

Smac mimetic overcomes resistance of bladder cancer cells to BCG-stimulated neutrophils through TNF-α.
Keywords: caspase, FADD, c-IAPs, XIAP, neutralizing antibodies
Abstract
BCG, the current gold standard immunotherapy for bladder cancer, exerts its activity via recruitment of neutrophils to the tumor microenvironment. Many patients do not respond to BCG therapy, indicating the need to understand the mechanism of action of BCG-stimulated neutrophils and to identify ways to overcome resistance to BCG therapy. Using isolated human neutrophils stimulated with BCG, we found that TNF-α is the key mediator secreted by BCG-stimulated neutrophils. RT4v6 human bladder cancer cells, which express TNFR1, CD95/Fas, CD95 ligand/FasL, DR4, and DR5, were resistant to BCG-stimulated neutrophil conditioned medium but effectively killed by the combination of conditioned medium and Smac mimetic. rhTNF-α and rhFasL, but not rhTRAIL, in combination with Smac mimetic, generated signature molecular events similar to those produced by BCG-stimulated neutrophils in combination with Smac mimetic. However, experiments using neutralizing antibodies to these death ligands showed that TNF-α secreted from BCG-stimulated neutrophils was the key mediator of anticancer action. These findings explain the mechanism of action of BCG and identified Smac mimetics as potential combination therapeutic agents for bladder cancer.
Introduction
BCG is the current gold standard immunotherapy for bladder cancer [1, 2]. Intravesical administration of BCG results in recruitment of neutrophils and subsequent cytokine secretion necessary for activation of the cell death program [3, 4]. Neutrophils are significant sources of TNF-α and TRAIL [3, 5]. However, to date, only TRAIL has been proposed to mediate the antitumor activity of BCG [6, 7]. Although BCG immunotherapy is the current standard for bladder cancer therapy, relapse and resistance to BCG are common; thus, there is a need to elucidate how exactly BCG-stimulated neutrophils exert their action and to find alternative therapies to overcome resistance to BCG immunotherapy [8].
Smac/direct IAP-binding protein with low pI (DIABLO) is a mitochondrial protein that binds to IAPs to release active caspases and effect cell death [9]. Smac mimetics are designed to inhibit IAPs and have been shown to work through DR (TNFR)-dependent mechanisms [10–12]. Hence, Smac mimetics represent an ideal choice for evaluation in combination with BCG for bladder cancer therapy.
Here, we show that RT4v6 bladder cancer cells are resistant to BCG-stimulated neutrophils. Use of Smac mimetic compound in combination with BCG-stimulated neutrophils effectively killed RT4v6 cells. BCG-stimulated neutrophils secreted increased TNF-α, 17 kDa FasL, and TRAIL. This raised the question: which of these death ligands mediate the anticancer action of BCG-stimulated neutrophils? Recombinant forms of all three death ligands were capable of killing RT4v6 cells in combination with Smac mimetic. Using the molecular signatures that are generated during cell death and neutralizing antibodies to these death ligands, we found that the key mediator of the action of BCG-stimulated neutrophils was TNF-α, but not TRAIL or FasL. Our study identified the mechanism of BCG immunotherapy and identified Smac mimetics as a potential target for enabling anticancer action of neutrophils in the context of BCG immunotherapy resistance.
MATERIALS AND METHODS
Cell lines and reagents
Human RT4v6 bladder cancer cells [generated by A.M.K. from the RT4 cell line from the American Type Culture Collection (Manassas, VA, USA) by serial passaging in mice six times and authenticated by DNA fingerprinting] were cultured in MEM with 10% FBS, penicillin, streptomycin, vitamins, L-glutamine, nonessential amino acids, and pyruvate supplements.
Smac mimetic compound-C was obtained from TetraLogic Pharmaceuticals (Malvern, PA, USA). Smac mimetics have been designed based on the four N-terminal amino acid sequences of Smac and caspase-9 [13, 14]. Smac mimetics antagonize XIAP-mediated inhibition of active caspases [13]. Compound-C (a proprietary Smac mimetic provided by TetraLogic Pharmaceuticals) is a bivalent Smac mimetic that has cellular and biophysical properties comparable with compound-A and other bivalent Smac mimetics [15]. The cellular activity of compound-A has been described by Vince et al. [16], and its biophysical behavior was reported recently [17] and used by various groups, including ours [16, 18, 19].
rhTNF-α (210-TA), rhTRAIL (375-TL), and rhFasL (126FL/CF) and neutralizing antibodies to TNF-α (MAB610), TRAIL (MAB375), and FasL (MAB126) were purchased from R&D Systems (Minneapolis, MN, USA). Antibodies to TNFR1 (3736), c-IAP1 (4952), XIAP (2045), FasL (4273) and cleaved caspase-8-Asp391/374 (9496) were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibody to c-IAP2 (51-9000062) was purchased from BD Biosciences (San Jose, CA, USA). Fas/CD95 (610197) and FADD (610399) antibodies were purchased from Transduction Labs (Lexington, KY, USA). Antibodies to DR4 (Sc-7863), caspase-8 anti-p18 (Sc-7890), cleaved caspase-9 (Sc-7885), and cleaved caspase-3 (Sc-7148) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). TICE BCG (live-attenuated) was purchased from Organon Teknika (the Netherlands). Histopaque-1119, Histopaque-1077, pepstatin-A, and ascorbic acid were purchased from Sigma (St. Louis, MO, USA). Bortezomib was purchased from ChemieTek (Indianapolis, IN, USA).
Neutrophil isolation and conditioned media preparation
Neutrophils were isolated from human buffy coats (obtained from the blood bank of The University of Texas MD Anderson Cancer Center, Houston, TX, USA). The buffy coats were diluted 1:1 with PBS and layered over gradients of Histopaque-1077 and Histopaque-1119. This set-up was centrifuged in a swing-out bucket rotor at 700 g for 30 min, and the neutrophil layer was aspirated for further purification using RBC lysis (0.2% NaCl) and recovery (1.6% NaCl), as described previously [20]. Neutrophil preparations were identity-verified for segmented nuclear morphology using Hoechst-33342 staining and viability-checked using trypan blue staining. Viable neutrophils (3×106) were cultured with or without 1 × 106 CFUs of live TICE BCG/ml RPMI for 24 h (for dilution calculations, 1–8×108 CFUs/vial was considered as 4×108 CFUs/vial), and the conditioned media were subjected to centrifugation at 3500 rpm for 5 min to pellet down the neutrophils and BCG. The supernatants were filtered through 0.22 μm filters (Nalgene) to avoid the interference of residual neutrophils and BCG in PI-FACS and Western immunoblotting procedures and to confirm that the secreted factors from BCG-stimulated neutrophils indeed mediated the cell death. Batches of conditioned media were calibrated by DNA fragmentation analysis using PI-FACS. Whereas we and others have used cycloheximide in the past for these experiments, we avoided its use in the current protocol, as it is not used to treat bladder cancer patients, and it is capable of modulating DR components.
