

Abstract
Passive limb movement elicits a robust increase in limb blood flow (LBF) and limb vascular conductance (LVC), but the peripheral vascular mechanisms associated with this increase in LBF and LVC are unknown. This study sought to determine the contribution of nitric oxide (NO) to movement-induced LBF and LVC and document the potential for passive-limb movement to assess NO-mediated vasodilatation and therefore NO bioavailability. Six subjects underwent passive knee extension with and without nitric oxide synthase (NOS) inhibition via intra-arterial infusion of NG-monomethyl-l-arginine (l-NMMA). LBF was determined second-by-second by Doppler ultrasound, and central haemodynamics were measured by finger photoplethysmography. Although l-NMMA did not alter the immediate increase (initial ∼9 s) in LBF and LVC, NOS blockade attenuated the peak increase in LBF (control: 653 ± 81; l-NMMA: 399 ± 112 ml−1 min−1, P= 0.03) and LVC (control: 7.5 ± 0.8; l-NMMA: 4.1 ± 1.1 ml min−1 mmHg−1, P= 0.02) and dramatically reduced the overall vasodilatory and hyperaemic response (area under the curve) by nearly 80% (LBF: control: 270 ± 51; l-NMMA: 75 ± 32 ml, P= 0.001; LVC: control: 2.9 ± 0.5; l-NMMA: 0.8 ± 0.3 ml mmHg−1, P < 0.001). Passive movement in control and l-NMMA trials evoked similar increases in heart rate, stroke volume, cardiac output and a reduction in mean arterial pressure. As movement-induced increases in LBF and LVC are predominantly NO dependent, passive limb movement appears to have significant promise as a new approach to assess NO-mediated vascular function, an important predictor of cardiovascular disease risk.
Key points
Passive limb movement elicits a robust increase in limb blood flow (LBF) and limb vascular conductance (LVC) without a concomitant increase in skeletal muscle metabolism.
The peripheral vascular mechanisms associated with the increase in LBF and LVC are unknown.
Using an intra-arterial infusion of NG-monomethyl-l-arginine (l-NMMA) to inhibit nitric oxide synthase (NOS) the hyperaemic and vasodilatory response to passive limb movement was attenuated by nearly 80%.
This finding demonstrates that the increases in LBF and LVC during passive limb movement are primarily NO dependent.
Passive limb movement appears to have significant promise as a new approach to assess NO-mediated vascular function, an important predictor of cardiovascular disease risk.
Introduction
The ability to accurately and reliably assess endothelial function is valuable from both a research and a clinical perspective, as endothelial dysfunction has been linked to the initial stages of hypertension and cardiovascular disease (Anderson et al. 1995; Dakak et al. 1998; Takase et al. 1998). Flow mediated dilatation (FMD) following ischemic cuff occlusion, first described by Celermajer et al. (1992), has been adopted by researchers to evaluate global endothelial function but has failed to be embraced by the clinical community despite early work that established a positive correlation between brachial artery FMD and invasively measured endothelial function of the coronary arteries (Anderson et al. 1995). A key impetus for the continued use of FMD as a measure of endothelial function in research is the concept that FMD can be used to evaluate endothelium-derived nitric oxide (NO) bioavailability in humans. However, the somewhat complex methodology and analysis in combination with recent evidence that challenges the notion that FMD is a reliable and selective method to determine NO-mediated endothelial function (Tschakovsky & Pyke, 2005; Pyke et al. 2010; Parker et al. 2011) have left some questioning the usefulness of FMD. This uncertainty regarding FMD as an in vivo bioassay of NO bioavailability, coupled with the vasoprotective and cardioprotective effects of NO, has prompted the search for a new methodological approach to assess NO-dependent endothelial function.
Recently, our group (Wray et al. 2005a; Hayman et al. 2010; McDaniel et al. 2010a,b; Trinity et al. 2010, 2011) and others (Gonzalez-Alonso et al. 2008; Hellsten et al. 2008; Hoier et al. 2010) have focused on the peripheral and central haemodynamic responses to passive limb movement as a reductionist model to better understand the factors controlling movement-induced hyperaemia. By removing the increase in metabolism associated with active exercise important findings related to the control of hyperaemia have been revealed. Specifically, in healthy humans, following the initial onset of passive movement, there is a transient, yet robust, increase in limb blood flow and vascular conductance that sets in motion a cascade of events that triggers additional peripheral haemodynamic changes likely to include flow mediated dilatation in addition to increases in heart rate and cardiac output that support the hyperaemia (McDaniel et al. 2010a; Trinity et al. 2010). Through various experimental designs, the roles of afferent feedback (Trinity et al. 2011; Venturelli et al. 2012), the muscle pump (Wray et al. 2005a), perfusion pressure (Trinity et al. 2011), age (McDaniel et al. 2010b), and cardiac innervation/denervation (Hayman et al. 2010) have all been reported to have an substantial impact on passive movement-induced hyperaemia. However, whether NO plays a fundamental role in movement-induced hyperaemia has yet to be determined.
At rest the inhibition of nitric oxide synthase (NOS) consistently decreases limb blood flow and vascular conductance by 30–40% indicating an essential role of NO in controlling basal blood flow (Taddei et al. 2001; Wray et al. 2011). However, during exercise the reduction in blood flow following NOS inhibition is typically less, falling in the range of 5–25% (Endo et al. 1994; Gilligan et al. 1994; Shoemaker et al. 1997; Dinenno & Joyner, 2003; Schrage et al. 2004; Green et al. 2005; Wray et al. 2011). This implies a reduced contribution of NO to exercise-induced hyperaemia, an experimental paradigm characterized by increased metabolism which is also likely to play a significant role in elevating blood flow during exercise. In contrast, passive limb movement does not invoke a peripheral metabolic response, and thus the hyperaemic response may be predominantly NO mediated. Establishing a critical role of NO in movement induced hyperaemia would lend credence to the potential use of passive limb movement as a means to non-invasively determine NO bioavailability and endothelial function.
Therefore, using a model devoid of metabolism, yet highly responsive in terms of hyperaemia, the purpose of this study was to determine the extent to which NO is involved in passive movement-induced blood flow and vasodilatation, exploring the potential of this model to assess NO-mediated endothelial function. We directly tested the hypothesis that movement-induced hyperaemia is NO mediated by performing passive limb movement in the absence and presence of high-dose endothelial NOS inhibition achieved by continuous intra-arterial infusion of l-NMMA.
Methods
Subjects
Six recreationally active men volunteered to participate in this research study (stature and blood characteristics presented in Table 1). Subjects were not taking any prescription medication and were free from overt cardiovascular disease. Protocol approval and written informed consent were obtained according to the University of Utah and Salt Lake City Veteran's Administration Medical Centre (VAMC) Institutional Review Boards, in accordance with the principles outlined in the Declaration of Helsinki. All data collection took place at the Salt Lake City VAMC Geriatric Research, Education, and Clinical Centre in the Utah Vascular Research Laboratory.
Table 1.
Stature and blood characteristics
| Age (years) | 24 ± 1 |
| Height (cm) | 175 ± 2 |
| Weight (kg) | 72 ± 3 |
| Body mass index (kg m−2) | 24 ± 1 |
| Leg volume (dl) | 72 ± 4 |
| Glucose (mg dl−1) | 66 ± 4 |
| Cholesterol (mg dl−1) | 162 ± 12 |
| Triglycerides (mg dl−1) | 90 ± 18 |
| HDL (mg dl−1) | 51 ± 4 |
| LDL (mg dl−1) | 99 ± 10 |
Values are means ± SEM.
