

Abstract
Microbial immune evasion can be achieved through the expression, or mimicry, of host-like carbohydrates on the microbial cell surface to hide from detection. However, disparate reports collectively suggest that evasion could also be accomplished through the modulation of the host glycosylation pathways, a mechanism that we call the “Glyco-Evasion Hypothesis”. Here, we will summarize the evidence in support of this paradigm by reviewing three separate bodies of work present in the literature. We review how infection and inflammation can lead to host glycosylation changes, how host glycosylation changes can increase susceptibility to infection and inflammation and how glycosylation impacts molecular and cellular function. Then, using these data as a foundation, we propose a unifying hypothesis in which microbial products can hijack host glycosylation to manipulate the immune response to the advantage of the pathogen. This model reveals areas of research that we believe could significantly improve our fight against infectious disease.
Keywords: glycosylation, immune evasion, infectious disease, major histocompatibility complex
Introduction
There is a wealth of data across a broad section of the primary literature that describes changes in glycosylation associated with infection and inflammation (Magalhaes and Reis 2010; Moran et al. 2011; Varki 2011). Likewise, abundant research exists describing how glycosylation is linked to molecular and cellular function within the immune system (Sato et al. 2009; Davicino et al. 2011; Varki 2011). With the exception of the congenital disorders of glycosylation, however, it is exceedingly difficult to find studies in which infectious and inflammatory disease-associated changes in glycosylation are experimentally linked to functional changes in the immune system. In contrast to molecular mimicry where pathogens hide from the immune system by alterations in the “microbial” carbohydrates to appear “host-like” (reviewed elsewhere), this article is intended to provide a conceptual framework that we call the “Glyco-Evasion Hypothesis” in which we propose that microbes can manipulate the immune response to their benefit through the modulation of “host” glycosylation. In order to reach this goal, we present sub-reviews on three bodies of research that independently provide evidence for the key cellular pathways we believe intersect within a single cohesive paradigm. These sub-reviews are then used as the basis for the formal presentation of our Glyco-Evasion Hypothesis and its potential implications for future research and the fight against infectious disease.
Glycome changes lead to infection/inflammation
In simple terms, the glycome represents the glycan structures present on a given tissue, cell or molecule population. Although it has been firmly established that changes in the glycome are linked to disease, including inflammation and infection, there continues to be a chicken-or-egg problem in our understanding. Do changes in host glycosylation lead to inflammation or does infection-induced inflammation/innate signaling cause changes in host glycosylation? Based on the published record thus far, the answer appears to be both.
Many glycosylation-related knockout mice have been generated over the last two decades, though not all of these genetic changes are tolerated. Some of these gene knockouts are embryonic or newborn lethal in mice (Marek et al. 1999; Shafi et al. 2000; Wang et al. 2001; Ye and Marth 2004), which is mirrored in the human population by the rarity of survival and severity of pathologies associated with the congenital disorder of glycosylation (CDG) diseases (Jaeken 2010; Freeze and Ng 2011). These data alone show the importance of glycans in physiology as a whole, but when we move beyond this fundamental truth, it is common to find that the ablation of glycosylation enzymes generates immune-associated pathologies.
A prominent example of immune disease arising from glycan-changing knockouts is α-mannosidase IIa1 (Man2a1). This is an enzyme in the N-linked glycosylation pathway that is responsible for the removal of the terminal α1-3- and α1-6-linked mannose residues prior to the addition of N-acetylglucosamine (GlcNAc) residues which are required for the synthesis of complex-type asparagine (N)-linked glycans (Figure 1). Remarkably, the Man2a1−/− mice develop a pathology that is similar to human systemic lupus erythematosus (SLE; Chui et al. 2001). Although lymphocyte development and the ability for B and T cells to proliferate are unchanged in these animals, antibody levels in the serum are increased and the animals develop nephritis that mirrors what is seen in SLE patients. Furthermore, anti-nuclear antibodies developed, which is another hallmark of human SLE. Perhaps even more remarkable is that the glomerulonephritis and resulting kidney failure were not due to cells of a hematopoietic origin since bone marrow transplants from wild-type donors failed to alter the kidney disease course. Instead, the phenotype appears to be at least in part due to the chronic activation of the innate immune pathways and systemic sterile inflammation (Green et al. 2007), although this does not fully explain why anti-nuclear antibodies develop in these animals.
Fig. 1.
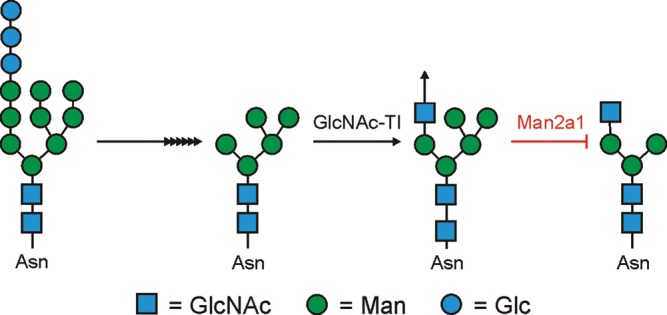
Man2a1 is an enzyme in the N-glycosylation pathway that removes the terminal α1-3Man and α1-6Man residues from the GlcNAcMan5GlcNAc1 structure. Without the removal of these two mannose residues, branched complex-type N-glycans are not formed. However, the terminal β1-2-linked GlcNAc can be extended (arrow) by the cohort of Golgi enzymes present.
Leukocyte homing and extravasation is driven by selectin–glycan interactions within the blood vessels near sites of inflammation and infection. During infection, inflammatory mediators such as tumor necrosis factor α (TNFα) increase the surface concentrations of selectin molecules on the surface of endothelial cells near the site of infection. These adhesion molecules then bind to leukocytes in circulation via glycans on the cell surface and facilitate recruitment of leukocytes to the infected site (Rosen 2004). This process is central to the function of the immune response and is dependent upon the glycan structures present on the leukocyte population. As such, it stands to reason that changes in the glycans would be expected to alter the ability of cells to home to these sites to mount the appropriate immune response. As another example of how glycan-altering knockouts impact the immune response, ablation of Gcnt1, a core 2 β1,6-GlcNAc transferase (Figure 2), results in reduced neutrophil rolling on surfaces expressing E-, L- and P-selectins and leads to deficient neutrophil homing to sites of inflammation (Ellies et al. 1998; Stone et al. 2009). Likewise, the α2,3-sialyltransferase family (six have been identified; ST3Gal-I to ST3Gal-VI) appear to play roles in the synthesis of selectin ligands. Specifically, the loss of ST3Gal-IV in mice reduced the number and increased the velocity of leukocytes actively rolling in an E-selectin-dependent fashion, indicating that 2,3-linked sialic acid containing glycans are important for E-selectin binding (Ellies et al. 2002) and that the modulation of ST3Gal-IV expression can regulate leukocyte homing to sites of infection.
Fig. 2.

The bovine herpesvirus 4-encoded β1,6-GlcNAc transferase (Bo17/β1,6GnT) shown in red is a homolog of Gcnt1, the host transferase that adds GlcNAc to the “core 2” O-glycan (core 2 GlcNAcT). The microbial expression of this enzyme could lead to significant increases in core 2 structures, which are known to decrease the interactions of a number of cells, including immune cells (Fukuda and Tsuboi 1999).
The epithelial barrier is of paramount importance as a first line of defense against pathogens, and glycans play a critical role in cell–cell adhesion and communication (Zhao et al. 2008). Within the mucosal surfaces, mucins also play a critical role in the physical barrier between host and microbe (Fukuda 2002) and are among the most heavily glycosylated proteins known. In mice lacking functional Gcnt2 (another O-glycan core 2 β1,6-GlcNAc transferase), the barrier of the gut is compromised. These animals show a significant leakage of intestinal molecules into the circulation and increased susceptibility to colitis (Stone et al. 2009). Although the expression level of mucin glycoproteins was the same, the nature of the mucin glycans was not studied in these knockouts, leaving the mechanism of leakage unclear. These animals show increased numbers of circulating neutrophils, possibly due to the leakiness of the gut, but also produce lower levels of antibodies (Stone et al. 2009). In fact, most antibody classes were reduced, including IgG, IgM and IgA (IgE levels were not reported), but the underlying mechanism(s) of this defect remains unexplained and unexplored.