MTT assay
Five thousand cells/well were seeded in 96-well plates and were treated 24 h after seeding, as indicated in the figures. Twenty-four hours after treatment, the MTT assay was performed as described previously [21]. The results were presented as mean ± se in terms of percentage (n=4).
DNA fragmentation analysis
Sixty thousand cells/well were seeded in six-well plates. Twenty-four hours later, the cells were treated as indicated in the figures. Twenty-four hours after treatment, the cells were subjected to PI-FACS analysis as described previously [21]. The results were presented as mean ± se in terms of percentage (n=3).
Western blotting
Cells were lysed (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1 mM EGTA, 25 mM NaF, 1% Triton-X 100, 1% Nonidet P-40, 0.1 mM Na3VO4, 12.5 mM β-glycerophosphate, 1 mM PMSF, and complete protease inhibitors) and were clarified at 13,000 rpm for 10 min before immunoblotting. For secretory protein profiling, the conditioned media were prepared as described in the quantitative ELISA section below, and 40 μl conditioned medium was subjected to Western blotting.
Microscopy
Six-well plates were seeded with 150,000 cells/well. Twenty-four hours later, cells were treated as indicated in the figures. Twenty-four hours after treatment, cells were imaged using an Olympus IX inverted-phase contrast microscope. For chromatin condensation experiments, the treated cells were subjected to Hoechst-33342 staining for 20 min before imaging. The images were acquired and exported as Tagged Image File Format files using SlideBook software (version SB 4.2.0.12) and composited using Adobe Photoshop (CS4 and CS5).
Quantitative ELISA for TNF-α and TRAIL
Unstimulated and BCG-stimulated neutrophil conditioned media were prepared as described above and were subjected to quantitative ELISA, per the manufacturer's instructions [R&D Systems Quantikine human TRAIL (DTRL00) and human TNF-α (DTA00C) ELISA kits]. For evaluation of autocrine loops of TNF-α and TRAIL, unstimulated or BCG-stimulated neutrophil conditioned medium was incubated with 50,000 RT4v6 cells/ml for 24 h as indicated in the figures. The conditioned media from RT4v6 cells were clarified at 3500 rpm for 5 min, and the supernatants were subjected to quantitative ELISA. The results were presented as mean ± sd (n=2).
Antibody-mediated blocking of TNF-α, TRAIL, and FasL secreted from BCG-stimulated neutrophils
To evaluate the specific role of TNF-α, TRAIL, and FasL secreted from BCG-stimulated neutrophils, respective neutralizing antibodies (1000 ng/ml) were incubated with BCG-stimulated neutrophil conditioned medium (undiluted) for 20 min in parallel with other treatment conditions at room temperature, just before a 1:1 dilution with MEM and treatment. The treated cells were incubated for 24 h and then subjected to PI-FACS DNA fragmentation analysis and microscopy.
Evaluation of TRAIL and FasL neutralizing antibodies for efficacy
To evaluate the efficacy of TRAIL and FasL neutralizing antibodies, 1 ng/ml TRAIL or FasL was incubated with or without 1000 ng/ml respective neutralizing antibody in MEM for 20 min at room temperature and used to treat RT4v6 cells for 24 h. The abilities of neutralizing antibodies to block cell death were assessed by PI-FACS DNA fragmentation analysis.
Statistical analyses
All statistical significance analyses were performed using Student's t test (two-tailed distribution, two-sample unequal variance) in Microsoft Excel 2010. Differences in mean values were considered significant at a P value of <0.05. Error bars were represented as se. All results with error bars were done in triplicate unless specified. All findings on Western blotting and microscopy were confirmed at least twice.
RESULTS
Smac mimetic enables the anticancer action of BCG-stimulated neutrophils
BCG is postulated to induce anticancer activity against bladder cancer cells by stimulating neutrophils and augmenting the secretion of TRAIL from azurophilic granules [3]. However, tumor cells often resist TRAIL-induced cell death through up-regulation of IAP proteins [22]. Smac mimetics work through a DR-dependent mechanism by overcoming this resistance, often through TNFR [12, 15, 16]. Our laboratory has shown recently that Smac mimetics reverse resistance to TRAIL in bladder cancer cells [18]. Here, we were interested in examining how neutrophils stimulated with BCG kill cancer cells and whether Smac mimetic could enable the anticancer action of BCG-stimulated neutrophils in bladder cancer cells that are resistant to BCG-stimulated neutrophils. We chose RT4v6 cells, as they represent the closest approximation of human papillary bladder cancer [23] (all other cell lines represent invasive bladder cancer, which is not treated with BCG). Furthermore, RT4v6 cells possess major DRs, such as TNFR, DR4, DR5, and Fas (see below in Figs. 3B and 6A), which is a prerequisite to examine which death ligands from BCG-stimulated neutrophils mediate the anticancer action.
Figure 3. TRAIL and TNF-α are capable of inducing cell death in combination with Smac mimetic, but TRAIL is not the major active ingredient of BCG-stimulated neutrophil conditioned medium.

(A) Microscopic visualization of cell death induced by Smac mimetic in combination with TRAIL or TNF-α. RT4v6 cells were treated as indicated in the figure and imaged 24 h later. (B) PI-FACS DNA fragmentation analysis and Western blotting analysis for TNFR1 and DR4 in RT4v6 cells, 24 h after treatment (FACS; n=3); *see text for P values. BP1, binding protein 1. (C) Quantitative ELISA for TNF-α (left panel) and TRAIL (right panel) in unstimulated and BCG-stimulated neutrophil conditioned media. (D) Evaluation of the ability of low-picogram levels of rhTNF-α to induce cell death in RT4v6 cells in combination with Smac mimetic. The TNF-α concentrations were achieved by serial dilution in MEM. The cell death induction was measured by PI-FACS DNA fragmentation analysis.
Figure 6. Cell death induced by the combination of Smac mimetic and BCG-stimulated neutrophil conditioned medium involves secretion of 17 kDa FasL and sFADD from neutrophils and down-regulation of FasL and FADD in RT4v6 cells.

(A) Western blotting showing down-regulation of intracellular FasL and FADD during cell death. (B) Western blotting showing secretion of 17 kDa FasL and sFADD in BCG-stimulated neutrophil conditioned media. The conditioned media from RT4v6 cells were collected after 24 h treatment (see Materials and Methods for more details). (C and D) Evaluation of TRAIL and TNF-α for their ability to down-regulate the expression of FADD, XIAP, and c-IAP2. *The media were diluted 1:1 with fresh complete MEM.