Experimental protocol
Before the experiment all subjects reported to the laboratory for a familiarization trial, fasting blood draw, and thigh volume measurement. During this session, passive knee extension and Doppler ultrasound imaging of the femoral artery were performed to both familiarize the subjects and ensure that acceptable images could be obtained at rest and during movement. Upon arrival at the laboratory, body mass and height were recorded and the right femoral artery was catheterized (18-gauge central venous catheter, Arrow International, Reading, PA, USA) using the Seldinger technique. Following a 30 min rest period, subjects were moved to the supine position. Due to lasting effects of l-NMMA, the control trials were always performed prior to l-NMMA infusion. Trials were separated by at least 30 min to ensure blood flow and central haemodynamic measures returned to baseline values. Before the commencement of passive movement, stable baseline central and peripheral haemodynamic measures were attained. An automated occlusion cuff (Hokansen, Bellevue, WA, USA) was placed just below the knee of the passively moved limb and inflated to 250 mmHg in order to ensure that the infusate did not perfuse the lower portion of the limb. Single leg passive movement was achieved by a member of the research team moving the leg through a 90 deg range of motion at 1 Hz. The starting position of the leg was the full extension at the knee (i.e. 180 deg) and the first movement served to passively flex the knee (i.e. move to a 90 deg knee joint angle). Real-time feedback was provided to the researcher by a digital display of knee joint angle. Before the start and throughout the protocol, subjects were encouraged to remain passive and resist the urge to assist with leg movement. To avoid the startle reflex and active resistance to the passive movement, subjects were made aware that passive movement would take place in ∼1 min, but to minimize the chance of an anticipatory response, they were not informed of exactly when this movement would initiate. Passive movement was performed for 2 min. A subset of three subjects performed a second passive movement trial as part of a separate investigation which provided evidence regarding the reproducibility of the blood flow response.
l-NMMA infusion
Thigh volume was determined anthropometrically and used for the calculation of drug dosing. l-NMMA (Bachem, Switzerland) was diluted from 250 mg lyophilised powder in normal saline to a concentration of 5 mg ml−1. l-NMMA was infused at a priming dose of 0.48 mg (dl thigh volume)−1 min−1 for 5 min prior to exercise. During the minute prior to passive movement, l-NMMA was infused at a maintenance dose 0.24 mg (dl thigh volume)−1 min−1 which was then maintained for the duration of passive movement. During control trials normal saline was infused intra-arterially at the same rate and duration as the l-NMMA. The l-NMMA infusion rates were based on previous dose–response curves that demonstrated a plateau in the reduction of arm blood flow at 0.24 mg (dl arm volume)−1 min−1 (Wray et al. 2011).
Measurements
Femoral blood flow
Measurements of femoral arterial blood velocity and vessel diameter were performed in the passively moved and the non-moved legs distal to the inguinal ligament and proximal to the deep and superficial femoral bifurcation with a Logic 7 and Logic e Doppler ultrasound systems (General Electric Medical Systems, Milwaukee, WI, USA), respectively. The ultrasound systems were equipped with a linear transducer operating at an imaging frequency of 10 MHz. Vessel diameter was determined at a perpendicular angle along the central axis of the scanned area both prior to and during passive movement. Blood velocity was measured using the same transducers with a frequency of 5 MHz. All blood velocity measurements were obtained with the probes appropriately positioned to maintain an insonation angle of 60 deg or less. The sample volume was maximized according to vessel size and was centred within the vessel. Arterial diameter was measured, and mean velocity (Vmean) (angle corrected, and intensity-weighted area under the curve) was automatically calculated (Logic 7 and Logic e). Using arterial diameter and Vmean, blood flow in the femoral artery was calculated as blood flow =Vmean×π× (vessel diameter/2)2× 60, where blood flow is in millilitres per minute.
Central haemodynamic variables
HR, SV, CO and MAP were determined with a finometer (Finapres Medical Systems, Amsterdam, the Netherlands). SV was calculated from beat-by-beat pressure waveforms assessed by photoplethysmography using the Modelflow method (Beatscope version 1.1; Finapres Medical Systems), which, in combination with HR, has been documented to accurately estimate CO during a variety of experimental protocols (Sugawara et al. 2003; van Lieshout et al. 2003; Azabji Kenfack et al. 2004; de Vaal et al. 2005; de Wilde et al. 2009).
Data acquisition
Throughout each protocol, HR, SV, CO, MAP and ECG signals underwent analog-to-digital conversion and were simultaneously acquired (200 Hz) using a data acquisition system, (AcqKnowledge; Biopac Systems, Goleta, CA, USA). In addition, this data acquisition system acquired the audio antegrade and retrograde signals (10,000 Hz) from the Doppler ultrasound system that served as a qualitative indicator of changes in blood velocity thereby facilitating the temporal processing of all variables.
Data and statistical analysis
The data acquisition software allowed second-by-second analyses of HR, SV, CO and MAP. All analyses were performed using a 5 s moving average. The second-by-second velocities were analysed on the ultrasound system (GE Logic 7 and Logic e) for the first 60 s of movement, and 12 s averages were assessed from 60 to 120 s of movement. Two-way repeated-measures ANOVA was used to determine significant differences between control and l-NMMA conditions. When a significant main effect (interaction of treatment by time) was observed, further post hoc analysis was performed to determine whether a significant change over time occurred within a treatment. Cumulative area under the curve (AUC) was calculated as the summed second-by-second response during the first 60 s of passive movement and used to identify how differences over time were affected by the treatment. Absolute and percentage change from baseline to peak between conditions were compared using Student's t test for paired samples. Significance was set at an α level of 0.05, and data are presented as means ± SEM.
Results
l-NMMA at rest
Intra-arterial inhibition of NOS by l-NMMA at rest decreased LBF by 32 ± 5% (control: 244 ± 30; l-NMMA: 163 ± 17 ml min−1, P= 0.005, Fig. 1A) and LVC by 34 ± 5% (control: 2.6 ± 03; l-NMMA 1.7 ± 0.1 ml min−1 mmHg−1, P= 0.007, Fig. 2A) but had no effect on femoral artery diameter (control: 0.88 ± 0.03 cm; l-NMMA: 0.88 ± 0.04 cm, P= 0.88). Centrally, l-NMMA infusion reduced resting HR (control: 54 ± 1; l-NMMA: 50 ± 2 bpm, P= 0.046) and CO (control: 5.5 ± 0.4; l-NMMA: 4.8 ± 0.3 l min−1, P= 0.038). No differences in SV (control: 101 ± 6; l-NMMA: 97 ± 6 ml beat−1, P= 0.25) or MAP (control: 91 ± 1; l-NMMA: 93 ± 3 mmHg, P= 0.52) were observed at rest.
Figure 1. Hyperaemic response of the passively moved limb over 2 min with and without intra-arterial NG-monomethyl-l-arginine infusion.