Sialylated core 1 O-glycans are a hallmark of naïve CD8+ T cells in the thymus, whereas the loss of the sialylated core 1 and the synthesis of core 2 O-glycans are seen upon activation in the periphery (Chervenak and Cohen 1982; Piller et al. 1988). Upon differentiation into CD8+ memory cells, the O-glycans revert to the core 1-dominated phenotype (Galvan et al. 1998) and those effector CD8+ T cells still carrying core 2 O-glycans progress into apoptosis. In 2000, Marth and colleagues reported that animals without the ST3Gal-I sialyltransferase enzyme lacked the sialylated core 1 O-glycans on naïve CD8+ T cells (Priatel et al. 2000). Indeed, the loss of ST3Gal-I promoted the synthesis of the core 2 O-glycans on naïve T cells in the periphery, leading to enhanced apoptosis in the absence of immune challenge (Priatel et al. 2000). These results led to the conclusion that glycoprotein sialylation is a homeostatic mechanism for the proper function, differentiation and resolution of CD8+ T cell-dependent immune responses. Perhaps more to the point of this review, these data clearly show that sialylation is an actively regulated pathway that is critical for proper T cell responses.
Continuing with the sialylation theme, ST6Gal-I is the lone enzyme known to attach α2,6-linked terminal sialic acids on N-linked glycans and the loss of this enzyme eliminates all α2,6-linked sialic acids (Hennet et al. 1998). CD22 is a member of the Siglec (sialic acid-binding Ig-like lectin) family of cell surface molecules and is an integral member of the B cell receptor complex. CD22 (a.k.a. Siglec-2) binds specifically to glycans containing an α2,6-linked terminal sialic acid (O'Reilly et al. 2011). The ablation of the ST6Gal-I enzyme in mice resulted in a severe B cell-centered phenotype whereby IgM levels were significantly reduced and B cell proliferation was attenuated upon stimulation via IgM or CD40 cross-linking (Hennet et al. 1998). Antibody responses were also reduced for both T-independent and T-dependent antigens. Remarkably, this phenotype was not due to defects in the B cell itself, in that subsequent investigation found that the co-deletion of CD22 eliminated the overall phenotype of the animal (Grewal et al. 2006). This suggests that the change in the CD22 ligand pool is altered in the ST6Gal-I knockout, sending aberrant signals into the B cells, but that the elimination of the receptor for those ligands eliminates those same aberrant signals. To date, the identity of these CD22 ligand(s) remains unclear.
Another interesting knockout model is the mouse lacking the Mgat5 (α-6-d-mannoside β1,6 N-GlcNAc transferase V; GlcNAc-TV) locus. The Mgat5 enzyme is responsible for creating highly branched (tetra-antennary) complex-type N-glycans (Figure 3). The first study that described these animals gave rise to notion that limiting the branching of N-glycans on T cells reduces T cell activation threshold (Demetriou et al. 2001). Contrary to earlier hypotheses concerning the role of N-glycans on the T cell receptor (TCR), which suggested that the glycans themselves limited TCR clustering by providing structural barriers between adjacent molecules (Rudd et al. 1999, 2001), the Mgat5 knockout animals showed that the β1,6 branch of complex N-glycans initiated by the GlcNAc-TV enzyme (Figure 3) is the ligand for galectin-3 (gal-3) (and many other galectins) which maintains TCR spacing. Loss of that ligand on the TCR induced clustering, lowering the threshold and promoting autoimmunity (Demetriou et al. 2001; Morgan et al. 2004). In agreement with this interpretation, risk factors for multiple sclerosis (MS) appear to converge on the N-glycosylation pathway to disrupt the normal homeostatic state of T cells in patients via glycan alterations (Mkhikian et al. 2011). Remarkably, under normal conditions, TCR signaling even promotes the changes in N-glycosylation that maximize Mgat5 efficiency and thus maximize the TCR signaling thresholds, demonstrating that this pathway is actively used by T cells to regulate antigen-dependent signaling (Chen et al. 2009).
Fig. 3.

GlcNAc transferases I, II, IV and V are necessary for the synthesis of mono-, bi-, tri- and tetra-antennary branched complex-type N-glycans, although mono-antennary structures are quite rare. This figure provides a framework of possible modifications; however, the glycans shown represent a very small fraction of the variety than can occur through differing the sugar linkages of those shown (e.g. α2,3- vs α2-6-linked sialic acids) and the action of a host of enzymes that include fucosyltransferases, galactosyltransferases, sulfotransferases and so on.
There are also models in which a glycosylation-associated transgene was inserted rather than ablated into the mouse genome. Perhaps the most pertinent to the present review is the mouse carrying a transgene of the human α1,2-fucosyltransferase 1 locus (Brown et al. 2004). The result was a striking and spontaneous colitis in which the intestinal permeability was not compromised. Instead, these mice showed profound defects in T cell development and it is now clear from other reports that fucosylation is a regulating factor in lymphocyte development via the Notch pathway. Although the role of glycans in Notch signaling has been reviewed in detail elsewhere (Stanley and Guidos 2009; Stanley and Okajima 2010), it is important to note that fucosylation is actively regulated and modulates Notch–ligand interactions and signal strength into developing lymphocytes. As such, the disruption of the normal regulation of Notch fucosylation has dramatic deleterious effects on the adaptive immune system.
Moving away from animal models, it is also clear that disease-associated changes in glycosylation are associated with increased susceptibility to infection. As a case in point, patients with cystic fibrosis carry a genetic lesion in the cystic fibrosis transmembrane conductance regulator (CFTR), an epithelial chloride channel. Although it is a matter of some debate in the field as to whether mutations in the CFTR are (Poschet et al. 2001) or are not (Leir et al. 2005) directly responsible for the glycan phenotype, it is clear that significant changes in the glycosylation of both cell surface (Poschet et al. 2001) and secreted proteins, especially the mucins (Wesley et al. 1983; Xia et al. 2005; Schulz et al. 2007), are the characteristic of cystic fibrosis (CF) patients. These changes in glycosylation are thought to be critical to promote chronic Pseudomonas aeruginosa lung infections in CF patients (van Heeckeren et al. 2004). In fact, augmenting N-glycosylation in CF-model animals via either exposure to a viral expression vector with mannose-6-phosphate isomerase to compensate for CF-associated loss of expression or a “hypermannose” water diet, partially normalized the glycosylation pattern in the animals and reduced colonization and bacterial burden of P. aeruginosa (Martino et al. 2011). These data demonstrate that disease-associated changes in glycosylation can lead directly to increased susceptibility to infection.
Infection/inflammation leads to glycome changes
The findings presented thus far paint a convincing argument that changes in the glycome can alter the immune system such that inflammation, autoimmunity and/or infection ensue, but what about the reverse scenario? Does inflammation and/or infection lead to changes in the host glycome, and if so, how and to what extent? While data on the “how” and “extent” part of that question is practically non-existent in the primary literature, it is clear that inflammation and infection do influence the host glycome.