Treatment of RT4v6 cells with vehicle (1 μl DMSO/ml MEM), single-agent Smac mimetic, single-agent BCG, or BCG in combination with Smac mimetic did not induce significant cell death, as evaluated by microscopy (Fig. 1A). In addition, unstimulated neutrophil conditioned medium alone, unstimulated neutrophil conditioned medium in combination with Smac mimetic, or BCG-stimulated neutrophil conditioned medium alone did not induce significant cell death. However, treatment with Smac mimetic in combination with BCG-stimulated neutrophil conditioned medium resulted in increased cell death (Fig. 1A). To avoid serum starvation as a result of the use of conditioned media, we diluted unstimulated and BCG-stimulated neutrophil conditioned media 1:1 with fresh complete MEM.
Figure 1. Smac mimetic sensitizes RT4v6 bladder cancer cells to the anticancer action of BCG-stimulated neutrophils.

(A) Microscopic visualization of cell death. RT4v6 cells were treated as indicated in the figure and imaged 24 h later. (B) Formazan formation ability measured by MTT assay, 24 h after treatment. *P = 0.015 compared with control. (C) DNA fragmentation analysis by PI-FACS, 24 h after treatment. *P = 0.0013 compared with control. Note that the BCG-stimulated neutrophil conditioned medium failed to induce cell death in the absence of Smac mimetic, and addition of Smac mimetic enabled cell death (A–C). PMN sup, Neutrophil conditioned medium; C.media, conditioned media diluted 1:1 with fresh complete MEM.
We further confirmed the cell death observed by microscopy using two additional tests: the MTT assay and DNA fragmentation assay. Consistent with the microscopy results, the MTT assay showed that cotreatment with Smac mimetic and BCG-stimulated neutrophil conditioned medium (but not other conditions, including treatment with Smac mimetic alone or BCG alone) significantly reduced the formazan-forming ability of RT4v6 cells (to 45% compared with control 100%; P=0.015; Fig. 1B). Furthermore, the combination of Smac mimetic and BCG-stimulated neutrophil conditioned medium induced significant (68.8%; P=0.0013) DNA fragmentation (Fig. 1C). Taken together, these data demonstrated that BCG stimulation of neutrophils is not sufficient to kill RT4v6 cells, but the addition of Smac mimetic enables the anticancer action of the BCG-stimulated neutrophil conditioned medium.
Sensitization of RT4v6 cells to death by the combination of Smac mimetic and BCG-stimulated neutrophil conditioned medium is associated with down-regulation of c-IAP2 and XIAP
Smac mimetics are known to modulate IAP protein levels during cell death [24]. Western blotting analysis revealed that c-IAP1 was down-regulated when cells were treated with Smac mimetic as a single agent or in combinations (Fig. 2, lanes 3, 5, 7, and 9). However, c-IAP1 was not down-regulated completely under cell death conditions induced by the combination of Smac mimetic and BCG-stimulated neutrophil conditioned medium, suggesting that complete down-regulation of c-IAP1 is not required for this Smac mimetic-induced cell death (Fig. 2, lane 9). On the other hand, c-IAP2 was degraded completely upon treatment with the combination of Smac mimetic and BCG-stimulated neutrophil conditioned medium (Fig. 2, lane 9). c-IAP2 has been reported to be degraded under various cell death conditions [15]. We also found complete degradation of c-IAP2 under other conditions, in which no significant cell death occurred, indicating the involvement of additional molecular events during cell death (Fig. 2, lanes 3, 5, and 7).
Figure 2. Effects of Smac mimetic in combination with BCG-stimulated neutrophil conditioned medium on DR regulatory components and IAPs.
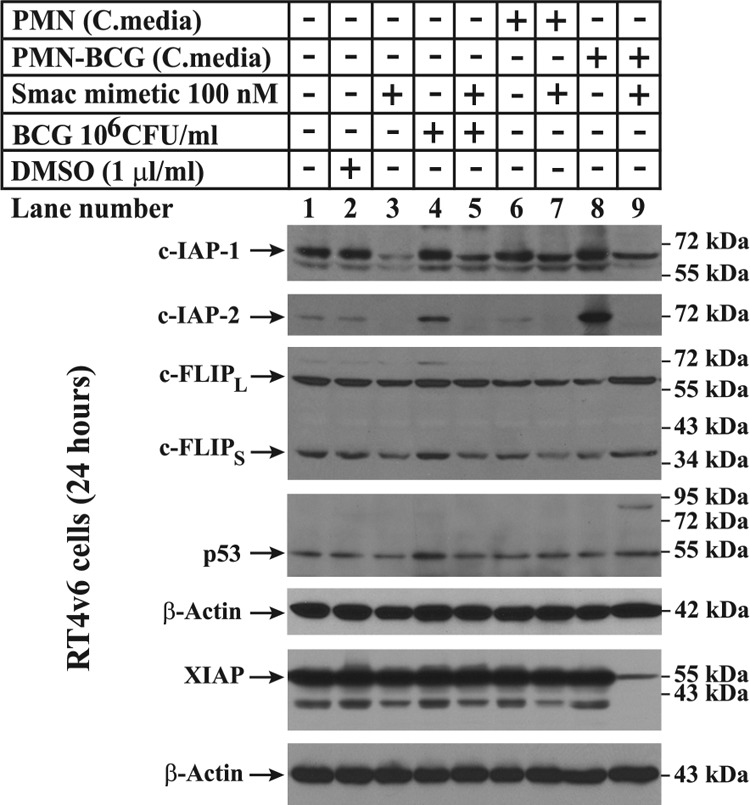
The cells were treated as indicated in the figure, lysed 24 h after treatment, and subjected to Western blotting for c-IAP1, c-IAP2, c-FLIP, p53, and XIAP. as described in Materials and Methods. Note that Smac mimetic as a single agent was capable of down-regulating c-IAP1 (lane 3) but was not down-regulated completely during cell death (lane 9). cFLIPL, c-FLIP long form; cFLIPS, c-FLIP short form.
DRs are known to be modulated by c-FLIP and are transcriptionally up-regulated by stabilized p53 [25, 26], but we did not observe any differences in the expression of c-FLIP or p53 between the different experimental conditions (Fig. 2). However, XIAP, a known inhibitor of active caspases [27], was down-regulated by the combination of Smac mimetic and BCG-stimulated neutrophil conditioned medium (Fig. 2).
Taken together, these data demonstrated that the cell death induced by the combination of Smac mimetic and BCG-stimulated neutrophil conditioned medium is associated with down-regulation of c-IAP2 and XIAP but not c-IAP1, c-FLIP, or p53.
