDNF at the site of repair restored axon regeneration in the absence of Schwann cell-derived BDNF and enhanced the length of regenerating axons into both wild-type and BDNF-null grafts dramatically. Thus, we concluded that BDNF derived from Schwann cells in the pathway surrounding regenerating axons is important for their elongation after nerve transection. If the host animal is treadmill-trained after injury, then motoneuron-derived BDNF can enhance axon regeneration regardless of the pathway surrounding the axons. Two weeks of daily treadmill training after nerve transection results in enhanced axon elongation, likely due to an increased expression of BDNF mRNA in motoneuron cells bodies. Selectively knocking out BDNF in neurons blocked the enhancing effect of treadmill training on axon elongation into grafts from Schwann cells lacking BDNF. We infer from these data that motoneuron-derived BDNF mediates the effect of treadmill training on elongation of regenerating axons after nerve injury.
We propose a model of axon regeneration in peripheral nerves in which two distinct mechanisms involving BDNF released from Schwann cells and neurons may be involved. Following peripheral axotomy, Schwann cells in the nerve segment distal to the injury synthesize and secrete BDNF into the Schwann cell basal laminae (Funakoshi et al., 1993). Regenerating axons entering endoneurial tubes in the distal segment initially use this Schwann cell-derived BDNF to begin elongation. Spinal motoneurons contain BDNF and trkB mRNAs (Al-Majed et al., 2000; Buck et al., 2000). After axotomy, spinal motoneurons increase the expression of BDNF relatively quickly, but expression levels decrease within 1 week (Funakoshi et al., 1993). Schwann cells in the distal stump, however, can synthesize and secrete BDNF for 2 months or longer (for review, see Gordon, 2009). Since the transformed Schwann cells in the pathway of regenerating axons do not contain a full-length trkB receptor (Frisén et al., 1993), the stimulation of axon elongation is via a classic retrograde signaling pathway from the Schwann cells via trkB receptors on the regenerating axons. Treadmill training after nerve transection results in an increased production of neuronal BDNF. We propose a second neurotrophin signaling mechanism whereby neuronal expression of BDNF produced by treadmill training enhances the elongation of axons through the cut site using an autocrine signaling mechanism. This second neurotrophin mechanism of elongation is sufficient to overcome a lack of BDNF in the distal stump.
Using Cre-lox gene deletion technology, we created mice in which BDNF was greatly reduced in CNTF-expressing Schwann cells. When tissue from these animals was used to repair nerves after transection, axon regeneration was inhibited. In previous studies, a transient upregulation of BDNF in the motoneuron cell bodies (Funakoshi et al., 1993), as well as skeletal muscles (Madison et al., 2009), has been reported after nerve transection; however, clearly these sources of BDNF are not sufficient to overcome the lack of BDNF in these grafts. Contributions from Schwann cells in the proximal stump also do not seem to play an important role in BDNF-mediated axonal elongation, as experiments in which both axons and grafts lacked BDNF resulted in little growth despite the presence of WT levels of BDNF in the Schwann cells in the proximal stump. Using RT-PCR analysis of CNTF-Cre::BDNFf/f nerves, we show that a small amount of BDNF mRNA may be present in the distal stumps of transected CF and TIB nerves. The source of the BDNF in this tissue is unknown. The most likely explanations are that it is due to incomplete deletion of the bdnf gene by Cre-recombinase in CNTF-expressing Schwann cells (Nagy, 2000) or to cells not expressing CNTF (and Cre). Regardless of the source, the remaining BDNF is 25-fold lower than levels found in WT nerves and is insufficient to encourage axon growth into the CNTF-Cre:: BDNFf/f nerve graft. Thus, we conclude that Schwann cell-derived BDNF in the pathway taken by regenerating axons acting on receptors on those axons must be the primary mechanism by which axons regenerate in cut nerves. When animals are treadmill-trained, however, a more pronounced and prolonged production of BDNF by neurons is sufficient to allow for autocrine signaling to occur independent of Schwann cell-derived BDNF.
Consistent with previous findings, we found that treadmill training produced longer axon profiles (Sabatier et al., 2008). Regardless of the presence or absence of BDNF in Schwann cells in the distal stump, these axon profiles grew almost as long as axons growing into grafts treated with BDNF at the time of surgical repair. This elongation of axons is likely due to the upregulation of BDNF mRNA in motoneuron cell bodies after training. Removing BDNF from neurons results in axons that can no longer grow into grafts lacking Schwann cell derived-BDNF when the host is treadmill-trained. This finding that motoneuron upregulation of BDNF can mediate axon elongation is supported by similar findings in experiments using electrical stimulation after nerve cut. Electrically stimulating the proximal stump of a cut nerve produces a large increase in motoneuron BDNF expression and a concomitant enhancement of axon elongation (Al-Majed et al., 2000). When the cut nerve is repaired with an acellular graft lacking neurotrophins, electrically stimulated axons can still regenerate (English et al., 2007). Thus, with training or electrical stimulation, the upregulation of BDNF in motoneurons is sufficient to promote regeneration of axons even in the absence of Schwann cell BDNF.