The host glycome is determined by a number of factors (Kornfeld and Kornfeld 1985; Herscovics 1999; Berninsone and Hirschberg 2000; Schachter 2000), most of which remain poorly understood in the sense that the outcome for any given molecule or cell remains unpredictable. The most obvious and easy to understand the determining factor is that the cohort of glycosidases, glycosyltransferases and nucleotide sugar transporters in the Golgi Apparatus to a large degree dictates the types of structures “possible” within a given cell (Brockhausen et al. 2009; Stanley et al. 2009); however, the composition and heterogeneity at the molecular level is more complicated than that. The modifying enzymes are known to segregate into regions of the Golgi cisternae (Colley 1997; Opat et al. 2001) and many of these compete for the same glycan substrates during trafficking, introducing significant heterogeneity in the final products (Brockhausen et al. 2009; Stanley et al. 2009). The length of time that a particular glycoprotein is present in the Golgi can strongly influence the nature of the complex glycans present (Kornfeld and Kornfeld 1985), possibly limiting the number of glycan modifications for glycoproteins that remain in the Golgi only for a short period of time. The structure of the underlying protein backbone can also play a significant site-specific role in the types of glycans found at varying sites within the same molecule, likely dictated by steric hindrance of the modifying enzyme(s) to approach each individual glycan substrate (Kornfeld and Kornfeld 1985). For example, gp120 (and other viral coat proteins) carries some N-glycosylation sites that are consistently occupied by traditional branched complex-type N-glycans, whereas other sites are consistently occupied by high-mannose structures (Zhu et al. 2000). Another contributing factor is a change in metabolic activity, thus altering the availability of the carbohydrate donor molecules (i.e. sugar nucleotides) that act as substrates for the Golgi enzymes (Berninsone and Hirschberg 2000). Likewise, it is possible that increased expression of glycoproteins due to increased and rapid proliferation during an immune response could overcome the limits of the glycosylation pathway, thus limiting the relative amount of modification and complexity. Thus, it is easy to imagine how a pathogen or the surrounding inflammatory response against that pathogen might affect host glycosylation through cellular activation and the associated changes in transcription, host cell proliferation, competition for nutrients in the environment and even competition for the synthetic pathways during replication within cells, yet few have actually connected the dots experimentally.
To date, the predominate form of evidence for infection-mediated glycome changes can be found in correlation studies, which lack mechanistic detail. One particularly strong example is that infection with Helicobacter pylori appears to induce changes in mucin oligosaccharide structures [e.g. increased expression of sialyl Lewis X (SLex); Mahdavi et al. 2002; Cooke et al. 2009; Linden et al. 2010] in the gut, presumably to promote colonization. Additional findings further demonstrated that H. pylori-induced gastritis increased α-1,4-GlcNAc transferase (α4GnT) to a degree proportional to the level of inflammation, but that clearance of the pathogen correlated with a return to normal levels of α4GnT expression (Matsuzwa et al. 2003). Here, the link between infection and glycome change is strong and the investigators might even have one part of how that could occur, but it remains unclear how or why α4GnT expression is increased, the degree to which other Golgi enzymes are altered in their expression, and what, if any, link exists between increased α4GnT and increased SLex on the secreted mucins.
Other examples of infection-mediated glycome changes include the observation that novel O-linked glycans on peptides derived from fibrinogen α1 chain in the urine have been associated with urinary tract infections (Pacchiarotta et al. 2012), whereas infection with the nematode Nippostrongylus brasiliensis appears to up-regulate 3-O-sulfotransferase 1, an enzyme that sulfates host carbohydrates, in gastric epithelial cells (Yamauchi et al. 2006). Another intriguing study from nearly 10 years ago showed a strong correlation between exposure to commensal bacteria in the gut and significant glycome changes (Nanthakumar et al. 2003). In this study, the investigators compared the relative levels of glycosyltransferase activities in intestinal brushborder cells derived from germ-free (GF) and conventional (CONV) mice. In CONV mice, suckling pups showed high α-2,3/6-sialyltransferase activity and low α1,2-fucosyltransferase activity, with those relative activity levels reversing by adulthood (i.e. 4 weeks). In contrast, the transition to the adult activity levels (i.e. low sialyltransferase, high fucosyltransferase) failed to occur in the absence of the microflora in GF animals. When colonized with gut microflora, however, the GF mice rapidly switched relative activities of these enzymes, showing a role for the microbiota in shaping the glycosyltransferase expression and activity levels in the host cells that are in contact to the microbes. These three studies demonstrate a strong correlation between microbial exposure and glycome changes, yet the mechanism of that influence continues to be a mystery.
The only reference we could find that directly shows a mechanistic connection between a specific pathway, a specific microbial product and glycome changes was published just last year. This study revealed that the stimulation of TLR9 on B cells with CpG resulted in increased galactosylation and reduced bisecting GlcNAc levels on IgG molecules (Wang et al. 2011), although it still remains to be seen whether this effect was due to direct NFκB-mediated signaling and transcriptional changes or some other secondary effect. Likewise, it is unclear what functional impact TLR9-mediated signaling had on the antibodies being produced.
The main quasi-mechanistic data on how inflammation (and presumably infection-mediated inflammation) alters the host glycome come from cancer research. For example, the level of circulating glycoproteins carrying SLex structures (Figure 4) increases with the expression of C-reactive protein in cancer patients (Mizuguchi, Nishiyama, Iwata, Nishida, Izumi, Tsukioka, Inoue, Kameyama, et al. 2007, Mizuguchi, Nishiyama, Iwata, Nishida, Izumi, Tsukioka, Inoue, Uenishi, et al. 2007; Saldova et al. 2007), and the resection of the tumor is accompanied by a reduction in SLex to normal levels in 60% of patients (Mizuguchi et al. 2006). The mediators associated with these observations include IL-1β, IL-6 and TNFα (Tricot 2000), all pro-inflammatory cytokines also seen in the context of infection. In fact, the human hepatoma cell line HuH-7 (Vecchi et al. 2010) shows increased branching and SLex structures on the N-glycans of secreted α1-acid glycoprotein when treated with either IL-1β or IL-6 (Mackiewicz et al. 1992; Azuma et al. 2000), whereas treatment with TNFα increases the expression of the sialotransferase ST3GalIV and the fucosyltransferase FUTIV in a NFκB-dependent manner (Ishibashi et al. 2005, 2006; Higai et al. 2006). Furthermore, differences in tri- and tetra-antennary glycans are seen with chronic inflammation such that the longer the inflammatory process, the more change is seen in serum proteins. These observations have been reported for alcoholic liver disease (Mann et al. 1994; Gravel et al. 1996), breast and ovarian cancer (Goodarzi and Turner 1995; Turner et al. 1995; Saldova et al. 2007), rheumatoid arthritis (Thompson et al. 1989, 1993) and Crohn's disease (Goodarzi and Turner 1998). At a minimum, these studies reveal that the two central mediators of inflammation common to cancer, autoimmunity and infection (i.e. IL-1 and TNFα) can have a direct effect on host glycosylation, though these studies also lack any follow-up to determine how IL-1β and TNFα lead to glycome changes.
Fig. 4.

Sialyl-Lewisx (SLex) antigen (boxed) is a tetrasaccharide motif composed of a sialic acid, galactose, GlcNAc and fucose residue. This motif can be found in both asparagine (N)-linked complex glycans, “core 2” and “core 4” serine or threonine (O)-linked glycans, as well as in glycolipids. Examples of both protein-based glycans are shown.
Outside of influencing the host cellular machinery through the transcriptional regulation of Golgi enzymes or other pathways, a stronger case can be made for direct alterations of host glycans by microbe-expressed enzymes. The first, and by far most well known, is the neuraminidase family. We will highlight several salient points using Haemophilus influenzae as the prototypical example, but it should be noted that H. influenzae neuraminidase and medicinal inhibitors have been reviewed many times elsewhere (e.g. Grienke et al. 2012). Hemagglutinin is a viral protein that binds to sialic acid-containing glycans on target cell surface glycoproteins and glycolipids, thereby facilitating viral adherence to the cell. The viral-encoded neuraminidase is responsible for cleaving these sialic acids from the host molecules upon entry, enabling the detachment of viral particles into the cell. This cleavage event is critical for the viral lifecycle, since neuraminidase inhibition can be an effective anti-viral drug (Grienke et al. 2012), but the removal of host sialic acids can also change the face of the cell by eliminating the negatively charged portion of the glycocalyx on the infected cell (the potential impact of this will be reviewed in the next section on function).