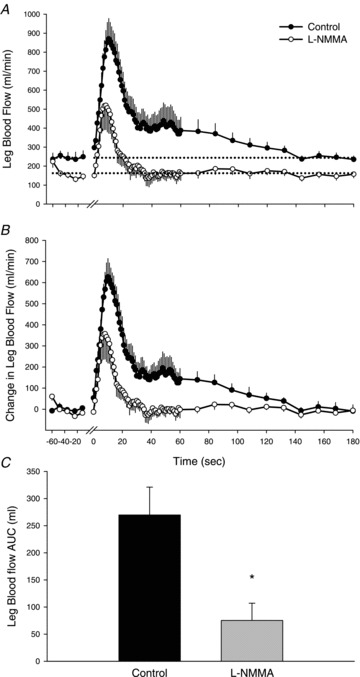
Values are means ± SEM. A, absolute leg blood flow (LBF, ml min−1). Main effect of interaction (condition × time), P < 0.01. B, change in LBF, normalized for the resting reduction in LBF due to l-NMMA. Main effect of interaction (condition × time), P < 0.01. C, area under the curve (AUC) calculated as the summed second-by-second response of LBF during the first 60 s of passive movement. *P < 0.05, significant difference between control and l-NMMA. One minute of resting data was collected before passive movement and the transition from rest to passive movement occurred at time 0 on the x-axis. Passive movement ended at 120 s and 1 min of recovery is presented (120–180 s).
Figure 2. Vasodilatory response of the passively moved limb over 2 min with and without intra-arterial NG-monomethyl-l-arginine (l-NMMA) infusion.
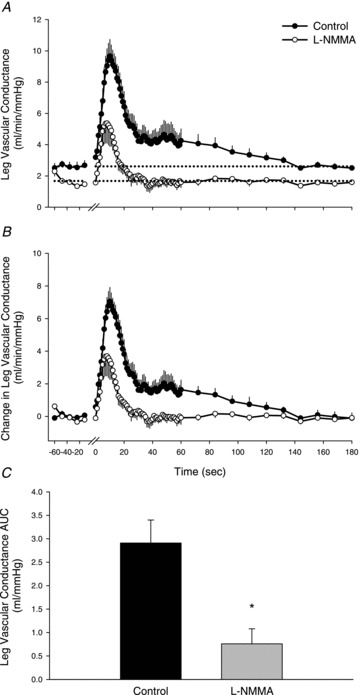
Values are means ± SEM. A, absolute leg vascular conductance (LVC, ml min−1 mmHg−1). Main effect of interaction (condition × time), P < 0.01. B, change in LVC, normalized for the resting reduction in LVC due to l-NMMA. Main effect of interaction (condition × time), P < 0.01. C, area under the curve (AUC) calculated as the summed second-by-second response of LVC during the first 60 s of passive movement. *P < 0.05, significant difference between control and l-NMMA. One minute of resting data was collected before passive movement and the transition from rest to passive movement occurred at time 0 on the x-axis. Passive movement ended at 120 s and 1 min of recovery is presented (120–180 s).
Peripheral haemodynamics during passive movement
At the onset of passive movement, LBF increased immediately in both control and l-NMMA conditions (Fig. 1A). During control, LBF reached a peak of 897 ± 101 ml min−1, which was attenuated during l-NMMA, 563 ± 120 ml min−1 (P= 0.01). Due to the resting reduction in LBF, the change in LBF from rest to peak was also determined and revealed a reduction in LBF due to l-NMMA during passive movement (control: 653 ± 81; l-NMMA: 399 ± 112 ml min−1, P= 0.03) (Fig. 1B). Both conditions exhibited a transient increase in LBF; however, during the control condition LBF remained elevated above baseline until the final 12 s of movement, whereas during l-NMMA, LBF returned to baseline 16 s after the onset of movement. The change in LBF during the first 9 s of movement was not different between conditions, signifying that NOS inhibition had no impact on the hyperaemic response during this time period. The AUC for the increase in LBF was reduced from 270 ± 51 to 75 ± 32 ml by l-NMMA (P= 0.001), indicating that 77 ± 7% of the hyperaemic response to passive movement was NO mediated (Fig. 1C). In a similar fashion to LBF, LVC reached a peak of 10.0 ± 1.1 ml min−1 mmHg−1, which was attenuated during l-NMMA, 6.0 ± 1.3 ml min−1 mmHg−1 (P= 0.01, Fig. 2A). The change in peak LVC was reduced by l-NMMA (control: 7.4 ± 0.9, l-NMMA: 4.3 ± 1.2 ml min−1 mmHg−1, P= 0.03) (Fig. 2B). The initial increase in LVC was not different between conditions during the first 7 s of movement. Overall, the AUC of the increase in LVC was reduced from 2.9 ± 0.5 to 0.8 ± 0.3 ml min−1 mmHg−1 (P < 0.001), indicating that 78 ± 8% of the increase in LVC during passive movement is NO mediated (Fig. 2C). In terms of the reproducibility of passive movement-induced hyperaemia, when a subset of subjects (n= 3) underwent passive movement under control conditions on a different day there were no discernible differences in the time course of the immediate hyperaemic response and the magnitude of the absolute peak in LBF (CV = 5%).
In the non-moved leg LBF and LVC were increased in the l-NMMA trial for the first minute of passive movement (main effect of interaction; LBF, P= 0.024 and LVC, P= 0.013) (Fig. 3A and B). During l-NMMA, LBF and LVC in the non-moved leg peaked at 29 s of passive movement (LBF: 516 ± 104 ml min−1 and LVC: 5.5 ± 1.2 ml min−1 mmHg−1). At the corresponding time of the control trial, LBF was significantly reduced (339 ± 42 ml min−1, P= 0.048) and LVC tended to be decreased (3.7 ± 0.5 ml min−1 mmHg−1, P= 0.06). In both legs there were no discernible changes in common femoral diameter throughout the passive movement protocols confirming previous findings that this conduit artery does not dilate in response to passive movement or exercise in healthy men (Wray et al. 2004, 2005b; Parker et al. 2008).
Figure 3. Absolute changes in leg blood flow (LBF; A) and leg vascular conductance (LVC; B) in the non-moved limb during 2 min of passive movement with and without intra-arterial NG-monomethyl-l-arginine (l-NMMA) infusion.
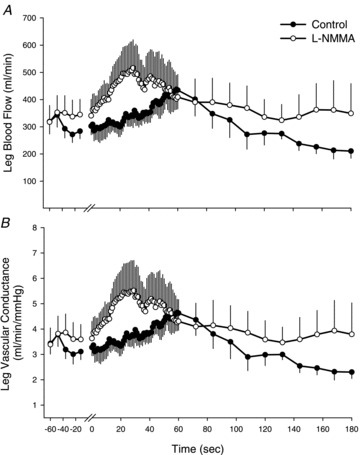
Values are means ± SEM. One minute of resting data was collected before passive movement and the transition from rest to passive movement occurred at time 0 on the x-axis. Passive movement ended at 120 s and 1 min of recovery is presented (120–180 s). Significant main effect of interaction (condition × time) for both LBF and LVC, P < 0.05.
