

Congenital long QT syndrome (LQTS) is a relatively common genetically determined condition with significant morbidity and mortality, particularly in young, otherwise healthy individuals (Goldenberg et al. 2011). Usually caused by genes encoding ion channel subunits controlling cardiac repolarisation, its clinical severity depends on a variety of factors, including the specific genotype, severity of repolarisation abnormalities (reflected by the degree of QT prolongation), sex, age and modifier genes (Goldenberg et al. 2011). The most common forms involve mutations in genes (KCNQ1, 30–35% of all cases, and KCNH2, 25–30% of cases) encoding the pore-forming α subunits of the principal cardiac repolarising K+ currents, the slow (IKs) and rapid (IKr) delayed rectifiers, respectively. Triggering events are known to precipitate arrhythmia episodes. In the KCNQ1 associated form LQT1, the majority of arrhythmic events are associated with exercise, with other episodes prompted by sudden arousal or occurring during sleep (Goldenberg et al. 2012). For the KCNH2 associated form LQT2, the most common precipitator is sudden arousal (e.g. loud noises, startle), responsible for almost half the events, with exercise implicated in about 10% (Kim et al. 2010). In both forms, β adrenergic receptor blockers, the mainstay of drug therapy for congenital LQTS, are highly effective for exercise induced events, but not for events triggered by other precipitants (Kim et al. 2010; Goldenberg et al. 2012).
Implantable cardioverter defibrillators (ICDs) are extremely effective in terminating potentially lethal events in LQTS patients. However, they are also expensive and have potential adverse physical and psychological consequences, so better patient selection criteria are needed, along with improved medical therapy options (Schwartz et al. 2010). Enhanced understanding of the mechanisms underlying arrhythmia event triggering has the potential to facilitate the development of more effective management approaches and better predictors of high risk patients.
Adrenergic stimulation plays an important role in LQTS triggering, as evidenced by the value of β-adrenoceptor blocking drugs. β-Receptor activation causes quantitatively similar enhancement of both L type Ca2+ current (ICaL), an inward current tending to prolong cardiac action potentials (APs), and IKs, tending to shorten APs (Han et al. 2001). The balance between them allows for enhanced cardiac contractility under physiological ‘fight or flight’ conditions (via increased ICaL) without excessive AP prolongation. When IKs is dysfunctional, as in LQT1, the resulting unbalanced ICaL effect causes excess AP prolongation (Han et al. 2001). The control of IKr by β-adrenoceptor stimulation is more complex than that of IKr. KCNH2 protein phosphorylation by cAMP-dependent protein kinase A reduces current amplitude, accelerates current deactivation, and shifts voltage-dependent activation to more positive voltages (Cui et al. 2000). On the other hand, cAMP directly binds to KCNH2, shifting activation voltage dependence to negative voltages. The net effect can be an increase or decrease in current, depending on stoichiometry between KCNH2 and accessory proteins (Cui et al. 2000). Differential adrenergic regulation of IKr/IKs is reflected in discrepant clinical responses. Intravenous administration of adrenaline (bolus followed by maintenance infusion) produces sustained QT prolongation in LQT1 patients, but transient QT increases in LQT2 subjects (Noda et al. 2002).
A lack of adequate animal models has been a major obstacle to understanding the fundamental determinants of LQTS. While gene defects can be precisely studied in genetically engineered mice, species differences in repolarising currents (especially delayed rectifiers) limit the utility of mouse models for human repolarisation disorders (Liu et al. 2004). Recently, transgenic LQTS rabbit models have been developed for both LQT1 (KCNQ1 knockout) and LQT2 (KCNH2 knockout) with features much closer to those of clinical LTQSs (Brunner et al. 2008).
In a recent issue of The Journal of Physiology, Liu et al. (2011) describe the use of LQT1 and LQT2 transgenic rabbits to examine LQTS triggering responses leading to the cellular initiators of LQTS arrhythmias, early afterdepolarisations (EADs). First, they examine the effect of hypokalaemia on repolarisation and EAD occurrence. Hypokalaemia, well known to delay repolarisation and promote LQTS by suppressing K+ currents like IKr and the background inward rectifier IK1, increased APD in both LQT1 and LQT2 cells, with greater effects on AP duration (APD) and EAD occurrence in LQT2 cells. The authors relate the larger response in LQT2 cells to a more important role of IKr than IKs in AP repolarisation in the absence of adrenergic stimulation.
They then go on to examine the effect of β-adrenergic stimulation with isoprenaline. They note strikingly greater APD prolongation and EAD induction in LQT1 hearts versus littermate controls, both in isolated cardiomyocytes and optically mapped Langendorff perfused hearts. However, in LQT2 cells they observe a particularly interesting and unanticipated response. At steady state, isoprenaline reduces APD in LQT2 cells, but over the first few seconds after exposure, EADs and APD prolongation occur. The authors relate this behaviour to differential time-dependent effects of isoprenaline on ICaL versus IKs: enhancement of ICaL appears rapidly, with a time constant of 9.2 s, whereas IKs increases are slower (time constant 43.6 s). The initial rapid increase in only ICaL causes EADs in the absence of countervailing IKr, which are then eliminated by the gradual build-up of countervailing IKs.