Another example is the opposite of the neuraminidases: the sialyltransferase family. There have been many bacterial and fungal microbes described that encode α2,3-sialyltransferases in their genome, including Neisseria meningitidis (Gilbert et al. 1996), Pasteurella multocida (Thon et al. 2011), Campylobacter jejuni (Lee et al. 2011), H. influenzae (Hood et al. 2001; Fox et al. 2006), Streptococcus agalactiae (Group B strep; Chaffin et al. 2002) and Cryptococcus neoformans (Rodrigues et al. 2002). In general, it is thought that addition of terminal sialic acids on microbial surfaces by these enzymes aids in colonization and immune evasion via molecular mimicry and there is little evidence that suggests that these enzymes are secreted and/or might affect host glycoconjugates, although this remains a possibility. However, there is at least one notable exception: the trans-sialidase expressed on the surface of Trypanosoma cruzi and Trypanosoma brucei (reviewed in Cross and Takle 1993). This enzyme has been shown to transfer sialic acid from host cell surface and soluble glycoconjugates to the trypanosome glycoconjugates, thereby altering both the host through the removal of terminal sialic acids and microbe's glycome through the addition of sialic acids at the parasite surface (Ferrero-Garcia et al. 1993). The modification of the microbial carbohydrates with sialic acid taken from the host presumably allows the microbe to hide from the immune response through mimicry as mentioned earlier, but it remains unclear what effect, if any, the removal of sialic acids from the host does to the immune response in the context of trypanosome infection.
On the other hand, viral sialyltransferases are far more likely to impact host proteins during their lifecycle because of the requirement for the host protein synthesis machinery during replication. The Myxoma virus is a rabbit pathogen of the Poxviridae family which expresses an α2,3-sialyltransferase (Jackson et al. 1999). Although the function of this enzyme is not well described, it is clear that it is important for virulence but not viral replication (Jackson et al. 1999). The substrate specificity of this enzyme shows that it is likely to affect host glycan structures (Sujino et al. 2000), although this has not yet been explored. Another instance of viral-encoded glycan-modifying enzymes is the Bo17 gene in bovine herpesvirus 4, which encodes a homolog of the core 2 β-1,6-GlcNAc transferase (β1,6GnT) enzyme, which adds GlcNAc sugars to the GalNAc core of O-linked glycans (Markine-Goriaynoff et al. 2004; Figure 2). This enzyme is believed to play an important role for the structural proteins of the virion, but could also impact the infected cells through alterations in antibody or complement-mediated lysis. This possibility and others can be proposed based on studies of host-encoded β1,6GnTs (Brockhausen 1999; Tsuboi and Fukuda 2001; Fukuda 2002). More specifically, the overexpression of GlcNAc transferase activity and the concomitant increased core 2 oligosaccharides is known to decrease interactions between cells, particularly those of the immune system (Fukuda and Tsuboi 1999), possibly providing a mechanism of immune evasion for HSV-infected cells. However, no studies have yet described whether the Bo17 gene can fulfill this role or not.
Outside of altering the enzyme complement of a given glycosylation pathway, there are other ways in which a pathogen can actively alter the host glycans. One interesting case is exemplified by Old World arenavirus infections, such as lymphocytic choriomeningitis virus and Lassa fever virus. Studies have revealed that viral glycoprotein associates with LARGE, a GlcNAc transferase found in the Golgi Apparatus (Peyrard et al. 1999). This binding sequesters LARGE from acting upon other substrates, thereby altering the glycosylation pattern of host molecules, particularly α-dystroglycan. These alterations perturb laminin binding and lead to altered cell–matrix interactions in the host (Rojek et al. 2007).
We acknowledge that the data for microbial-influenced glycome manipulation remain highly descriptive and lack mechanism, but the evidence is strong for microbes making an impact on host glycosylation. Likewise, it is clear that in disease-associated inflammation (e.g. autoimmunity, cancer etc.), changes in the glycome follow as a function of the inflammatory mediators like TNFα and IL-1β, which are also present during many (most?) infections. It appears that the investigators interested in glycosylation and those interested in pathogenesis are not commonly the same individuals; thus, we propose that the effect of microbial exposure on the host glycome is vastly understudied and is an area of research ripe with opportunity. But before we more specifically describe our proposed paradigm, we must first deal with glycan functionality in the immune system.
Glycome changes lead to molecular and cellular functional differences
It is reasonable to review the correlation of infection/inflammation with the glycome, but in the absence of functional consequences associated with those changes, the point is moot. Unfortunately, none of the referenced findings above reported even an attempt to explain what changes in biology might accompany the observed host glycosylation differences, so we must move to another set of findings in the literature on the role glycans play in the function of the immune system to make our argument.
Simply based on the fact that the ablation of glycosylation-related genes leads to immune pathology, as mentioned earlier, it logically follows that the function of at least some glycoproteins and glycolipids is altered when the glycans change, otherwise defects would not occur. The fucosylation and Mgat5 stories represent two clear examples of where glycan changes alter the function of the underlying protein, in those cases Notch and the TCR, respectively, but there are many more to support the notion that the nature and composition of protein glycosylation directly impacts function in ways that are grossly underappreciated by the general research community. Furthermore, it is important to differentiate between studies where sites of glycosylation are removed by mutagenesis and studies where the “nature of the attached glycan” changes. Although the removal of a glycosylation site from a protein may arise due to genetic mutation associated with cancer or other insults, this event is not biologically equivalent and rare in vivo compared with the actively regulated changes in glycan composition that are in focus here. Our emphasis is how changes in glycan composition affect the function of the glycoprotein as a whole.
The best studied and highly regulated glycan change known to alter function is sialylation. The fundamental biology of sialic acids have been reviewed previously (Troy 1992; Vimr et al. 2004; Severi et al. 2007; Lewis et al. 2009; Schauer 2009; Schauer et al. 2011), but the central theme is that sialylation tends to alter a molecule's binding partners and/or affinity for a given ligand. This can take more forms than can be adequately covered here, so we will limit the discussion to a few canonical and recent immune system-specific findings that will serve as the basis for our unifying hypothesis. It might also strike the reader that sialylation is the glycosylation step most obviously manipulated by microbes (e.g. by microbial neuraminidases, sialyltransferases and trans-sialidases), as discussed in the previous section.
One of the more established examples within the immunological setting is the impact of sialylation upon the synthesis of selectin ligands (Schauer 2009). As already mentioned, selectins require the negative charge provided by terminal 2,3-linked sialic acids on cell surface molecules like P-selectin glycoprotein ligand-1 (PSGL-1) and mucin-like glycoproteins (Cummings and Smith 1992; Ellies et al. 2002; Lowe 2003). Removal of these residues through mutation or neuraminidase treatment alters the selectin-dependent cellular homing properties such that pathogens can evade the brunt of the immune response. In this sense, the presence or the absence of sialic acids alters the function of PSGL-1 (for example) in that it can no longer bind well to the necessary selectin to promote appropriate leukocyte homing to sites of infection and inflammation. Indeed, this provides a nice example whereby a microbe-encoded neuraminidase could dramatically impact the host's immune response through the direct enzymatic modulation of selectin ligands.