We found that treadmill training enhances the elongation of BDNF-lacking axons growing into wild-type grafts but not into BDNF-lacking grafts. One explanation of this finding is that the training could also increase the expression of trkB receptors on the regenerating axons. Such an increase in trkB expression would make the regenerating axons more sensitive to Schwann cell-derived BDNF, thereby increasing the length of axons. Consistent with this idea, axon regeneration in trkB+/− mice is limited (Boyd and Gordon, 2001; MacGillivray, 2003; Lee et al., 2004), where the regenerating axons would be presumed to be less sensitive to the growth promoting effects of Schwann cell-derived BDNF.
Other mechanisms also exist that could account for the changes seen with treadmill training. The activation of motoneurons during the training could upregulate BDNF enough to cause a shift in the balance between growth promotion and growth inhibition, allowing axons to overcome growth inhibitory molecules such as chondroitin sulfate proteoglycans at the surgical repair site. Once the axons enter the distal stump, BDNF-secreting Schwann cells or the axons themselves could create a gradient to facilitate the growth toward target tissues. The upregulation of BDNF by treadmill training may enhance this process by releasing more BDNF at the growth cones and activating trkB receptors in an autocrine fashion.
It is likely that complex interactions between regenerating axons and the regenerative environment exist. Previously, parsing apart the contributions of each to the regenerative process has been difficult and interpretation complicated. Using emerging genetic technologies to manipulate the cellular sources of neurotrophins and other growth molecules has led to a better understanding of the important role of each of these in axonal regeneration. Utilizing exercise to increase neuronal BDNF could help to improve axon regeneration and patient outcome after traumatic nerve injury.
Footnotes
This work was funded by NIH Grants NS057190 (A.W.E.), K12GM000680 (J.C.W.), and HD055835 (G.J.B.). We thank Dr. Michael Sendtner for the generous gift of floxed BDNF and CNTF-Cre transgenic mice, and Dr. Jingsheng Gu, Dr. Christina Gross, and Ms. Amanda Mulligan for their technical assistance.
The authors declare no competing financial interests.
References
- Al-Majed AA, Brushart TM, Gordon T. Electrical stimulation accelerates and increases expression of BDNF and trkB mRNA in regenerating rat femoral motoneurons. Eur J Neurosci. 2000;12:4381–4390. [PubMed] [Google Scholar]
- Boyd JG, Gordon T. The neurotrophin receptors, trkB and p75, differentially regulate motor axonal regeneration. J Neurobiol. 2001;49:314–325. doi: 10.1002/neu.10013. [DOI] [PubMed] [Google Scholar]
- Buck CR, Seburn KL, Cope TC. Neurotrophin expression by spinal motoneurons in adult and developing rats. J Comp Neurol. 2000;416:309–318. doi: 10.1002/(sici)1096-9861(20000117)416:3<309::aid-cne3>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Conover JC, Yancopoulos GD. Neurotrophin regulation of the developing nervous system: analyses of knockout mice. Rev Neurosci. 1997;8:13–27. doi: 10.1515/revneuro.1997.8.1.13. [DOI] [PubMed] [Google Scholar]
- de Vries J, Menovsky T, van Gulik S, Wesseling P. Histological effects of fibrin glue on nervous tissue: a safety study in rats. Surg Neurol. 2002;57:415–422. doi: 10.1016/s0090-3019(02)00736-x. discussion 422. [DOI] [PubMed] [Google Scholar]
- Donnelly CJ, Willis DE, Xu M, Tep C, Jiang C, Yoo S, Schanen NC, Kirn-Safran CB, van Minnen J, English A, Yoon SO, Bassell GJ, Twiss JL. Limited availability of ZBP1 restricts axonal mRNA localization and nerve regeneration capacity. EMBO J. 2011;30:4665–4677. doi: 10.1038/emboj.2011.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English AW, Meador W, Carrasco DI. Neurotrophin-4/5 is required for the early growth of regenerating axons in peripheral nerves. Eur J Neurosci. 2005;21:2624–2634. doi: 10.1111/j.1460-9568.2005.04124.x. [DOI] [PubMed] [Google Scholar]