Central haemodynamics during passive movement
Peak HR was higher during control than l-NMMA (control: 61 ± 3; l-NMMA: 58 ± 3 bpm, P= 0.047); however, the change in HR from baseline to peak was not different between conditions (control: 7 ± 2; l-NMMA: 8 ± 3 bpm, P= 0.83, Fig. 4). Neither the peak (control: 107 ± 7; l-NMMA: 102 ± 4 ml beat−1, P= 0.21) nor the change in SV (control: 7 ± 2; l-NMMA: 6 ± 2 ml beat−1, P= 0.15) was different between conditions (Fig. 4). Similar to HR, peak CO was greater during control (6.2 ± 0.5 l min−1) compared to the l-NMMA condition (5.7 ± 0.6 l min−1, P= 0.05); however, the change in CO was not different between conditions (control: 0.8 ± 0.2; l-NMMA: 0.8 ± 0.3, P= 0.86, Fig. 4). MAP was transiently reduced during passive movement in both control (−4.1 ± 0.8 mmHg, P= 0.004) and l-NMMA (−5.2 ± 2.0 mmHg, P= 0.046) conditions, but the magnitude of the decrease was not different between conditions (P= 0.72, Fig. 4). When expressed as AUC, none of the central haemodynamics variables (HR, SV, CO and MAP) revealed a significant difference between conditions (data not shown).
Figure 4. Absolute peak changes in central haemodynamics heart rate (A), stroke volume (B), cardiac output (C), and mean arterial pressure (MAP) (D), during 2 min of passive movement with and without intra-arterial NG-monomethyl-l-arginine (l-NMMA) infusion.
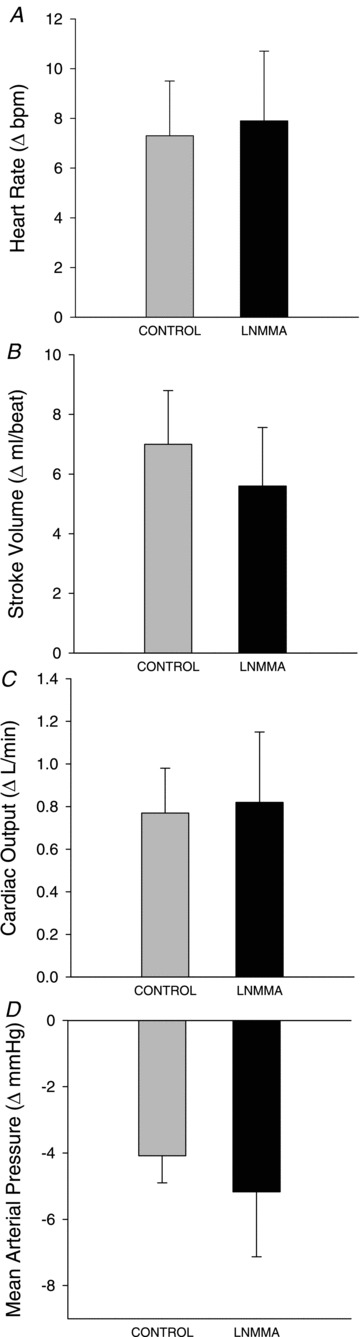
All variables exhibited significant changes from rest; however, there were no significant differences between control and l-NMMA conditions.
Discussion
Using passive movement as a model devoid of an exercise-induced increase in metabolism, yet highly responsive in terms of increased LBF and LVC, we have demonstrated that NO plays an essential role in the magnitude and duration of movement-induced hyperaemia and vasodilatation. Inhibition of NOS via intra-arterial infusion of l-NMMA revealed that NO contributes to ∼80% of the hyperaemia and vasodilatation associated with passive movement. Additionally, it should be noted that in the presence of l-NMMA, following a significant, yet reduced, transient peak, which cannot be explained by NO, both LBF and LVC decayed rapidly to resting levels (within 20 s) after the onset of movement. In contrast, in the control condition, LBF and LVC remained partially elevated above rest for nearly the full duration of the passive movement (2 min). Both with and without l-NMMA, passive movement elicited similar changes in central haemodynamics (HR, SV, CO and MAP). The robust and relatively easily measured reduction in NO-mediated vasodilatation and hyperaemia suggests that passive movement has significant promise as a new approach to assess NO-mediated vascular function, an important predictor of cardiovascular disease risk.
Passive movement: a potential approach to assess NO-dependant vascular function
Flow mediated dilatation (FMD) following ischaemic cuff occlusion, introduced nearly 20 years ago (Celermajer et al. 1992), has been routinely used in research to assess NO-dependent vascular function (Anderson et al. 1995; Joannides et al. 1995; Herrington et al. 2001; Corretti et al. 2002; Gokce et al. 2003; Tschakovsky & Pyke, 2005; Harris et al. 2010). However, although still having value in terms of cardiovascular event prediction (Green et al. 2011), recent findings have challenged the notion that this form of FMD is a valid and reliable measure of endothelial NO function (Tschakovsky & Pyke, 2005; Pyke et al. 2010). As a primary goal of this study we aimed to determine if passive movement can serve as a functional bioassay for endothelium-derived NO bioavailability in humans. Inhibition of NOS during passive movement reduced LVC, and therefore vasodilatation, within the passively moved limb by ∼80%, indicating that NO is the primary mechanism involved in this movement-induced vasodilatation. Inhibition of NOS during FMD has yielded equivocal results with investigators reporting complete ablation, partial reduction, or no change in FMD (Joannides et al. 1995; Doshi et al. 2001; Mullen et al. 2001; Kooijman et al. 2008; Pyke et al. 2010). Discrepancies between these studies may be due to the artery that is interrogated (brachial, radial, femoral), occlusion cuff placement (proximal or distal to the site of FMD assessment; Green et al. 2011), duration of cuff occlusion, and health/age/disease state of the individuals being tested. Compared to cuff occlusion-induced FMD, passive movement does not require precise measurement of conduit artery diameter as the common femoral artery does not dilate during exercise (Wray et al. 2004, 2005b; Parker et al. 2008), which greatly reduces the technical requirements in terms of both sonography and analysis. Additionally, acute tissue ischaemia, which is required to induce cuff occlusion FMD and may be uncomfortable or contraindicated in some individuals, is not a prerequisite for the assessment of vascular function by passive movement.
While not documented directly in the current study, we have previously determined that with age (McDaniel et al. 2010b), spinal cord injury (Venturelli et al. 2012), and heart transplantation (Hayman et al. 2010) vasodilatation following passive movement is reduced. These conditions are typically associated with reductions in vascular function (DeSouza et al. 2000; Taddei et al. 2000; Roig et al. 2009). These previous findings combined with the present results suggest that passive movement may be useful to determine differences in NO-dependent vascular function between groups and individuals. Further research is required to directly determine if ageing and other factors that alter NO bioavailability impact the NO dependency of passive movement-induced vasodilatation, as indicated in our prior work (Hayman et al. 2010; McDaniel et al. 2010b; Venturelli et al. 2012).
Contribution of NO to movement-induced hyperaemia
Inhibition of NOS via l-NMMA reduced the magnitude of the peak, overall hyperaemic response, and duration of the increase in LBF in response to passive movement. In fact, it is evident that movement-induced hyperaemia is primarily NO mediated as l-NMMA reduced the increase in LBF by approximately 80%. At rest, l-NMMA decreased LBF by only 32%, a basal reduction that agrees with earlier studies (Taddei et al. 2001; Wray et al. 2011). When normalized for this resting reduction, the attenuation in LBF due to NOS inhibition remains significant (Fig. 1B), providing evidence that the reduction in LBF during movement is not simply due to the initial reduction in resting LBF. The duration of the hyperaemic response was also affected by the reduction in NO. Specifically, following the immediate transient peak in LBF, which occurs in both conditions thereby likely being NO independent, LBF decays to resting levels. The rate of decay was greatly accelerated with l-NMMA and occurred completely within 20 s following the onset of passive movement. Conversely, LBF did not return to resting levels until the end of the second minute of movement in the control condition. The contrasting time courses between control and l-NMMA conditions provides in vivo evidence that the initial transient vasodilatation is largely NO independent while the peak and sustained vasodilatory and hyperaemic response is NO dependent.