TNF-α is the predominant active component of BCG-stimulated neutrophil conditioned medium
BCG is thought to induce TRAIL secretion from the azurophilic granules of neutrophils to induce antitumor activity [3]. Smac mimetics are thought to induce cell death through autocrine secretion of TNF-α [10]. Neutrophils are good sources of TRAIL and TNF-α. Hence, we examined whether rhTRAIL or rhTNF-α can induce cell death in combination with Smac mimetic, in the absence of BCG-stimulated neutrophil conditioned medium. Treatment of RT4v6 cells with TRAIL and TNF-α as single agents did not induce significant cell death, but the combination of TRAIL or TNF-α with Smac mimetic induced significant cell death, as visualized by microscopy (Fig. 3A) and quantified by DNA fragmentation (TRAIL: 56.4%, P=0.00046; TNF-α, 55.4%, P=0.00066; Fig. 3B). Analysis of TNF-α and TRAIL receptors by Western blotting indicated that TNFR1 and DR4 (TRAIL receptor) are present in RT4v6 cells (Fig. 3B). Our laboratory previously reported the expression of DR5, another TRAIL receptor, in RT4v6 cells [21].
As rhTRAIL or rhTNF-α, in combination with Smac mimetic, can induce cell death in RT4v6 cells significantly, we measured the levels of these cytokines in BCG-stimulated and unstimulated neutrophil conditioned media by quantitative ELISA. The results showed that stimulating neutrophils with BCG (without cycloheximide, which was used in prior reports) resulted in significant increase in secretion of TRAIL (P=0.029) and TNF-α (P=0.004) compared with unstimulated neutrophils. However, the increase in TNF-α was greater than the increase in TRAIL (Fig. 3C). It should be noted that RT4v6 cells can withstand 17 ng/ml TNF-α and 3 ng/ml TRAIL without significant cell death (Fig. 3A and B). To examine whether 150 pg/ml TNF-α (the quantity of TNF-α detected in BCG-stimulated neutrophils) could induce cell death in combination with Smac mimetic, we tested 150 pg/ml rhTNF-α in parallel with 17 ng/ml rhTNF-α, with or without Smac mimetic. We found that 150 pg/ml rhTNF-α, in combination with Smac mimetic, killed RT4v6 cells with efficiency similar to that of 17 ng/ml TNF-α in combination with Smac mimetic (Fig. 3D). These results suggested that TNF-α comprises the predominant active component of BCG-stimulated neutrophil conditioned medium. The role of TRAIL was not certain at this point and was examined further below, along with TNF-α.
Combination of Smac mimetic and BCG-stimulated neutrophil conditioned medium works through caspase-8 and caspase-3 but not through caspase-9 processing
As BCG-stimulated neutrophil conditioned medium had only picogram levels of TRAIL and TNF-α (Fig. 3C), we examined the caspase activation status in RT4v6 cells to understand the mechanism behind the observed cell death. Western blotting for Asp-391/374 (isoform A/B)-specific, cleaved caspase-8 showed that the generation of an ∼26-kDa caspase-8 band (p26) was specific to the cell death induced by Smac mimetic in combination with BCG-stimulated neutrophil conditioned medium (Fig. 4A). Furthermore, generation of caspase-8 p18 was a function of Smac mimetic, as this band was detected in all conditions in which Smac mimetic was included (Fig. 4A). These results suggested that the generation of a caspase-8 p26 fragment is correlated with the induction of significant cell death and that such death likely involves the extrinsic cell death pathway. Further reprobing of the same membrane with an antibody that detects caspase-8 p18 showed that p18 was constitutively generated in RT4v6 cells (Fig. 4A). This indicated that RT4v6 cells have constitutive caspase-8 processing for nonapoptotic purposes, but this does not involve caspase-8-Asp391/374 cleavage-mediated p26 generation.
Figure 4. Combination of Smac mimetic and BCG-stimulated neutrophil conditioned medium activates extrinsic cell death through activation of caspase-8 Asp-391/374 and caspase-3 but inhibition of caspase-9 processing.

All Western blots were performed using RT4v6 cells treated for 24 h as indicated. (A) Western blotting showing caspase-8 Asp-391/374 cleavage-generated p26 (upper panel). The lower panel shows constitutive activation of caspase-8 (reprobed in the same membrane used in the upper panel). (B) Western blotting showing inhibition of caspase-9 processing and activation of caspase-3 under cell death conditions. Caspase-3 was reprobed in the same membrane used for caspase-8 in A. (C) Hoechst-33342 live staining to show the chromatin condensation in response to caspase-3-mediated cell death. *The conditioned media were diluted 1:1 with fresh complete MEM.
Analysis of caspase-9 revealed that caspase-9 also underwent constitutive processing in RT4v6 cells, and interestingly, caspase-9 processing was blocked under cell death conditions induced by the combination of Smac mimetic and BCG-stimulated neutrophil conditioned medium (Fig. 4B). Caspase-3, on the other hand, had no constitutive cleavage but had significant cleavage (p17) only under the cell death condition (Fig. 4B). The functional activation of caspase-3 was demonstrated by Hoechst-33342 staining, showing that the combination of Smac mimetic and BCG-stimulated neutrophil conditioned medium caused chromatin condensation (Fig. 4C).
Together, these results suggested that RT4v6 cells have constitutive, nonapoptotic caspase-8 and caspase-9 processing and that under cell death conditions, these cleavages are altered to caspase-8-Asp391/374 p26 cleavage and inhibition of caspase-9 processing, resulting in caspase-3 activation. Thus, these results strongly indicated involvement of the extrinsic pathway of cell death.
Cell death induced by the combination of Smac mimetic and BCG-stimulated neutrophil conditioned medium is not dependent on TRAIL or cathepsin-D and is independent of TNF-α and TRAIL autocrine loops
Although the results so far stressed involvement of the extrinsic cell death pathway, neutrophils are not known to be a good source of FasL, another major death ligand other than TRAIL and TNF-α. In addition, as neutrophils are known to undergo granule exocytosis in response to BCG stimulation, it is possible that cathepsin-D (one of the granule content capable of activating caspase-8) could be involved in caspase-8 cleavage [28]. Furthermore, caspase-8 may undergo proteasome-mediated processing. We thus investigated the generation of the caspase-8 p26 fragment by treatment with the combination of Smac mimetic and BCG-stimulated neutrophil conditioned medium in the presence of ascorbic acid, a known inhibitor of FasL-induced caspase-8 activity [29]; pepstatin-A, a known inhibitor of cathepsin-D [28]; and bortezomib, a known inhibitor of proteasome [30]. We found that ascorbic acid could effectively inhibit processing of caspase-8 into the p26 fragment, whereas neither pepstatin-A nor bortezomib was able to do so, which suggested involvement of FasL or Fas/CD95 but not cathepsin-D or proteasome in caspase-8 p26 generation (Fig. 5A). As Smac mimetics are known to be active in DR (TNFR)-related pathways [10], we investigated whether rhTRAIL or rhTNF-α, in combination with Smac mimetic, could induce caspase-8 p26 generation. The results revealed that TNF-α, in combination with Smac mimetic, could effectively generate caspase-8 p26 to a greater degree than TRAIL, which suggested that TNF-α has a more predominant role in this process. To further investigate the involvement of autocrine TNF-α, which is known to kill cells in response to Smac mimetics [10], we tested the conditioned media from RT4v6 cells for TNF-α by quantitative ELISA. The results showed no significant increase in TNF-α levels in the RT4v6 conditioned medium under cell death conditions (P=0.173; Fig. 5C). The slight elevation in the TNF-α level was a result of the TNF-α, which was present in the BCG-stimulated neutrophil conditioned medium (the conditioned medium was diluted 1:1 with complete MEM; hence, it was expected to have one-half of the TNF-α concentration of the original, BCG-stimulated neutrophil conditioned medium). These results ruled out the possibility of autocrine TNF-α secretion in the induction of cell death. As it is not known whether TRAIL has a similar autocrine loop, we tested TRAIL concentrations in the RT4v6 conditioned media in a similar manner. The results showed lack of involvement of the TRAIL autocrine loop in cell death, especially in RT4v6 conditioned medium, where BCG-stimulated neutrophil conditioned medium was used (P=0.349; Fig. 5D).