These results provide plausible physiological explanations for the discrepancies in triggering events between LQT1 and LQT2, as illustrated in the Fig. 1. Arousal triggers, prominent in LQT2, cause a sudden surge of adrenergic tone, comparable to the early and transient EAD inducing responses noted by Liu et al. in LQT2 rabbit hearts. Exercise triggers, predominant in LQT1, are associated with sustained increases in adrenergic tone, paralleling the increases in APD and EADs induced by continued isoprenaline infusion in LQT1 rabbit cells.
Figure 1. Fundamental mechanisms by which arousal and exercise triggers differentially affect LQT1 vs. LQT2.
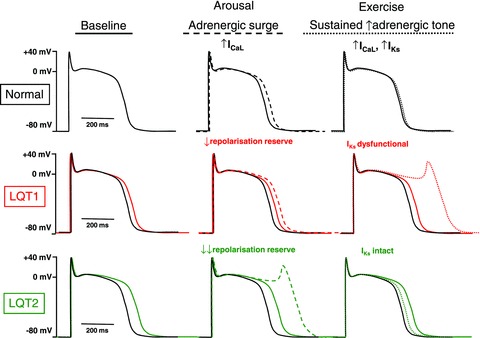
The normal cardiac AP is shown in black in all panels, whereas APs in LQT1 or LQT2 are shown in red and green, respectively (baseline APs are shown as continuous lines in each panel). Dashed and dotted lines in middle and right panels indicate changes resulting respectively from arousal (adrenergic surge) and exercise (sustained increase in adrenergic tone). Since IKr is the predominant repolarizing current under normal conditions, APs are more prolonged in LQT2 at baseline. With sudden arousal stimuli, there is a transient surge of adrenergic tone, which increases ICaL before any change in IKs. In LQT2, with major loss in repolarisation reserve due to loss of IKr, APs are strongly prolonged and EADs arise. In LQT1, intact IKr prevents excess AP prolongation. With exercise, sustained increases in adrenergic tone increase both ICaL and IKs, producing little net change in AP duration when IKs is intact. However, when IKs is dysfunctional and cannot increase to compensate for ICaL enhancement (LQT1), strong AP prolongation and EADs occur.
Despite these intriguing parallels suggesting that Liu et al. have discovered the basis of previously mysterious clinical responses, their observations leave some questions unanswered. For example, if arousal triggers induce LQTS events via transient surges of β-adrenergic stimulation, why are β-blockers relatively ineffective in preventing arousal related episodes (Kim et al. 2010; Goldenberg et al. 2012)? If sustained adrenergic drive shortens APD without inducing EADs in LQT2 cells, as observed by Liu et al., why does exercise sometimes induce arrhythmic events in LQT2 patients? What is the basis for LQTS events occurring during sleep? Of course, no studies answer all questions and good studies raise new ones. In addition to raising questions, the Liu et al. study introduces a powerful new tool, the LQTS transgenic rabbit, with which to answer them. We look forward to many future answers (and stimulating questions) from this exciting system.
Acknowledgments
Research support in this area is provided to S.N. by the Canadian Institutes of Health Research (MOP 68929), by the Quebec Heart and Stroke Foundation, by the MITACS Network and by the Fondation Leducq (ENAFRA Network, 07/CVD/03).
References
- Brunner M, Peng X, Liu GX, et al. J Clin Invest. 2008;118:2246–2259. doi: 10.1172/JCI33578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Melman Y, Palma E, Fishman GI, McDonald TV. Curr Biol. 2000;10:671–674. doi: 10.1016/s0960-9822(00)00516-9. [DOI] [PubMed] [Google Scholar]
- Goldenberg I, Horr S, Moss AJ, et al. J Am Coll Cardiol. 2011;57:51–59. doi: 10.1016/j.jacc.2010.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenberg I, Thottathil P, Lopes CM, et al. Heart Rhythm. 2012;9:49–56. doi: 10.1016/j.hrthm.2011.08.020. [DOI] [PubMed] [Google Scholar]
- Han W, Wang Z, Nattel S. Am J Physiol Heart Circ Physiol. 2001;280:H1075–H1080. doi: 10.1152/ajpheart.2001.280.3.H1075. [DOI] [PubMed] [Google Scholar]
- Kim JA, Lopes CM, Moss AJ, et al. Heart Rhythm. 2010;7:1797–1805. doi: 10.1016/j.hrthm.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GX, Choi BR, Ziv O, Li W, de Lange E, Qu Z, Koren G. J Physiol. 2011;590:1171–1180. doi: 10.1113/jphysiol.2011.218164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GX, Zhou J, Nattel S, Koren G. J Physiol. 2004;556:401–413. doi: 10.1113/jphysiol.2003.059303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda T, Takaki H, Kurita T, et al. Eur Heart J. 2002;23:975–983. doi: 10.1053/euhj.2001.3079. [DOI] [PubMed] [Google Scholar]
- Schwartz PJ, Spazzolini C, Priori SG, et al. Circulation. 2010;122:1272–1282. doi: 10.1161/CIRCULATIONAHA.110.950147. [DOI] [PubMed] [Google Scholar]