The 80% reduction in LBF reported here for passive movement is far greater than the reduction in hyperaemia reported as a consequence of NOS inhibition during exercise. Indeed, NOS inhibition during exercise has only been reported to attenuate hyperaemia by 5–25% (Endo et al. 1994; Gilligan et al. 1994; Shoemaker et al. 1997; Radegran & Saltin, 1999; Frandsen et al. 2001; Gordon et al. 2002; Schrage et al. 2004; Green et al. 2005; Wray et al. 2011). Recently, our group (Wray et al. 2011) reported a 20–25% reduction in brachial artery blood flow during progressive handgrip exercise using an identical loading dose of l-NMMA as the current study. During active exercise, other vasodilatory mechanisms such as prostaglandins, endothelial derived hyperpolarizing factors and ATP appear to compensate for the reduction in NO bioavailability induced by NOS inhibition (Mortensen et al. 2007). These compensatory mechanisms appear to play a much more minor role during passive movement when an increase in metabolic rate is not evoked.
Despite the substantial 80% reduction in LBF, the immediate increase in the hyperaemic response appears to be largely NO independent. In fact, during the first 9 s of passive movement the change in LBF was nearly identical between conditions. The immediate and rapid response documented here supports previous findings, using an active exercise model, that the increase in vasodilatation occurs following the first muscle contraction without a measurable delay and without a contribution from NO (Saunders & Tschakovsky, 2004; Saunders et al. 2005). Vascular deformation, caused by contraction, or in the case of passive movement, the changes in muscle length, may stimulate a mechanosensitive mechanism causing subsequent vasodilatation and hyperaemia (Mohrman & Sparks, 1974a,b; Hamann et al. 2004; Clifford et al. 2006; Kirby et al. 2007). Clifford et al. (2006) reported that mechanical compression caused immediate dilatation in isolated rat feed arteries. Importantly, in reference to the current study, increasing the number of compressions increased the magnitude of the dilatation (Clifford et al. 2006; Kirby et al. 2007). The passive movement at 1 Hz, employed in this study, may be analogous to multiple compressions which increases the vasodilatory response. Additionally, removal of the feed artery endothelium reduced, but did not abolish, the vasodilatation implicating both endothelium-dependent and independent mechanisms (Clifford et al. 2006). In agreement with this finding (Clifford et al. 2006), in the present study NOS inhibition is likely to have effectively removed NO-dependent vasodilatation, yet 20% of the vasodilatory response was unaffected and remains unexplained.
Although the specific vasodilator or combination of vasodilators and myogenic regulation involved in the immediate vasodilatory response is not known, our findings speak specifically against a role for NO. Others have also indicated that local endothelium-dependent vasodilators (NO and prostaglandins) in addition to neural mechanisms do not appear essential for immediate dilatation (Shoemaker et al. 1996; Brock et al. 1998; Dyke et al. 1998; Buckwalter & Clifford, 1999; Saunders et al. 2005). A potential mechanism may involve smooth muscle hyperpolarization associated with muscle activation which immediately increases interstitial and plasma potassium (K+) concentration following a single muscle contraction (Mohrman & Sparks, 1974b; Hamann et al. 2004; Armstrong et al. 2007). Inhibition of the K+ channels reduced the contraction-induced hyperaemia thereby determining that K+ is a significant contributor to the immediate vasodilatory response (Hamann et al. 2004). Given the lack of voluntary muscle activation with passive movement the contribution of K+ to the immediate hyperaemic response in the current study is uncertain; however, mechanosensitive ion channels are present in endothelial cells and may contribute to the stretch and shear induced changes in vascular tone (Lansman et al. 1987; Olesen et al. 1988). Myogenic vasodilatation may play a role as we have previously reported an increase in perfusion pressure augmented LBF and LVC immediately following the onset of movement (Trinity et al. 2011). Further research, in this regard, is warranted to determine the mechanism involved in the rapid movement-induced vasodilatory response.
Central haemodynamics and passive movement-induced hyperaemia
Passive movement elicits a reflex-mediated increase in cardio-acceleration and central haemodynamics (Nobrega & Araujo, 1993; Wray et al. 2005a; Hayman et al. 2010; McDaniel et al. 2010a; Trinity et al. 2010, 2011). At the onset of passive movement, group III mechanosensitive skeletal afferent fibres are stimulated by the movement itself. This afferent feedback is relayed to the cardiovascular control centre and causes subsequent increases in HR and CO (Trinity et al. 2010). As the passive movement was identical between conditions and the afferent stimulus was intact, the subsequent increase in HR and CO would be expected to be similar with and without l-NMMA. However, previously, we have reported that a greater increase in LBF is associated with a greater CO response (Hayman et al. 2010; McDaniel et al. 2010b; Trinity et al. 2010, 2011). The small number of subjects may have influenced our ability to observe a significant difference in the CO response between conditions; however, when viewed across the entire passive movement protocol, the central haemodynamic responses (HR, CO and MAP) were also very similar between conditions (data not shown). In the current study, acute reductions in vascular conductance, induced by l-NMMA infusion, during passive movement impacted LBF, but not the magnitude of the central haemodynamic response. Previously, in agreement with this concept, complete occlusion of the passively moved limb tended to increase blood flow to the non-moved leg despite having no impact on HR and a minimal influence on CO (McDaniel et al. 2010a). Interestingly, in the current study, l-NMMA infusion reduced LBF and LVC in the passively moved limb and immediately after the onset of movement, the non-moved leg experienced a significantly faster increase in LBF and LVC compared to the control condition. This finding indicates that when the increase in LVC of the passively moved leg is inhibited, the increased CO is directed to other vascular beds, thereby facilitating the ‘absorption’ of the increased CO without increasing MAP.
Conclusion
Passive movement-induced increases in vasodilatation and subsequent hyperaemia are ∼80% NO-dependent. Inhibition of NOS with l-NMMA attenuated the magnitude and duration of the hyperaemic and vasodilatory response. The second-by-second resolution of the measurements of LBF and LVC revealed that the immediate and sustained vasodilatory and hyperaemic responses are governed by distinct mechanisms. The small, but immediate increase in LBF and LVC appears to be primarily NO independent while the majority of the movement-induced response beyond the first 10 s is largely NO dependent. Given the high NO dependency of the overall vasodilatory response, and the relatively simple method of assessment and analysis, passive movement appears to be a novel approach to the in vivo determination of NO bioavailability and endothelium-dependent vascular function.