A somewhat more esoteric example is CD22/Siglec-2. This B cell-specific surface glycoprotein is a member of the B cell receptor complex and is now recognized to be a sialic acid-binding lectin (O'Reilly et al. 2011). What is particularly remarkable about CD22 is that it binds to as of yet unidentified ligands (possibly in cis and trans orientations) carrying α2,6-linked sialic acids, rather than the more common α2,3-linked sialic acids (Grewal et al. 2006). To date, it is unknown whether specific molecules carrying such glycan structures are critical, or whether any specific glycoprotein or glycolipid with 2,6-linked sialic acids are particularly important; however, the ablation of the lone enzyme that creates this linkage (ST6Gal1) creates significant B cell defects that include suppressed B cell receptor signaling, reduced serum IgM and reduced antibody responses to both T cell-dependent and T cell-independent antigens (Hennet et al. 1998). Remarkably, removing CD22 from the ST6Gal1 knockout background normalized B cell function (Collins et al. 2006). This story provides evidence that unknown ligands carrying α2,6-linked sialic acids are necessary for proper B cell function via recognition by CD22, yet it remains a mystery as to the identity or nature of these ligands outside of the presence of the sialic acid-containing glycans. Likewise, the other members of the Siglec family are still described as “cellular adhesion” or “cellular interaction” molecules, and specific targets have not yet been fully explored, though it is clear that ligand activity is exquisitely sensitive to sialic acid content.
Although the CD22/Siglec and selectin data show that ligands and their interactions with key immune proteins are modulated by sialylation, there is evidence to show that the function of immune-associated glycoproteins themselves are altered by sialylation and glycan composition. Two examples, where the functional differences are largely attributed to the galectin family of molecules, can be found in studies of CD45 and the TCR complex. For both of these systems, it is important to note that galectins bind to LacNAc disaccharide units in N- and O-linked glycans, but this binding can be strongly inhibited for some galectins by the presence of terminal sialic acids on those glycans (Liu and Rabinovich 2010). As such, the TCR complex could be an example for how the addition or the removal of terminal sialic acids on the glycans decorating the TCR might be used to modulate T cell responsiveness. More specifically, the increased expression of the appropriate sialyltransferase in a T cell leading to increased TCR sialylation could conceivably reduce the formation of the galectin-TCR lattice at the cell surface through the inhibition of galectin binding, thereby mimicking the loss of Mgat5 and the associated reduction in TCR threshold discussed earlier (Demetriou et al. 2001). This could be equally true in the reverse, with decreased sialylation leading to increased threshold for signaling. As a result, the level of sialylation and the presence of the LacNAc motif on the TCR glycans could potentially act as a rheostat for T cell activation via galectin-mediated effects, which makes this pathway an attractive target for pathogens seeking to evade the immune response.
CD45 serves as another prominent example of the galectin-mediated glycosylation-dependent functional effect. While this has been reviewed in detail recently (Earl and Baum 2008), several salient points are of specific interest in terms of the present line of reasoning. CD45 is a cell surface receptor expressed in hematopoietic cells in a number of isoforms (RA, RB, RBC, RABC and RO) and is a critical factor in cellular development, activation and cell death through its tyrosine phosphatase activity (Hermiston et al. 2003). All isoforms of CD45 include both O- (primarily core 1 and core 2) and complex N-linked glycans. Importantly, during T cell development and within the various T cell lineages (memory, activated, naïve etc.), the glycosylation of CD45 and other cell surface glycoproteins changes in a way that is characteristic for each lineage (Earl and Baum 2008). For example, Th2 cells express ST6Gal1, whereas Th1 cells do not, resulting in Th2 cells (but not Th1 cells) carrying CD45 with α2,6-linked sialic acids (Toscano et al. 2007). This is a critical observation because of the impact of sialylation on gal-1 binding to CD45. Without the terminal sialic acids, gal-1 binds and cross-links CD45, whereas the presence of terminal sialic acids inhibits this interaction (Earl et al. 2010). Gal-1-mediated cross-linking of CD45 can have a number of effects, depending on the context, but often leads to the apoptosis of Th1 and Th17 cells which lack terminal 2,6-linked sialic acids on CD45, whereas Th2 cells are protected from this effect due to the sialic acids on surface glycans (Toscano et al. 2007). In a biological setting, gal-1 can skew the cytokine response of T cells toward Th2, possibly through the cell death pathway of Th1 cells. Furthermore, changes in CD45 and CD43 glycosylation, specifically sialylation, are associated with aging and the age-related reduction in CD4+ T cell responsiveness (Abdul-Salam et al. 2000). In total, it is now clear that the active modulation of CD45 glycosylation in T cells is a galectin-dependent regulatory pathway during T cell development and apoptosis.
Outside of the galectin effects that almost certainly include many other cell surface receptor molecules not yet identified or studied, it has been recently discovered that antibodies themselves depend on glycosylation for the determination of their function. Textbooks teach that the function of an antibody is determined by the constant (Fc) domain of antibodies (Abbas et al. 2000). The Fc domain carries a single but highly conserved site (asparagine 297) of N-linked glycosylation that has long been recognized as critical for antibody structural stability (Arnold et al. 2007). Although a few early studies hinted at this, a number of recent findings now reveal that the composition of the glycans at this site in the Fc domain has a dramatic impact on the overall function of antibodies. As early as 1987 (Roitt and Cooke 1987), it was found that IgG molecules isolated from patients with rheumatoid arthritis have galactose-deficient Fc domain glycans (Roitt et al. 1988, 1992; Bond et al. 1990). Since galactose is the target for ST6Gal1, these antibodies also must have lacked terminal 2,6-linked sialic acids on these structures. Indeed, this interpretation was only recently confirmed 2 years ago (van de Geijn et al. 2009). More recently, changes in Fc glycosylation were aligned with another autoimmune disease, Wegener's granulomatosis, where the level of 2,6-linked sialic acid-containing IgG molecules was significantly reduced (Espy et al. 2011).
These observations are highly significant in light of the findings that have focused upon the biological activity of IVIg, where it was shown that terminal 2,6-linked sialic acids decorating the IgG Fc glycan (2,6-sialyl-IgG) alter the Fc receptor affinity and therefore antibody function (Kaneko et al. 2006; Anthony, Nimmerjahn, et al. 2008; Anthony, Wermeling, et al. 2008; Anthony et al. 2011). This discovery was observed within the context of IVIg, which is a treatment approach used for over two decades as an effective means to suppress autoimmunity. This pioneering work has now shown that the anti-inflammatory activity of IVIg in autoimmune patients can be highly enriched by lectin affinity using Sambucus nigra lectin (SNA), which binds to 2,6-linked sialic acids. The enriched sialic acid-containing IgG pool showed a 100-fold increase in activity over IVIg by weight (Kaneko et al. 2006). In addition, the presence of 2,6-linked sialic acid shifted the affinities of the IgG molecules for various Fc receptors such that asialo-IgG bound tighter to the activating pro-inflammatory FcγRIII receptor whereas 2,6-sialyl-IgG preferentially associated with FcγRIIB (Kaneko et al. 2006; Anthony, Nimmerjahn, et al. 2008; Anthony et al. 2011) as well as DC-SIGN (or SIGN-R1, the murine homolog; Wieland et al. 2007; Anthony, Wermeling, et al. 2008), both of which send inhibitory signals into responding cells. Thus, IgG molecules can be pro-inflammatory (asialo-IgG) or anti-inflammatory (2,6-sialyl-IgG), and this is exquisitely regulated by the glycan composition of the Fc domain.
The alignment of autoimmunity and aberrant antibody glycosylation was also recently seen in IgA molecules from autoimmune glomerulonephritis patients. IgA nephropathy is an autoimmune disease characterized by mesangial immunodeposits containing high concentrations of IgA1. Investigators have now discovered that the IgA1 from these deposits are deficient in galactose in the hinge-region O-glycans (Novak et al. 2011). This altered that IgA1 is bound by glycan-specific antibodies and seems to arise from plasma cells with aberrant glycosyltransferase expression. Although this appears to affect IgA through a different mechanism than described for IgG molecules earlier, these data further support the notion that glycosylation composition alters antibody function and fitness.