Figure 5. Screening of TRAIL, TNF-α, FasL, cathepsin-D, and proteasome for their role in cell death induced by the combination of Smac mimetic and BCG-stimulated neutrophil conditioned medium.

All Western blots were performed using cells treated for 24 h as indicated. (A and B) Western blotting for caspase-8 Asp-391/374. (Note the p26 band; TRAIL is not efficient in generating p26, which indicates that the p26, generated by Smac mimetic in combination with BCG-stimulated neutrophil conditioned medium in Fig. 4A, is not a result of TRAIL.) (C) Quantitative ELISA to evaluate autocrine secretion of TNF-α. (D) Quantitative ELISA to evaluate autocrine secretion of TRAIL. *The conditioned media were diluted 1:1 with fresh complete MEM.
Taken together, these results show that TRAIL may not be involved in cell death induced by the combination of Smac mimetic and BCG-stimulated neutrophil conditioned medium, whereas cathepsin-D is not involved in caspase-8 p26 generation. Furthermore, these results demonstrated lack of involvement of autocrine TNF-α and TRAIL loops in cell death induction, but these cytokines from BCG-stimulated neutrophil conditioned medium could still play a role.
Combination of Smac mimetic and BCG-stimulated neutrophil conditioned medium down-regulates FasL and FADD in RT4v6 cells
Whereas neutrophils are not known to be a significant source of FasL, Mycobacterium is capable of inducing FasL [31]. Furthermore, many cancer cells express FasL for self-defense. Hence, we investigated whether RT4v6 cells express FasL or Fas/CD95. Western blotting of RT4v6 cell lysates revealed that RT4v6 cells expressed a significant amount of FasL in all conditions except cotreatment with Smac mimetic and BCG-stimulated neutrophil conditioned medium (Fig. 6A). These cells also expressed Fas/CD95, but there were no significant differences in Fas/CD95 expression between any of the conditions tested (Fig. 6A). Interestingly, Western blotting showed that FADD (an adapter protein for FAS receptor) was down-regulated specifically during cell death induced by Smac mimetic in combination with BCG-stimulated neutrophil conditioned medium (Fig. 6A).
BCG-stimulated neutrophils secrete 17 kDa FasL and sFADD
To examine whether FasL is involved in cell death, we tested neutrophil conditioned media before and after treatment of RT4v6 cells for FasL secretion by Western blotting. We found that 17 kDa FasL was secreted specifically in the BCG-stimulated neutrophil conditioned medium (Fig. 6B). FasL is known to be cleaved into a 17-kDa fragment, reportedly via a mechanism involving a disintegrin and metalloproteinase domain-containing protein 10 (ADAM-10) [32]. Neither full-length FasL nor glycosylated FasL was detected in conditioned media, but sFasL was detected (sFasL could be from the FBS, as it was also detected in MEM, which is not incubated with cells). Interestingly, FADD was also secreted by the neutrophils, and a low molecular-weight form of secreted FADD was detected in BCG-stimulated neutrophil conditioned medium (Fig. 6B). These results suggested that 17 kDa FasL, sFADD, and Fas/CD95 signaling may be involved in cell death induced by Smac mimetic in combination with BCG-stimulated neutrophil conditioned medium.
rhTNF-α and rhFasL but not rhTRAIL mimic BCG-stimulated neutrophil conditioned medium in combination with Smac mimetic
Based on the data so far, BCG-stimulated neutrophil conditioned medium resulted in c-IAP2 up-regulation in the absence of Smac mimetic and down-regulation of FADD, c-IAP2, and XIAP in combination with Smac mimetic. Hence, we examined whether rhTNF-α or rhTRAIL, in combination with Smac mimetic, would also result in similar molecular changes. Western blotting for FADD, c-IAP2, and XIAP had revealed that rhTNF-α but not rhTRAIL mimicked the BCG-stimulated neutrophil conditioned medium when combined with Smac mimetic in terms of FADD and XIAP down-regulation and c-IAP2 up-regulation (Fig. 6C and D).
As we also found 17 kDa FasL in BCG-stimulated neutrophil conditioned medium, we tested whether rFasL could generate molecular events similar to those observed in cells treated with BCG-stimulated neutrophil conditioned medium (Fig. 2), such as caspase-8 p26 generation in the presence of Smac mimetic and c-IAP2 up-regulation in the absence of Smac mimetic. Western blotting analysis revealed that rhFasL, in combination with Smac mimetic, could generate caspase-8 p26 fragment as efficiently as rhTNF-α, in combination with Smac mimetic, and the p26 generation by FasL was inhibited by the addition of ascorbic acid (Fig. 7A). Furthermore, rhTNF-α and rhFasL, as single agents, could up-regulate c-IAP2, and this up-regulation was blocked by the addition of Smac mimetic (Fig. 7A); these findings were similar to our earlier finding that BCG-stimulated neutrophil conditioned medium could stabilize c-IAP2 and that this stabilization was blocked by Smac mimetic (Fig. 2). Taken together, these results raised the possibility that TRAIL may not be the major mediator of cell death induced by the combination of Smac mimetic and BCG-stimulated neutrophil conditioned medium.
Figure 7. Evaluation of the role of FasL, TNF-α, and TRAIL in induction of cell death in combination with Smac mimetic.

(A) Evaluation of rhFasL and rhTNF-α for caspase-8 p26 generation and c-IAP2 stabilization (the signature events generated by BCG-stimulated neutrophils in Fig. 2 and Fig. 4A) by Western blotting. (B) Evaluation of the role of TNF-α, TRAIL, and FasL, secreted from BCG-stimulated neutrophils by blocking of these cytokines using neutralizing antibodies (N-Ab; 1000 ng/ml final concentration after 1:1 dilution with MEM—i.e., 2000 ng/ml before dilution with MEM; 50,000 cells/ml). The blocking duration was 20 min at room temperature before dilution of the conditioned medium with MEM, and the 1:1 diluted medium was used for treatments. Twenty-four hours after treatment, cell death was assessed by PI-FACS DNA fragmentation analysis (upper panel) and microscopy (selected conditions only in lower panels; n=3) *Media were diluted 1:1 with fresh complete MEM. **Note that TRAIL and FasL neutralizing antibody combination has no blocking activity (B, lower panels). (C) Evaluation of neutralizing abilities of TRAIL and FasL antibodies using rhTRAIL and rhFasL in combination with Smac mimetic. Note that the impact of neutralizing antibodies on DNA fragmentation levels was similar to the impact of single-agent Smac mimetic (see text for details).