Acknowledgments
The work was supported by National Institutes of Health Grant P01-H1-091830 (to R.S. Richardson), Veterans Affairs Rehabilitation Research and Development Service Grant E6910R (to R.S. Richardson), and Veterans Affairs Advanced Fellowship in Geriatrics (to J.D. Trinity)
Glossary
Abbreviations
- FMD
flow mediated dilatation
- LBF
limb blood flow
- LVC
limb vascular conductance
- NOS
nitric oxide synthase
Author contributions
J.D.T: Conception and design of experiments. Collection, analysis and interpretation of the data and drafting of the manuscript. H.J.G: Conception and design of experiments. Collection, analysis and interpretation of the data G.L: Conception and design of experiments. Collection, analysis and interpretation of the data M.J.R: Conception and design of experiments. Collection, analysis and interpretation of the data S.J.I: Conception and design of experiments. Collection, analysis and interpretation of the data S.R: Collection and interpretation of the data and provided medical oversight. B.G: Collection and interpretation of the data and provided medical oversight. A.B: Collection and interpretation of the data and provided medical oversight. R.S.R: Conception and design of experiments. Collection, analysis and interpretation of the data and drafting of the manuscript. All authors were involved in revising the manuscript, providing intellectual content, and approving the final version.
References
- Anderson TJ, Uehata A, Gerhard MD, Meredith IT, Knab S, Delagrange D, Lieberman EH, Ganz P, Creager MA, Yeung AC. Close relation of endothelial function in the human coronary and peripheral circulations. J Am Coll Cardiol. 1995;26:1235–1241. doi: 10.1016/0735-1097(95)00327-4. [DOI] [PubMed] [Google Scholar]
- Armstrong ML, Dua AK, Murrant CL. Potassium initiates vasodilatation induced by a single skeletal muscle contraction in hamster cremaster muscle. J Physiol. 2007;581:841–852. doi: 10.1113/jphysiol.2007.130013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azabji Kenfack M, Lador F, Licker M, Moia C, Tam E, Capelli C, Morel D, Ferretti G. Cardiac output by Modelflow method from intra-arterial and fingertip pulse pressure profiles. Clin Sci (Lond) 2004;106:365–369. doi: 10.1042/CS20030303. [DOI] [PubMed] [Google Scholar]
- Brock RW, Tschakovsky ME, Shoemaker JK, Halliwill JR, Joyner MJ, Hughson RL. Effects of acetylcholine and nitric oxide on forearm blood flow at rest and after a single muscle contraction. J Appl Physiol. 1998;85:2249–2254. doi: 10.1152/jappl.1998.85.6.2249. [DOI] [PubMed] [Google Scholar]
- Buckwalter JB, Clifford PS. Autonomic control of skeletal muscle blood flow at the onset of exercise. Am J Physiol Heart Circ Physiol. 1999;277:H1872–1877. doi: 10.1152/ajpheart.1999.277.5.H1872. [DOI] [PubMed] [Google Scholar]
- Celermajer DS, Sorensen KE, Gooch VM, Spiegelhalter DJ, Miller OI, Sullivan ID, Lloyd JK, Deanfield JE. Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet. 1992;340:1111–1115. doi: 10.1016/0140-6736(92)93147-f. [DOI] [PubMed] [Google Scholar]
- Clifford PS, Kluess HA, Hamann JJ, Buckwalter JB, Jasperse JL. Mechanical compression elicits vasodilatation in rat skeletal muscle feed arteries. J Physiol. 2006;572:561–567. doi: 10.1113/jphysiol.2005.099507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corretti MC, Anderson TJ, Benjamin EJ, Celermajer D, Charbonneau F, Creager MA, Deanfield J, Drexler H, Gerhard-Herman M, Herrington D, Vallance P, Vita J, Vogel R. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: A report of the International Brachial Artery Reactivity Task Force. J Am Coll Cardiol. 2002;39:257–265. doi: 10.1016/s0735-1097(01)01746-6. [DOI] [PubMed] [Google Scholar]
- Dakak N, Husain S, Mulcahy D, Andrews NP, Panza JA, Waclawiw M, Schenke W, Quyyumi AA. Contribution of nitric oxide to reactive hyperemia: impact of endothelial dysfunction. Hypertension. 1998;32:9–15. doi: 10.1161/01.hyp.32.1.9. [DOI] [PubMed] [Google Scholar]
- de Vaal JB, de Wilde RBP, van den Berg PCM, Schreuder JJ, Jansen JRC. Less invasive determination of cardiac output from the arterial pressure by aortic diameter-calibrated pulse contour. Br J Anaesth. 2005;95:326–331. doi: 10.1093/bja/aei189. [DOI] [PubMed] [Google Scholar]
- de Wilde RB, Geerts BF, Cui J, van den Berg PC, Jansen JR. Performance of three minimally invasive cardiac output monitoring systems. Anaesthesia. 2009;64:762–769. doi: 10.1111/j.1365-2044.2009.05934.x. [DOI] [PubMed] [Google Scholar]
- DeSouza CA, Shapiro LF, Clevenger CM, Dinenno FA, Monahan KD, Tanaka H, Seals DR. Regular aerobic exercise prevents and restores age-related declines in endothelium-dependent vasodilation in healthy men. Circulation. 2000;102:1351–1357. doi: 10.1161/01.cir.102.12.1351. [DOI] [PubMed] [Google Scholar]
- Dinenno FA, Joyner MJ. Blunted sympathetic vasoconstriction in contracting skeletal muscle of healthy humans: is nitric oxide obligatory? J Physiol. 2003;553:281–292. doi: 10.1113/jphysiol.2003.049940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doshi SN, Naka KK, Payne N, Jones CJ, Ashton M, Lewis MJ, Goodfellow J. Flow-mediated dilatation following wrist and upper arm occlusion in humans: the contribution of nitric oxide. Clin Sci (Lond) 2001;101:629–635. [PubMed] [Google Scholar]
- Dyke CK, Dietz NM, Lennon RL, Warner DO, Joyner MJ. Forearm blood flow responses to handgripping after local neuromuscular blockade. J Appl Physiol. 1998;84:754–758. doi: 10.1152/jappl.1998.84.2.754. [DOI] [PubMed] [Google Scholar]
- Endo T, Imaizumi T, Tagawa T, Shiramoto M, Ando S, Takeshita A. Role of nitric oxide in exercise-induced vasodilation of the forearm. Circulation. 1994;90:2886–2890. doi: 10.1161/01.cir.90.6.2886. [DOI] [PubMed] [Google Scholar]
- Frandsen U, Bangsbo J, Sander M, Hoffner L, Betak A, Saltin B, Hellsten Y. Exercise-induced hyperaemia and leg oxygen uptake are not altered during effective inhibition of nitric oxide synthase with NG-nitro-L-arginine methyl ester in humans. J Physiol. 2001;531:257–264. doi: 10.1111/j.1469-7793.2001.0257j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilligan DM, Panza JA, Kilcoyne CM, Waclawiw MA, Casino PR, Quyyumi AA. Contribution of endothelium-derived nitric oxide to exercise-induced vasodilation. Circulation. 1994;90:2853–2858. doi: 10.1161/01.cir.90.6.2853. [DOI] [PubMed] [Google Scholar]