Finally, our own work on the MHCII-dependent presentation of bacterial polysaccharide “glycoantigens” has recently revealed that the nature of the N-linked glycans on MHCII modulates antigen-binding properties. We previously discovered that zwitterionic polysaccharides isolated from the capsules of commensal bacteria are processed and presented by MHCII to T cells for recognition and activation (Cobb et al. 2004; Cobb and Kasper 2008; Kreisman and Cobb 2011; Ryan et al. 2011). Through our attempts to better understand how these unusual glycoantigens associate with MHCII, we found that preventing the formation of complex-type N-glycans on MHCII using both pharmacologic and genetic approaches resulted in defects in both the amount of presented antigen at the cell surface as well as the overall T cell response to these antigens (Ryan et al. 2011). In vitro binding experiments with recombinant MHCII confirm that this presentation defect was due to a loss of interactions between glycoantigens and MHCII when the MHCII N-glycans were comprised of only high-mannose or hybrid structures. Moreover, mimicking the CDG-IIa in vitro through ablation of the Mgat2 locus, which encodes the GlcNAc transferase II enzyme responsible for initiating branched complex N-glycan synthesis (Figure 3), resulted in a lack of T cell response to commensal glycoantigens (Ryan et al. 2011). These data show that the function of MHCII and the nature of the presented antigens at the cell surface can be modulated or regulated via changes in the N-glycosylation pathway of antigen presenting cells.
Collectively, the data are strong in support the general conclusion that glycosylation impacts the function and binding interactions of immune proteins. In most cases, changes in glycosylation alter the interactions between various molecules, be they lectins that associate directly with the glycans or other ligands whose affinity changes for reasons that remain unclear (e.g. the glycoantigen binding to MHCII). The biophysics of how glycosylation alters this second group of examples is a major challenge to dissect since most structural work relies on crystallography, which often fails with fully glycosylated molecules and is further confounded by the natural heterogeneity of the system. Still, it is clear that glycosylation is linked to glycoprotein function.
A Glyco-Evasion Hypothesis
The data summarized here provide strong support for the three underlying components of our Glyco-Evasion Hypothesis: (i) inflammation and infection can lead to changes in host glycosylation; (ii) modulation of host glycosylation can lead to changes in host protein and cell function and (iii) changes in host protein and cell function through alterations in glycosylation can lead to inflammation, susceptibility to infection and overall immune dysfunction. Once we take the next logic step in our reasoning, the Glyco-Evasion Hypothesis readily emerges. If pathogens modulate host glycosylation (A → B) and host glycosylation modulates the immune response (B → C), then pathogens can modulate the immune response through control over host glycosylation (A → C). The Glyco-Evasion Hypothesis formally states that microbes manipulate host glycosylation to promote infection through glycan-mediated immune dysfunction.
In bringing together the findings highlighted in this review, one must acknowledge that any unifying hypothesis will contain evidentiary holes. In fact, there is no example we could find in the literature where a change in host glycosylation was associated with an infection and where the mechanism of that same change was determined and where the resulting functional differences that follow the glycosylation change were shown to benefit the pathogen. Although these types of observations exist independently (reviewed here), a unifying model is needed to systematically determine whether these events are truly linked or not. As such, our Glyco-Evasion Hypothesis is proposed in order to highlight a potential new and exciting avenue for investigation in our fight against infectious disease.
Within the representative examples in this review, the Glyco-Evasion Hypothesis can be applied to identify important gaps in our understanding. Consider the bovine herpesvirus 4 case where virally encoded core 2 GlcNAc transferase is expressed in infected cells (Markine-Goriaynoff et al. 2004). Studies unrelated to herpes demonstrate that the overexpression of core 2 GlcNAc transferase activity leads to increased core 2 glycans and a reduction in cell–cell interactions within the immune system (Fukuda and Tsuboi 1999). The Glyco-Evasion Hypothesis would therefore predict that the virus could evade immune attack through the expression of core 2 GlcNAc transferase activity, leading to increased core 2 glycans on host cells and decreased immune cell activation by limiting cell–cell adhesion.
Consider another example where we found that presentation of bacterial glycoantigens by MHCII proteins was remarkably sensitive to the composition of the branched complex N-glycans on MHCII (Ryan et al. 2011). We have also demonstrated that these antigens normally induce potent anti-inflammatory Treg responses that dampen inflammation and the adaptive immune system (Kreisman and Cobb 2011). Unrelated studies have revealed that inflammation (Mackiewicz et al. 1992; Azuma et al. 2000) and exposure to TLR agonists (Wang et al. 2011) are strongly correlated with branching changes (mostly increased branching) in host N-glycans. The Glyco-Evasion Hypothesis would therefore predict that pathogens carrying glycoantigens, such as Staphylococcus aureus (Tzianabos et al. 2001), which also stimulate the innate/inflammatory response through TLR activation (i.e. by S. aureus lipoteichoic acid; Takeuchi et al. 1999) could increase the MHCII-dependent presentation of bacterial glycoantigen by modulating MHCII N-glycan branching, thereby increasing the induction of inhibitory Tregs and reducing immune-mediated microbial killing.
As a final illustration, consider the CD45-galectin pathway where gal-1-mediated cross-linking of CD45 leads to T cell apoptosis when terminal sialic acids are lacking (Toscano et al. 2007). Th1 and Th17 cells are susceptible to this pathway, but Th2 cells are protected due to the presence of sialic acids on CD45 (Toscano et al. 2007). Studies demonstrate that many pathogens (e.g. H. influenzae) remove host sialic acids through the expression of neuraminidases (Grienke et al. 2012). The Glyco-Evasion Hypothesis would therefore predict that pathogens with the ability to remove host sialic acids could potentially evade Th2-mediated immunity by de-sialylating CD45 on responding T cells, thus promoting their apoptosis and ultimately microbial survival.
There are other conceivable scenarios in which a microbe may manipulate the immune response for their benefit through changes in host glycosylation, including the promotion of pathogen escape from leukocyte homing via alterations in selectin ligands, depletion of CD22 ligands to promote B cell dysfunction and many others. The three specific examples described earlier are provided to illustrate the potential impact of the pathway proposed by the Glyco-Evasion Hypothesis, while also revealing a number of investigative opportunities. How does inflammation and infection lead to glycome changes in the host? What are the functional consequences of those changes. Do the changes in host glycosylation mediated by microbial signals or enzymes lead to an advantage during infection? All of these issues are essentially unexplored, and it is imperative that we begin work to better understand the mechanisms by which host glycosylation is regulated. In addition, we must determine how glycosylation actually impacts the function of all glycoproteins and therefore all cells. Systems biology is not truly “systemic” without integrating the role of carbohydrates (and lipids) into the greater picture, and we must begin to embrace the heterogeneity inherent to the non-template-driven synthesis of these molecules and what it tells us about biological function. Thus, we readily acknowledge that the Glyco-Evasion Hypothesis provides far more questions than answers, but the concept is built upon many years of observations and we believe that it represents a new frontier in our fight against infectious disease through the application of glycoscience to investigations of infection and immunity.
Funding
Work in B.A. Cobb's laboratory is support by grants from the National Institute of General Medical Science (GM082916), the National Institutes of Health Office of the Director (OD004225) and the American Asthma Foundation.
Conflict of interest
None declared.
Abbreviations
α4GnT, α-1,4-GlcNAc transferase; β1,6GnT, β-1,6-GlcNAc transferase; CDG, congenital disorders of glycosylation; CF, cystic fibrosis; CFTR, cystic fibrosis transmembrane conductance regulator; CONV, conventional; gal-1, galectin-1; GF, germ-free; GlcNAc, N-acetylglucosamine: GlcNAc-TV, α-6-d-mannoside β1,6 N-GlcNAc transferase V; Man2a1, α-mannosidase IIa1; MHC, major histocompatibility complex; MS, multiple sclerosis; MYXV, Myxoma virus; PSGL-1, P-selectin glycoprotein ligand-1; SLE, systemic lupus erythematosus; SLex, sialyl Lewis X; SNA, Sambucus nigra lectin; TCR, T cell receptor; TNFα, tumor necrosis factor α.