- English AW, Schwartz G, Meador W, Sabatier MJ, Mulligan A. Electrical stimulation promotes peripheral axon regeneration by enhanced neuronal neurotrophin signaling. Dev Neurobiol. 2007;67:158–172. doi: 10.1002/dneu.20339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernfors P, Van De Water T, Loring J, Jaenisch R. Complementary roles of BDNF and NT-3 in vestibular and auditory development. Neuron. 1995;14:1153–1164. doi: 10.1016/0896-6273(95)90263-5. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Frisén J, Verge VM, Fried K, Risling M, Persson H, Trotter J, Hökfelt T, Lindholm D. Characterization of glial trkB receptors: differential response to injury in the central and peripheral nervous systems. Proc Natl Acad Sci U S A. 1993;90:4971–4975. doi: 10.1073/pnas.90.11.4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funakoshi H, Frisén J, Barbany G, Timmusk T, Zachrisson O, Verge VM, Persson H. Differential expression of mRNAs for neurotrophins and their receptors after axotomy of the sciatic nerve. J Cell Biol. 1993;123:455–465. doi: 10.1083/jcb.123.2.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Pinilla F, Ying Z, Opazo P, Roy RR, Edgerton VR. Differential regulation by exercise of BDNF and NT-3 in rat spinal cord and skeletal muscle. Eur J Neurosci. 2001;13:1078–1084. doi: 10.1046/j.0953-816x.2001.01484.x. [DOI] [PubMed] [Google Scholar]
- Gómez-Pinilla F, Ying Z, Roy RR, Molteni R, Edgerton VR. Voluntary exercise induces a BDNF-mediated mechanism that promotes neuroplasticity. J Neurophysiol. 2002;88:2187–2195. doi: 10.1152/jn.00152.2002. [DOI] [PubMed] [Google Scholar]
- Gordon T. The role of neurotrophic factors in nerve regeneration. Neurosurg Focus. 2009;26:E3. doi: 10.3171/FOC.2009.26.2.E3. [DOI] [PubMed] [Google Scholar]
- Groves ML, McKeon R, Werner E, Nagarsheth M, Meador W, English AW. Axon regeneration in peripheral nerves is enhanced by proteoglycan degradation. Exp Neurol. 2005;195:278–292. doi: 10.1016/j.expneurol.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–318. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- Jones KR, Fariñas I, Backus C, Reichardt LF. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SB, Geroski DH, Prausnitz MR, Edelhauser HF. Drug delivery through the sclera: effects of thickness, hydration, and sustained release systems. Exp Eye Res. 2004;78:599–607. doi: 10.1016/s0014-4835(03)00211-2. [DOI] [PubMed] [Google Scholar]
- MacGillivray TE. Fibrin sealants and glues. J Card Surg. 2003;18:480–485. doi: 10.1046/j.0886-0440.2003.02073.x. [DOI] [PubMed] [Google Scholar]
- Madison RD, Sofroniew MV, Robinson GA. Schwann cell influence on motor neuron regeneration accuracy. Neuroscience. 2009;163:213–221. doi: 10.1016/j.neuroscience.2009.05.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muddashetty RS, Kelić S, Gross C, Xu M, Bassell GJ. Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J Neurosci. 2007;27:5338–5348. doi: 10.1523/JNEUROSCI.0937-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- Nagy A. Cre recombinase: the universal reagent for genome tailoring. Genesis. 2000;26:99–109. [PubMed] [Google Scholar]
- Rauskolb S, Zagrebelsky M, Dreznjak A, Deogracias R, Matsumoto T, Wiese S, Erne B, Sendtner M, Schaeren-Wiemers N, Korte M, Barde YA. Global deprivation of brain-derived neurotrophic factor in the CNS reveals an area-specific requirement for dendritic growth. J Neurosci. 2010;30:1739–1749. doi: 10.1523/JNEUROSCI.5100-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatier MJ, Redmon N, Schwartz G, English AW. Treadmill training promotes axon regeneration in injured peripheral nerves. Exp Neurol. 2008;211:489–493. doi: 10.1016/j.expneurol.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger TJ, Calderon GA, Bradley LC, Sena-Esteves M, Rios M. Selective deletion of BDNF in the ventromedial and dorsomedial hypothalamus of adult mice results in hyperphagic behavior and obesity. J Neurosci. 2007;27:14265–14274. doi: 10.1523/JNEUROSCI.3308-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm JC, Cucoranu D, Gu J, Mulligan A, English AW. Limited peripheral axon regeneration in conditional BDNF knockout mice. Soc Neurosci Abstr. 2009 35510.8. [Google Scholar]
- Willis DE, Xu M, Donnelly CJ, Tep C, Kendall M, Erenstheyn M, English AW, Schanen NC, Kirn-Safran CB, Yoon SO, Bassell GJ, Twiss JL. Axonal localization of transgene mRNA in mature PNS and CNS neurons. J Neurosci. 2011;31:14481–14487. doi: 10.1523/JNEUROSCI.2950-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood K, Wilhelm JC, Sabatier MJ, Liu K, Gu J, English AW. Sex differences in the effectiveness of treadmill training in enhancing axon regeneration in injured peripheral nerves. Dev Neurobiol. 2011 doi: 10.1002/dneu.20960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young P, Qiu L, Wang D, Zhao S, Gross J, Feng G. Single-neuron labeling with inducible Cre-mediated knockout in transgenic mice. Nat Neurosci. 2008;11:721–728. doi: 10.1038/nn.2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JY, Luo XG, Xian CJ, Liu ZH, Zhou XF. Endogenous BDNF is required for myelination and regeneration of injured sciatic nerve in rodents. Eur J Neurosci. 2000;12:4171–4180. [PubMed] [Google Scholar]