- Gokce N, Keaney JF, Jr, Hunter LM, Watkins MT, Nedeljkovic ZS, Menzoian JO, Vita JA. Predictive value of noninvasively determined endothelial dysfunction for long-term cardiovascular events inpatients with peripheral vascular disease. J Am Coll Cardiol. 2003;41:1769–1775. doi: 10.1016/s0735-1097(03)00333-4. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Alonso J, Mortensen SP, Jeppesen TD, Ali L, Barker H, Damsgaard R, Secher NH, Dawson EA, Dufour SP. Haemodynamic responses to exercise, ATP infusion and thigh compression in humans: insight into the role of muscle mechanisms on cardiovascular function. J Physiol. 2008;586:2405–2417. doi: 10.1113/jphysiol.2008.152058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon MB, Jain R, Beckman JA, Creager MA. The contribution of nitric oxide to exercise hyperemia in the human forearm. Vasc Med. 2002;7:163–168. doi: 10.1191/1358863x02vm439oa. [DOI] [PubMed] [Google Scholar]
- Green DJ, Bilsborough W, Naylor LH, Reed C, Wright J, O’Driscoll G, Walsh JH. Comparison of forearm blood flow responses to incremental handgrip and cycle ergometer exercise: relative contribution of nitric oxide. J Physiol. 2005;562:617–628. doi: 10.1113/jphysiol.2004.075929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DJ, Jones H, Thijssen D, Cable NT, Atkinson G. Flow-mediated dilation and cardiovascular event prediction: does nitric oxide matter? Hypertension. 2011;57:363–369. doi: 10.1161/HYPERTENSIONAHA.110.167015. [DOI] [PubMed] [Google Scholar]
- Hamann JJ, Buckwalter JB, Clifford PS. Vasodilatation is obligatory for contraction-induced hyperaemia in canine skeletal muscle. J Physiol. 2004;557:1013–1020. doi: 10.1113/jphysiol.2004.062836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RA, Nishiyama SK, Wray DW, Richardson RS. Ultrasound assessment of flow-mediated dilation. Hypertension. 2010;55:1075–1085. doi: 10.1161/HYPERTENSIONAHA.110.150821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayman MA, Nativi JN, Stehlik J, McDaniel J, Fjeldstad AS, Ives SJ, Wray DW, Bader F, Gilbert EM, Richardson RS. Understanding exercise-induced hyperemia: central and peripheral hemodynamic responses to passive limb movement in heart transplant recipients. Am J Physiol Heart Circ Physiol. 2010;299:H1653–1659. doi: 10.1152/ajpheart.00580.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellsten Y, Rufener N, Nielsen JJ, Hoier B, Krustrup P, Bangsbo J. Passive leg movement enhances interstitial VEGF protein, endothelial cell proliferation, and NOS mRNA content in human skeletal muscle. Am J Physiol Regulatory Integrative Comp Physiol. 2008;294:R975–982. doi: 10.1152/ajpregu.00677.2007. [DOI] [PubMed] [Google Scholar]
- Herrington DM, Fan L, Drum M, Riley WA, Pusser BE, Crouse JR, Burke GL, McBurnie MAD, Morgan TM, Espeland MA. Brachial flow-mediated vasodilator responses in population-based research: methods, reproducibility and effects of age, gender and baseline diameter. J Cardiovasc Risk. 2001;8:319–328. doi: 10.1177/174182670100800512. [DOI] [PubMed] [Google Scholar]
- Hoier B, Rufener N, Bojsen-Moller J, Bangsbo J, Hellsten Y. The effect of passive movement training on angiogenic factors and capillary growth in human skeletal muscle. J Physiol. 2010;588:3833–3845. doi: 10.1113/jphysiol.2010.190439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joannides R, Haefeli WE, Linder L, Richard V, Bakkali EH, Thuillez C, Luscher TF. Nitric oxide is responsible for flow-dependent dilatation of human peripheral conduit arteries in vivo. Circulation. 1995;91:1314–1319. doi: 10.1161/01.cir.91.5.1314. [DOI] [PubMed] [Google Scholar]
- Kirby BS, Carlson RE, Markwald RR, Voyles WF, Dinenno FA. Mechanical influences on skeletal muscle vascular tone in humans: insight into contraction-induced rapid vasodilatation. J Physiol. 2007;583:861–874. doi: 10.1113/jphysiol.2007.131250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooijman M, Thijssen DHJ, de Groot PCE, Bleeker MWP, van Kuppevelt HJM, Green DJ, Rongen GA, Smits P, Hopman MTE. Flow-mediated dilatation in the superficial femoral artery is nitric oxide mediated in humans. J Physiol. 2008;586:1137–1145. doi: 10.1113/jphysiol.2007.145722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansman JB, Hallam TJ, Rink TJ. Single stretch-activated ion channels in vascular endothelial cells as mechanotransducers? Nature. 1987;325:811–813. doi: 10.1038/325811a0. [DOI] [PubMed] [Google Scholar]
- McDaniel J, Fjeldstad AS, Ives S, Hayman M, Kithas P, Richardson RS. Central and peripheral contributors to skeletal muscle hyperemia: response to passive limb movement. J Appl Physiol. 2010a;108:76–84. doi: 10.1152/japplphysiol.00895.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel J, Hayman MA, Ives S, Fjeldstad AS, Trinity JD, Wray DW, Richardson RS. Attenuated exercise induced hyperaemia with age: mechanistic insight from passive limb movement. J Physiol. 2010b;588:4507–4517. doi: 10.1113/jphysiol.2010.198770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohrman DE, Sparks HV. Myogenic hyperemia following brief tetanus of canine skeletal muscle. Am J Physiol. 1974a;227:531–535. doi: 10.1152/ajplegacy.1974.227.3.531. [DOI] [PubMed] [Google Scholar]
- Mohrman DE, Sparks HV. Role of potassium ions in the vascular response to a brief tetanus. Circ Res. 1974b;35:384–390. doi: 10.1161/01.res.35.3.384. [DOI] [PubMed] [Google Scholar]
- Mortensen SP, Gonzalez-Alonso J, Damsgaard R, Saltin B, Hellsten Y. Inhibition of nitric oxide and prostaglandins, but not endothelial-derived hyperpolarizing factors, reduces blood flow and aerobic energy turnover in the exercising human leg. J Physiol. 2007;581:853–861. doi: 10.1113/jphysiol.2006.127423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen MJ, Kharbanda RK, Cross J, Donald AE, Taylor M, Vallance P, Deanfield JE, MacAllister RJ. Heterogenous nature of flow-mediated dilatation in human conduit arteries in vivo: relevance to endothelial dysfunction in hypercholesterolemia. Circ Res. 2001;88:145–151. doi: 10.1161/01.res.88.2.145. [DOI] [PubMed] [Google Scholar]
- Nobrega AC, Araujo CG. Heart rate transient at the onset of active and passive dynamic exercise. Med Sci Sports Exerc. 1993;25:37–41. [PubMed] [Google Scholar]
- Olesen SP, Clapham DE, Davies PF. Haemodynamic shear stress activates a K+ current in vascular endothelial cells. Nature. 1988;331:168–170. doi: 10.1038/331168a0. [DOI] [PubMed] [Google Scholar]
- Parker BA, Smithmyer SL, Pelberg JA, Mishkin AD, Proctor DN. Sex-specific influence of aging on exercising leg blood flow. J Appl Physiol. 2008;104:655–664. doi: 10.1152/japplphysiol.01150.2007. [DOI] [PubMed] [Google Scholar]