References
- Abbas AK, Lichtman AH, Pober JS. Cellular and Molecular Immunology. 4th ed. 2000. W.B. Saunder Company, Philadelphia, PA. [Google Scholar]
- Abdul-Salam F, Moulana MG, Mansour MH. Age-related structural modulation of T lymphocyte-associated CD45 isoforms. Mech Ageing Dev. 2000;114:21–35. doi: 10.1016/s0047-6374(99)00114-1. doi:10.1016/S0047-6374(99)00114-1. [DOI] [PubMed] [Google Scholar]
- Anthony RM, Kobayashi T, Wermeling F, Ravetch JV. Intravenous gammaglobulin suppresses inflammation through a novel T(H)2 pathway. Nature. 2011;475:110–113. doi: 10.1038/nature10134. doi:10.1038/nature10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony RM, Nimmerjahn F, Ashline DJ, Reinhold VN, Paulson JC, Ravetch JV. Recapitulation of IVIG anti-inflammatory activity with a recombinant IgG Fc. Science. 2008;320:373–376. doi: 10.1126/science.1154315. doi:10.1126/science.1154315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony RM, Wermeling F, Karlsson MC, Ravetch JV. Identification of a receptor required for the anti-inflammatory activity of IVIG. Proc Natl Acad Sci USA. 2008;105:19571–19578. doi: 10.1073/pnas.0810163105. doi:10.1073/pnas.0810163105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold JN, Wormald MR, Sim RB, Rudd PM, Dwek RA. The impact of glycosylation on the biological function and structure of human immunoglobulins. Annu Rev Immunol. 2007;25:21–50. doi: 10.1146/annurev.immunol.25.022106.141702. doi:10.1146/annurev.immunol.25.022106.141702. [DOI] [PubMed] [Google Scholar]
- Azuma Y, Murata M, Matsumoto K. Alteration of sugar chains on α1-acid glycoprotein secreted following cytokine stimulation of HuH-7 cells in vitro. Clin Chim Acta. 2000;294:93–103. doi: 10.1016/s0009-8981(99)00248-x. doi:10.1016/S0009-8981(99)00248-X. [DOI] [PubMed] [Google Scholar]
- Berninsone PM, Hirschberg CB. Nucleotide sugar transporters of the Golgi apparatus. Curr Opin Struct Biol. 2000;10:542–547. doi: 10.1016/s0959-440x(00)00128-7. doi:10.1016/S0959-440X(00)00128-7. [DOI] [PubMed] [Google Scholar]
- Bond A, Cooke A, Hay FC. Glycosylation of IgG, immune complexes and IgG subclasses in the MRL-lpr/lpr mouse model of rheumatoid arthritis. Eur J Immunol. 1990;20:2229–2233. doi: 10.1002/eji.1830201011. doi:10.1002/eji.1830201011. [DOI] [PubMed] [Google Scholar]
- Brockhausen I. Pathways of O-glycan biosynthesis in cancer cells. Biochim Biophys Acta. 1999;1473:67–95. doi: 10.1016/s0304-4165(99)00170-1. doi:10.1016/S0304-4165(99)00170-1. [DOI] [PubMed] [Google Scholar]
- Brockhausen I, Schachter H, Stanley P. O-GalNAc Glycans. 2009. pp. 115–127. In: Varki A, Cummings RD, Esko JD, et al., editors. Essentials of Glycobiology, 2nd edition. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press. [PubMed] [Google Scholar]
- Brown SJ, Miller AM, Cowan PJ, Slavin J, Connell WR, Moore GT, Bell S, Elliott PR, Desmond PV, d'Apice AJ. Altered immune system glycosylation causes colitis in α1,2-fucosyltransferase transgenic mice. Inflamm Bowel Dis. 2004;10:546–556. doi: 10.1097/00054725-200409000-00008. doi:10.1097/00054725-200409000-00008. [DOI] [PubMed] [Google Scholar]
- Chaffin DO, McKinnon K, Rubens CE. CpsK of Streptococcus agalactiae exhibits α2,3-sialyltransferase activity in Haemophilus ducreyi. Mol Microbiol. 2002;45:109–122. doi: 10.1046/j.1365-2958.2002.02988.x. doi:10.1046/j.1365-2958.2002.02988.x. [DOI] [PubMed] [Google Scholar]
- Chen HL, Li CF, Grigorian A, Tian W, Demetriou M. T cell receptor signaling co-regulates multiple Golgi genes to enhance N-glycan branching. J Biol Chem. 2009;284:32454–32461. doi: 10.1074/jbc.M109.023630. doi:10.1074/jbc.M109.023630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chervenak R, Cohen JJ. Peanut lectin binding as a marker for activated T-lineage lymphocytes. Thymus. 1982;4:61–67. [PubMed] [Google Scholar]
- Chui D, Sellakumar G, Green R, Sutton-Smith M, McQuistan T, Marek K, Morris H, Dell A, Marth J. Genetic remodeling of protein glycosylation in vivo induces autoimmune disease. Proc Natl Acad Sci USA. 2001;98:1142–1147. doi: 10.1073/pnas.98.3.1142. doi:10.1073/pnas.98.3.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb BA, Kasper DL. Characteristics of carbohydrate antigen binding to the presentation protein HLA-DR. Glycobiology. 2008;18:707–718. doi: 10.1093/glycob/cwn050. doi:10.1093/glycob/cwn050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb BA, Wang Q, Tzianabos AO, Kasper DL. Polysaccharide processing and presentation by the MHCII pathway. Cell. 2004;117:677–687. doi: 10.016/j.cell.2004.05.001. doi:10.1016/j.cell.2004.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colley KJ. Golgi localization of glycosyltransferases: More questions than answers. Glycobiology. 1997;7:1–13. doi: 10.1093/glycob/7.1.1-b. doi:10.1093/glycob/7.1.1-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins BE, Smith BA, Bengtson P, Paulson JC. Ablation of CD22 in ligand-deficient mice restores B cell receptor signaling. Nat Immunol. 2006;7:199–206. doi: 10.1038/ni1283. doi:10.1038/ni1283. [DOI] [PubMed] [Google Scholar]
- Cooke CL, An HJ, Kim J, Canfield DR, Torres J, Lebrilla CB, Solnick JV. Modification of gastric mucin oligosaccharide expression in rhesus macaques after infection with Helicobacter pylori. Gastroenterology. 2009;137:1061–1071. doi: 10.1053/j.gastro.2009.04.014. doi:10.1053/j.gastro.2009.04.014. [DOI] [PubMed] [Google Scholar]
- Cross GA, Takle GB. The surface trans-sialidase family of Trypanosoma cruzi. Annu Rev Microbiol. 1993;47:385–411. doi: 10.1146/annurev.mi.47.100193.002125. doi:10.1146/annurev.mi.47.100193.002125. [DOI] [PubMed] [Google Scholar]
- Cummings RD, Smith DF. The selectin family of carbohydrate-binding proteins: Structure and importance of carbohydrate ligands for cell adhesion. Bioessays. 1992;14:849–856. doi: 10.1002/bies.950141210. doi:10.1002/bies.950141210. [DOI] [PubMed] [Google Scholar]
- Davicino RC, Elicabe RJ, Di Genaro MS, Rabinovich GA. Coupling pathogen recognition to innate immunity through glycan-dependent mechanisms. Int Immunopharmacol. 2011;11:1457–1463. doi: 10.1016/j.intimp.2011.05.002. doi:10.1016/j.intimp.2011.05.002. [DOI] [PubMed] [Google Scholar]
- Demetriou M, Granovsky M, Quaggin S, Dennis JW. Negative regulation of T-cell activation and autoimmunity by Mgat5N-glycosylation. Nature. 2001;409:733–739. doi: 10.1038/35055582. doi:10.1038/35055582. [DOI] [PubMed] [Google Scholar]