TNF-α but not TRAIL or FasL is the mediator of cell death secreted by BCG-stimulated neutrophils in combination with Smac mimetic
As TNF-α and FasL but not TRAIL generated molecular events similar to those generated by the combination of Smac mimetic and BCG-stimulated neutrophil conditioned medium, we examined the specific role of these cytokines using neutralizing antibody approach. As we had detected constitutive caspase-8 and caspase-9 activation in RT4v6 cells, we included neutralizing antibody controls to check whether mere addition of these neutralizing antibodies will influence the survival of RT4v6 cells. The results showed that addition of 1000 ng/ml neutralizing antibodies to TNF-α, TRAIL, or FasL, in any combination, did not induce cell death in RT4v6 cells (Fig. 7B, upper panel), indicating the suitability of these neutralizing antibodies for the neutralization test. Addition of various combinations of these neutralizing antibodies at equal concentrations, in addition to Smac mimetic and BCG-stimulated neutrophil conditioned medium, revealed that only TNF-α neutralizing antibody was able to inhibit cell death (P=0.0000173), whereas TRAIL and FasL neutralizing antibodies had no inhibitory effect (P=0.504 and 0.942, respectively; Fig. 7B, upper panel). The combination of TRAIL and FasL neutralizing antibodies had absolutely no inhibitory effect, which raised the possibility of inactive neutralizing antibodies (Fig. 7B). To rule out this possibility, we examined the neutralizing capacity of these antibodies using 1 ng/ml rTRAIL or rFasL (which are higher concentrations than detected in BCG-stimulated neutrophil conditioned medium) in combination with Smac mimetic. We found that these neutralizing antibodies, at the concentration of 1000 ng/ml, were capable of complete neutralization of rhTRAIL and rhFasL at 1 ng/ml, as assessed by PI-FACS DNA fragmentation (Fig. 7C). The 20% DNA fragmentation observed in the presence of neutralizing antibodies was a result of single-agent Smac mimetic (Fig. 7C).
Together, these results demonstrated that although BCG-stimulated neutrophils secrete TNF-α, TRAIL, and FasL, TNF-α is the key cytokine that mediates cell death in combination with Smac mimetic.
DISCUSSION
Our study is the first to evaluate the efficacy of Smac mimetic in the context of BCG-stimulated neutrophils against cancer cells. Although BCG administration results in the recruitment of multiple cell types in vivo, neutrophils account for ∼75% of these recruited cells and are capable of self-priming through IFN release in response to bacterial pathogens [33, 34]; hence, BCG stimulation does not require other inflammatory cells for neutrophil priming in our experimental set-up. Our findings demonstrate that Smac mimetic exerts its effects through TNF-α secreted from BCG-stimulated neutrophils but independently of TNF-α and TRAIL autocrine loops from RT4v6 cancer cells.
A previous study by Kemp et al. [3] showed significant secretion of TRAIL (∼500 pg/ml) from BCG-stimulated neutrophils; however, in our study, we detected only 20–40 pg/ml TRAIL because of the technical alterations that we performed to mimic bladder cancer treatment conditions. This difference is possibly due to the fact that Kemp et al. [3] used heat-killed BCG strain MV261, which is not used for bladder cancer therapy, whereas we used live attenuated TICE BCG. TICE-BCG is commonly used for bladder cancer therapy [35]. Furthermore, Kemp et al. [3] used cycloheximide and actinomycin-D for their studies. Both of these drugs can kill cells when administered as single agents [36, 37], and cycloheximide can modulate DR components, leading to neutrophil cell death [11, 38], resulting in the leakage of granule contents rather than secretion. [We have observed that RT4v6 cells undergo cell death in response to single-agent cycloheximide treatment (data not shown)]. Of note, we used 3 ng/ml TRAIL, which is higher than the concentration Kemp et al. [3] obtained from BCG-stimulated neutrophils, but still TRAIL was unable to generate caspase-8 p26 as efficiently as TNF-α or FasL did (Figs. 5B and 7A). It should be noted that rhTRAIL and rhTNF-α were equally efficient in inducing cell death (Fig. 3B); hence, the differences in generation of the 26-kDa caspase-8 fragment are not a result of variations in the amounts of death ligands (Fig. 5B).
The inability of rhTRAIL (when compared with BCG-stimulated neutrophil conditioned medium, rhTNF-α) to down-regulate FADD and XIAP with Smac mimetic (Fig. 6C and D), inability to stabilize c-IAP2 without Smac mimetic (Fig. 6D), and inability of TRAIL neutralizing antibody to reduce cell death induced by BCG-stimulated neutrophil conditioned medium (Fig. 7B) demonstrated that the TRAIL secreted from BCG-stimulated neutrophils has a minor or no role in the cell death induced in combination with Smac mimetic. In addition, single-agent Smac mimetic could not induce significant cell death, which demonstrated the lack of TRAIL or TNF-α autocrine loops in this cell system.
Although rhFasL generated molecular events similar to BCG-stimulated neutrophils and rhTNF-α (in terms of c-IAP2 stabilization and caspase-8 p26 generation), the neutralization test clearly revealed that the 17-kDa FasL, detected in BCG-stimulated neutrophil conditioned medium, was not active. Furthermore, our finding that unstimulated or BCG-stimulated neutrophil conditioned medium as a single agent could not kill RT4v6 cells is in agreement with the fact that FasL on tumor cells is known to inactivate neutrophils [39].
Smac mimetics are known to work through degradation of c-IAP1 [15], and in our study, Smac mimetic as a single agent did result in c-IAP1 degradation. However, complete degradation of c-IAP1 was not observed in the presence of BCG-stimulated neutrophil conditioned medium (Fig. 2, lane 9), which suggests that Smac mimetic-induced c-IAP1 degradation is not required for BCG-stimulated neutrophil-induced cell death in this system. Our data on c-IAP1 are in agreement with a previous report that certain cells can undergo death without c-IAP1 degradation [40].
In summary, our findings identify the mechanism of action of BCG and suggest that the combination of Smac mimetic with BCG may represent a viable mechanism-based combination therapy for bladder cancer.