- Parker BA, Tschakovsky ME, Augeri AL, Polk DM, Thompson PD, Kiernan FJ. Heterogenous vasodilator pathways underlie flow-mediated dilation in men and women. Am J Physiol Heart Circ Physiol. 2011;301:H1118–1126. doi: 10.1152/ajpheart.00400.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyke K, Green DJ, Weisbrod C, Best M, Dembo L, O’Driscoll G, Tschakovsky M. Nitric oxide is not obligatory for radial artery flow-mediated dilation following release of 5 or 10 min distal occlusion. Am J Physiol Heart Circ Physiol. 2010;298:H119–126. doi: 10.1152/ajpheart.00571.2009. [DOI] [PubMed] [Google Scholar]
- Radegran G, Saltin B. Nitric oxide in the regulation of vasomotor tone in human skeletal muscle. Am J Physiol Heart Circ Physiol. 1999;276:H1951–1960. doi: 10.1152/ajpheart.1999.276.6.H1951. [DOI] [PubMed] [Google Scholar]
- Roig E, Cuppoletti A, Masotti M, Kianco R, Vallejos I, Sitges M, Ortiz J, Perez-Villa F. Assessment of peripheral endothelial-dependent vasodilatation within the first year after heart transplantation. J Heart Lung Transplant. 2009;28:299–304. doi: 10.1016/j.healun.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Saunders NR, Dinenno FA, Pyke KE, Rogers AM, Tschakovsky ME. Impact of combined NO and PG blockade on rapid vasodilation in a forearm mild-to-moderate exercise transition in humans. Am J Physiol Heart Circ Physiol. 2005;288:H214–220. doi: 10.1152/ajpheart.00762.2004. [DOI] [PubMed] [Google Scholar]
- Saunders NR, Tschakovsky ME. Evidence for a rapid vasodilatory contribution to immediate hyperemia in rest-to-mild and mild-to-moderate forearm exercise transitions in humans. J Appl Physiol. 2004;97:1143–1151. doi: 10.1152/japplphysiol.01284.2003. [DOI] [PubMed] [Google Scholar]
- Schrage WG, Joyner MJ, Dinenno FA. Local inhibition of nitric oxide and prostaglandins independently reduces forearm exercise hyperaemia in humans. J Physiol. 2004;557:599–611. doi: 10.1113/jphysiol.2004.061283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker JK, Halliwill JR, Hughson RL, Joyner MJ. Contributions of acetylcholine and nitric oxide to forearm blood flow at exercise onset and recovery. Am J Physiol Heart Circ Physiol. 1997;273:H2388–2395. doi: 10.1152/ajpheart.1997.273.5.H2388. [DOI] [PubMed] [Google Scholar]
- Shoemaker JK, Naylor HL, Pozeg ZI, Hughson RL. Failure of prostaglandins to modulate the time course of blood flow during dynamic forearm exercise in humans. J Appl Physiol. 1996;81:1516–1521. doi: 10.1152/jappl.1996.81.4.1516. [DOI] [PubMed] [Google Scholar]
- Sugawara J, Tanabe T, Miyachi M, Yamamoto K, Takahashi K, Iemitsu M, Otsuki T, Homma S, Maeda S, Ajisaka R, Matsuda M. Non-invasive assessment of cardiac output during exercise in healthy young humans: comparison between Modelflow method and Doppler echocardiography method. Acta Physiol Scand. 2003;179:361–366. doi: 10.1046/j.0001-6772.2003.01211.x. [DOI] [PubMed] [Google Scholar]
- Taddei S, Galetta F, Virdis A, Ghiadoni L, Salvetti G, Franzoni F, Giusti C, Salvetti A. Physical activity prevents age-related impairment in nitric oxide availability in elderly athletes. Circulation. 2000;101:2896–2901. doi: 10.1161/01.cir.101.25.2896. [DOI] [PubMed] [Google Scholar]
- Taddei S, Virdis A, Ghiadoni L, Salvetti G, Bernini G, Magagna A, Salvetti A. Age-related reduction of no availability and oxidative stress in humans. Hypertension. 2001;38:274–279. doi: 10.1161/01.hyp.38.2.274. [DOI] [PubMed] [Google Scholar]
- Takase B, Uehata A, Akima T, Nagai T, Nishioka T, Hamabe A, Satomura K, Ohsuzu F, Kurita A. Endothelium-dependent flow-mediated vasodilation in coronary and brachial arteries in suspected coronary artery disease. Am J Cardiol. 1998;82:1535–1539. A1537–1538. doi: 10.1016/s0002-9149(98)00702-4. [DOI] [PubMed] [Google Scholar]
- Trinity JD, Amann M, McDaniel J, Fjeldstad AS, Barret-O’Keefe Z, Runnels S, Morgan DE, Wray DW, Richardson RS. Limb movement-induced hyperemia has a central hemodynamic component. Evidence from a neural blockade study. Am J Physiol Heart Circ Physiol. 2010;299:H1693–1700. doi: 10.1152/ajpheart.00482.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinity JD, McDaniel J, Venturelli M, Fjeldstad AS, Ives SJ, Witman MAH, Barrett-O’Keefe Z, Amann M, Wray DW, Richardson RS. Impact of body position on central and peripheral hemodynamic contributions to movement-induced hyperemia: implications for rehabilitative medicine. Am J Physiol Heart Circ Physiol. 2011;300:H1885–1891. doi: 10.1152/ajpheart.00038.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschakovsky ME, Pyke KE. Counterpoint: Flow-mediated dilation does not reflect nitric oxide-mediated endothelial function. J Appl Physiol. 2005;99:1235–1237. doi: 10.1152/japplphysiol.00607.2005. [DOI] [PubMed] [Google Scholar]
- van Lieshout JJ, Toska K, van Lieshout EJ, Eriksen M, Walloe L, Wesseling KH. Beat-to-beat noninvasive stroke volume from arterial pressure and Doppler ultrasound. Eur J Appl Physiol. 2003;90:131–137. doi: 10.1007/s00421-003-0901-8. [DOI] [PubMed] [Google Scholar]
- Venturelli M, Amann MK, McDaniel J, Trinity JD, Fjeldstad AS, Richardson RS. Central and peripheral hemodynamic responses to passive-limb movement: the role of arousal. Am J Physiol Heart Circ Physiol. 2012 doi: 10.1152/ajpheart.00851.2011. (in press: ajpheart.00851.02011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray DW, Donato AJ, Uberoi A, Merlone JP, Richardson RS. Onset exercise hyperaemia in humans: partitioning the contributors. J Physiol. 2005a;565:1053–1060. doi: 10.1113/jphysiol.2005.084327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray DW, Fadel PJ, Smith ML, Raven P, Sander M. Inhibition of α-adrenergic vasoconstriction in exercising human thigh muscles. J Physiol. 2004;555:545–563. doi: 10.1113/jphysiol.2003.054650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray DW, Uberoi A, Lawrenson L, Richardson RS. Heterogeneous limb vascular responsiveness to shear stimuli during dynamic exercise in humans. J Appl Physiol. 2005b;99:81–86. doi: 10.1152/japplphysiol.01285.2004. [DOI] [PubMed] [Google Scholar]
- Wray DW, Witman MAH, Ives SJ, McDaniel J, Fjeldstad AS, Trinity JD, Conklin JD, Supiano MA, Richardson RS. Progressive handgrip exercise: evidence of nitric oxide-dependent vasodilation and blood flow regulation in humans. Am J Physiol Heart Circ Physiol. 2011;300:H1101–1107. doi: 10.1152/ajpheart.01115.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]