ACKNOWLEDGMENTS
G.J.G. was supported in part by the Clayton Foundation for Research and in part by MD Anderson Cancer Center. This research was supported in part by U.S. National Institutes of Health through MD Anderson's Cancer Center Support Grant CA016672. The authors thank Drs. David J. McConkey, Bharat B. Aggarwal, and Joya Chandra for their generosity in providing various reagents, Stephanie Deming (Department of Scientific Publications, MD Anderson Cancer Center) for editorial assistance with the manuscript, and Drs. Woonyoung Choi, Aron Mobley, I-ling Lee, Rian J. Dickstein, Eugene K. Lee, Neema Navai, Mathew White, Lauren Marquis, Mai Tran, Wei Qi, and Aditi Das for various forms of help.
Footnotes
- BCG
- bacillus Calmette-Guérin
- c-FLIP
- cellular FLIP
- c-IAP
- cellular inhibitor of apoptosis
- DR
- death receptor
- FasL
- Fas ligand
- IAP
- inhibitor of apoptosis
- rh
- recombinant human
- s
- soluble
- Smac
- secondary mitochondria-derived activator of caspases
- XIAP
- X-linked inhibitor of apoptosis
AUTHORSHIP
G.J.G. designed the study, performed all experiments, and wrote the manuscript. A.M.K. supervised the entire study and provided expert opinion during experiments and writing of the manuscript. S.C. contributed the Smac mimetic.
DISCLOSURES
S.C. is a shareholder of TetraLogic Pharmaceuticals. A.M.K. and G.J.G. have no conflicts of interest. A.M.K. has previously received funding from TetraLogic Pharmaceuticals for studies related to Smac mimetic; however, the current study was not supported by TetraLogic Pharmaceuticals, except for their kind gift of the Smac mimetic compound for use in our studies.
REFERENCES
- 1. Martinez-Pineiro J. A., Martinez-Pineiro L. (1997) BCG update: intravesical therapy. Eur. Urol. 31 (Suppl. 1), 31–41 [DOI] [PubMed] [Google Scholar]
- 2. Kamat A. M., Lamm D. L. (2000) Intravesical therapy for bladder cancer. Urology 55, 161–168 [DOI] [PubMed] [Google Scholar]
- 3. Kemp T. J., Ludwig A. T., Earel J. K., Moore J. M., Vanoosten R. L., Moses B., Leidal K., Nauseef W. M., Griffith T. S. (2005) Neutrophil stimulation with Mycobacterium bovis bacillus Calmette-Guerin (BCG) results in the release of functional soluble TRAIL/Apo-2L. Blood 106, 3474–3482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Simons M. P., Nauseef W. M., Griffith T. S. (2007) Neutrophils and TRAIL: insights into BCG immunotherapy for bladder cancer. Immunol. Res. 39, 79–93 [DOI] [PubMed] [Google Scholar]
- 5. Oliveira R. B., Moraes M. O., Oliveira E. B., Sarno E. N., Nery J. A., Sampaio E. P. (1999) Neutrophils isolated from leprosy patients release TNF-α and exhibit accelerated apoptosis in vitro. J. Leukoc. Biol. 65, 364–371 [DOI] [PubMed] [Google Scholar]
- 6. Rosevear H. M., Lightfoot A. J., O'Donnell M. A., Griffith T. S. (2009) The role of neutrophils and TNF-related apoptosis-inducing ligand (TRAIL) in bacillus Calmette-Guerin (BCG) immunotherapy for urothelial carcinoma of the bladder. Cancer Metastasis Rev. 28, 345–353 [DOI] [PubMed] [Google Scholar]
- 7. Simons M. P., O'Donnell M. A., Griffith T. S. (2008) Role of neutrophils in BCG immunotherapy for bladder cancer. Urol. Oncol. 26, 341–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dovedi S. J., Davies B. R. (2009) Emerging targeted therapies for bladder cancer: a disease waiting for a drug. Cancer Metastasis Rev. 28, 355–367 [DOI] [PubMed] [Google Scholar]
- 9. Shiozaki E. N., Shi Y. (2004) Caspases, IAPs and Smac/DIABLO: mechanisms from structural biology. Trends Biochem. Sci. 29, 486–494 [DOI] [PubMed] [Google Scholar]
- 10. Petersen S. L., Wang L., Yalcin-Chin A., Li L., Peyton M., Minna J., Harran P., Wang X. (2007) Autocrine TNFα signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell 12, 445–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang L., Du F., Wang X. (2008) TNF-α induces two distinct caspase-8 activation pathways. Cell 133, 693–703 [DOI] [PubMed] [Google Scholar]
- 12. Wu H., Tschopp J., Lin S. C. (2007) Smac mimetics and TNFα: a dangerous liaison? Cell 131, 655–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Condon S. M. (2011) The discovery and development of Smac mimetics—small molecule antagonists of the inhibitor of apoptosis proteins. In Annual Reports in Medicinal Chemistry (Macor J. A. ed.), American Chemical Society, Washington, DC, 46, Chapter 13 [Google Scholar]
- 14. Zobel K., Wang L., Varfolomeev E., Franklin M. C., Elliott L. O., Wallweber H. J., Okawa D. C., Flygare J. A., Vucic D., Fairbrother W. J., Deshayes K. (2006) Design, synthesis, and biological activity of a potent Smac mimetic that sensitizes cancer cells to apoptosis by antagonizing IAPs. ACS Chem. Biol. 1, 525–533 [DOI] [PubMed] [Google Scholar]
- 15. Varfolomeev E., Blankenship J. W., Wayson S. M., Fedorova A. V., Kayagaki N., Garg P., Zobel K., Dynek J. N., Elliott L. O., Wallweber H. J., Flygare J. A., Fairbrother W. J., Deshayes K., Dixit V. M., Vucic D. (2007) IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell 131, 669–681 [DOI] [PubMed] [Google Scholar]
- 16. Vince J. E., Wong W. W., Khan N., Feltham R., Chau D., Ahmed A. U., Benetatos C. A., Chunduru S. K., Condon S. M., McKinlay M., Brink R., Leverkus M., Tergaonkar V., Schneider P., Callus B. A., Koentgen F., Vaux D. L., Silke J. (2007) IAP antagonists target cIAP1 to induce TNFα-dependent apoptosis. Cell 131, 682–693 [DOI] [PubMed] [Google Scholar]
- 17. Feltham R., Bettjeman B., Budhidarmo R., Mace P. D., Shirley S., Condon S. M., Chunduru S. K., McKinlay M. A., Vaux D. L., Silke J., Day C. L. (2011) Smac mimetics activate the E3 ligase activity of cIAP1 protein by promoting RING domain dimerization. J. Biol. Chem. 286, 17015–17028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Metwalli A. R., Khanbolooki S., Jinesh G., Sundi D., Shah J. B., Shrader M., Choi W., Lashinger L. M., Chunduru S., McConkey D. J., McKinlay M., Kamat A. M. (2010) Smac mimetic reverses resistance to TRAIL and chemotherapy in human urothelial cancer cells. Cancer Biol. Ther. 10, 885–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Griffith T. S., Kucaba T. A., O'Donnell M. A., Burns J., Benetatos C., McKinlay M. A., Condon S., Chunduru S. (2011) Sensitization of human bladder tumor cells to TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis with a small molecule IAP antagonist. Apoptosis 16, 13–26 [DOI] [PubMed] [Google Scholar]
- 20. Clark R. A., Nauseef W. M. (2001) Isolation and functional analysis of neutrophils. Curr. Protoc. Immunol. Chapter 7, Unit 7.23 [DOI] [PubMed]
- 21. Kamat A. M., Tharakan S. T., Sung B., Aggarwal B. B. (2009) Curcumin potentiates the antitumor effects of bacillus Calmette-Guerin against bladder cancer through the downregulation of NF-κB and upregulation of TRAIL receptors. Cancer Res. 69, 8958–8966 [DOI] [PubMed] [Google Scholar]
- 22. Jonsson G., Paulie S., Grandien A. (2003) cIAP-2 block apoptotic events in bladder cancer cells. Anticancer Res. 23, 3311–3316 [PubMed] [Google Scholar]
- 23. Tang Y., Simoneau A. R., Xie J., Shahandeh B., Zi X. (2008) Effects of the kava chalcone flavokawain A differ in bladder cancer cells with wild-type versus mutant p53. Cancer Prev. Res. (Phila) 1, 439–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bertrand M. J., Milutinovic S., Dickson K. M., Ho W. C., Boudreault A., Durkin J., Gillard J. W., Jaquith J. B., Morris S. J., Barker P. A. (2008) cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 30, 689–700 [DOI] [PubMed] [Google Scholar]
- 25. Liu X., Yue P., Chen S., Hu L., Lonial S., Khuri F. R., Sun S. Y. (2007) The proteasome inhibitor PS-341 (bortezomib) up-regulates DR5 expression leading to induction of apoptosis and enhancement of TRAIL-induced apoptosis despite up-regulation of c-FLIP and survivin expression in human NSCLC cells. Cancer Res. 67, 4981–4988 [DOI] [PubMed] [Google Scholar]
- 26. Takimoto R., El-Deiry W. S. (2000) Wild-type p53 transactivates the KILLER/DR5 gene through an intronic sequence-specific DNA-binding site. Oncogene 19, 1735–1743 [DOI] [PubMed] [Google Scholar]
- 27. Scott F. L., Denault J. B., Riedl S. J., Shin H., Renatus M., Salvesen G. S. (2005) XIAP inhibits caspase-3 and -7 using two binding sites: evolutionarily conserved mechanism of IAPs. EMBO J. 24, 645–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Conus S., Perozzo R., Reinheckel T., Peters C., Scapozza L., Yousefi S., Simon H. U. (2008) Caspase-8 is activated by cathepsin D initiating neutrophil apoptosis during the resolution of inflammation. J. Exp. Med. 205, 685–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Perez-Cruz I., Carcamo J. M., Golde D. W. (2003) Vitamin C inhibits FAS-induced apoptosis in monocytes and U937 cells. Blood 102, 336–343 [DOI] [PubMed] [Google Scholar]
- 30. Amiri K. I., Horton L. W., LaFleur B. J., Sosman J. A., Richmond A. (2004) Augmenting chemosensitivity of malignant melanoma tumors via proteasome inhibition: implication for bortezomib (VELCADE, PS-341) as a therapeutic agent for malignant melanoma. Cancer Res. 64, 4912–4918 [DOI] [PubMed] [Google Scholar]
- 31. Mustafa T., Phyu S., Nilsen R., Bjune G., Jonsson R. (1999) Increased expression of Fas ligand on Mycobacterium tuberculosis infected macrophages: a potential novel mechanism of immune evasion by Mycobacterium tuberculosis? Inflammation 23, 507–521 [DOI] [PubMed] [Google Scholar]
- 32. Kirkin V., Cahuzac N., Guardiola-Serrano F., Huault S., Luckerath K., Friedmann E., Novac N., Wels W. S., Martoglio B., Hueber A. O., Zornig M. (2007) The Fas ligand intracellular domain is released by ADAM10 and SPPL2a cleavage in T-cells. Cell Death Differ. 14, 1678–1687 [DOI] [PubMed] [Google Scholar]
- 33. De Boer E. C., de Jong W. H., van der Meijden A. P., Steerenberg P. A., Witjes F., Vegt P. D., Debruyne F. M., Ruitenberg E. J. (1991) Leukocytes in the urine after intravesical BCG treatment for superficial bladder cancer A flow cytofluorometric analysis. Urol. Res. 19, 45–50 [DOI] [PubMed] [Google Scholar]
- 34. Ethuin F., Gerard B., Benna J. E., Boutten A., Gougereot-Pocidalo M. A., Jacob L., Chollet-Martin S. (2004) Human neutrophils produce interferon γ upon stimulation by interleukin-12. Lab. Invest. 84, 1363–1371 [DOI] [PubMed] [Google Scholar]
- 35. De Jager R., Guinan P., Lamm D., Khanna O., Brosman S., De Kernion J., Williams R., Richardson C., Muenz L., Reitsma D.., et al. (1991) Long-term complete remission in bladder carcinoma in situ with intravesical TICE bacillus Calmette Guerin. Overview analysis of six phase II clinical trials. Urology 38, 507–513 [DOI] [PubMed] [Google Scholar]
- 36. Alessenko A. V., Boikov P., Filippova G. N., Khrenov A. V., Loginov A. S., Makarieva E. D. (1997) Mechanisms of cycloheximide-induced apoptosis in liver cells. FEBS Lett. 416, 113–116 [DOI] [PubMed] [Google Scholar]
- 37. Kleeff J., Kornmann M., Sawhney H., Korc M. (2000) Actinomycin D induces apoptosis and inhibits growth of pancreatic cancer cells. Int. J. Cancer 86, 399–407 [DOI] [PubMed] [Google Scholar]
- 38. Krakstad C., Christensen A. E., Doskeland S. O. (2004) cAMP protects neutrophils against TNF-α-induced apoptosis by activation of cAMP-dependent protein kinase, independently of exchange protein directly activated by cAMP (Epac). J. Leukoc. Biol. 76, 641–647 [DOI] [PubMed] [Google Scholar]
- 39. Chen Y. L., Chen S. H., Wang J. Y., Yang B. C. (2003) Fas ligand on tumor cells mediates inactivation of neutrophils. J. Immunol. 171, 1183–1191 [DOI] [PubMed] [Google Scholar]
- 40. Diessenbacher P., Hupe M., Sprick M. R., Kerstan A., Geserick P., Haas T. L., Wachter T., Neumann M., Walczak H., Silke J., Leverkus M. (2008) NF-κB inhibition reveals differential mechanisms of TNF versus TRAIL-induced apoptosis upstream or at the level of caspase-8 activation independent of cIAP2. J. Invest. Dermatol. 128, 1134–1147 [DOI] [PubMed] [Google Scholar]