- Earl LA, Baum LG. CD45 glycosylation controls T-cell life and death. Immunol Cell Biol. 2008;86:608–615. doi: 10.1038/icb.2008.46. doi:10.1038/icb.2008.46. [DOI] [PubMed] [Google Scholar]
- Earl LA, Bi S, Baum LG. N- and O-glycans modulate galectin-1 binding, CD45 signaling, and T cell death. J Biol Chem. 2010;285:2232–2244. doi: 10.1074/jbc.M109.066191. doi:10.1074/jbc.M109.066191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellies LG, Sperandio M, Underhill GH, Yousif J, Smith M, Priatel JJ, Kansas GS, Ley K, Marth JD. Sialyltransferase specificity in selectin ligand formation. Blood. 2002;100:3618–3625. doi: 10.1182/blood-2002-04-1007. doi:10.1182/blood-2002-04-1007. [DOI] [PubMed] [Google Scholar]
- Ellies LG, Tsuboi S, Petryniak B, Lowe JB, Fukuda M, Marth JD. Core 2 oligosaccharide biosynthesis distinguishes between selectin ligands essential for leukocyte homing and inflammation. Immunity. 1998;9:881–890. doi: 10.1016/s1074-7613(00)80653-6. doi:10.1016/S1074-7613(00)80653-6. [DOI] [PubMed] [Google Scholar]
- Espy C, Morelle W, Kavian N, Grange P, Goulvestre C, Viallon V, Chereau C, Pagnoux C, Michalski JC, Guillevin L, et al. Sialylation levels of anti-proteinase 3 antibodies are associated with the activity of granulomatosis with polyangiitis (Wegener's) Arthritis Rheum. 2011;63:2105–2115. doi: 10.1002/art.30362. doi:10.1002/art.30362. [DOI] [PubMed] [Google Scholar]
- Ferrero-Garcia MA, Trombetta SE, Sanchez DO, Reglero A, Frasch AC, Parodi AJ. The action of Trypanosoma cruzi trans-sialidase on glycolipids and glycoproteins. Eur J Biochem. 1993;213:765–771. doi: 10.1111/j.1432-1033.1993.tb17818.x. doi:10.1111/j.1432-1033.1993.tb17818.x. [DOI] [PubMed] [Google Scholar]
- Fox KL, Cox AD, Gilbert M, Wakarchuk WW, Li J, Makepeace K, Richards JC, Moxon ER, Hood DW. Identification of a bifunctional lipopolysaccharide sialyltransferase in Haemophilus influenzae: Incorporation of disialic acid. J Biol Chem. 2006;281:40024–40032. doi: 10.1074/jbc.M602314200. doi:10.1074/jbc.M602314200. [DOI] [PubMed] [Google Scholar]
- Freeze HH, Ng BG. Golgi glycosylation and human inherited diseases. Cold Spring Harb Perspect Biol. 2011;3:a005371. doi: 10.1101/cshperspect.a005371. doi:10.1101/cshperspect.a005371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M. Roles of mucin-type O-glycans in cell adhesion. Biochim Biophys Acta. 2002;1573:394–405. doi: 10.1016/s0304-4165(02)00409-9. doi:10.1016/S0304-4165(02)00409-9. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Tsuboi S. Mucin-type O-glycans and leukosialin. Biochim Biophys Acta. 1999;1455:205–217. doi: 10.1016/s0925-4439(99)00067-8. [DOI] [PubMed] [Google Scholar]
- Galvan M, Murali-Krishna K, Ming LL, Baum L, Ahmed R. Alterations in cell surface carbohydrates on T cells from virally infected mice can distinguish effector/memory CD8+ T cells from naive cells. J Immunol. 1998;161:641–648. [PubMed] [Google Scholar]
- Gilbert M, Watson DC, Cunningham AM, Jennings MP, Young NM, Wakarchuk WW. Cloning of the lipooligosaccharide α-\2,3-sialyltransferase from the bacterial pathogens Neisseria meningitidis and Neisseria gonorrhoeae. J Biol Chem. 1996;271:28271–28276. doi: 10.1074/jbc.271.45.28271. doi:10.1074/jbc.271.45.28271. [DOI] [PubMed] [Google Scholar]
- Goodarzi MT, Turner GA. Decreased branching, increased fucosylation and changed sialylation of alpha-1-proteinase inhibitor in breast and ovarian cancer. Clin Chim Acta. 1995;236:161–171. doi: 10.1016/0009-8981(95)06049-j. doi:10.1016/0009-8981(95)06049-J. [DOI] [PubMed] [Google Scholar]
- Goodarzi MT, Turner GA. Reproducible and sensitive determination of charged oligosaccharides from haptoglobin by PNGase F digestion and HPAEC/PAD analysis: Glycan composition varies with disease. Glycoconj J. 1998;15:469–475. doi: 10.1023/a:1006930902625. doi:10.1023/A:1006930902625. [DOI] [PubMed] [Google Scholar]
- Gravel P, Walzer C, Aubry C, Balant LP, Yersin B, Hochstrasser DF, Guimon J. New alterations of serum glycoproteins in alcoholic and cirrhotic patients revealed by high resolution two-dimensional gel electrophoresis. Biochem Biophys Res Commun. 1996;220:78–85. doi: 10.1006/bbrc.1996.0360. doi:10.1006/bbrc.1996.0360. [DOI] [PubMed] [Google Scholar]
- Green RS, Stone EL, Tenno M, Lehtonen E, Farquhar MG, Marth JD. Mammalian N-glycan branching protects against innate immune self-recognition and inflammation in autoimmune disease pathogenesis. Immunity. 2007;27:308–320. doi: 10.1016/j.immuni.2007.06.008. doi:10.1016/j.immuni.2007.06.008. [DOI] [PubMed] [Google Scholar]
- Grewal PK, Boton M, Ramirez K, Collins BE, Saito A, Green RS, Ohtsubo K, Chui D, Marth JD. ST6Gal-I restrains CD22-dependent antigen receptor endocytosis and Shp-1 recruitment in normal and pathogenic immune signaling. Mol Cell Biol. 2006;26:4970–4981. doi: 10.1128/MCB.00308-06. doi:10.1128/MCB.00308-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grienke U, Schmidtke M, von GS, Kirchmair J, Liedl KR, Rollinger JM. Influenza neuraminidase: A druggable target for natural products. Nat Prod Rep. 2012;29:11–36. doi: 10.1039/c1np00053e. doi:10.1039/c1np00053e. [DOI] [PubMed] [Google Scholar]
- Hennet T, Chui D, Paulson JC, Marth JD. Immune regulation by the ST6Gal sialyltransferase. Proc Natl Acad Sci USA. 1998;95:4504–4509. doi: 10.1073/pnas.95.8.4504. doi:10.1073/pnas.95.8.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermiston ML, Xu Z, Weiss A. CD45: A critical regulator of signaling thresholds in immune cells. Annu Rev Immunol. 2003;21:107–137. doi: 10.1146/annurev.immunol.21.120601.140946. doi:10.1146/annurev.immunol.21.120601.140946. [DOI] [PubMed] [Google Scholar]
- Herscovics A. Importance of glycosidases in mammalian glycoprotein biosynthesis. Biochim Biophys Acta. 1999;1473:96–107. doi: 10.1016/s0304-4165(99)00171-3. doi:10.1016/S0304-4165(99)00171-3. [DOI] [PubMed] [Google Scholar]
- Higai K, Miyazaki N, Azuma Y, Matsumoto K. Interleukin-1beta induces sialyl Lewis X on hepatocellular carcinoma HuH-7 cells via enhanced expression of ST3Gal IV and FUT VI gene. FEBS Lett. 2006;580:6069–6075. doi: 10.1016/j.febslet.2006.09.073. doi:10.1016/j.febslet.2006.09.073. [DOI] [PubMed] [Google Scholar]
- Hood DW, Cox AD, Gilbert M, Makepeace K, Walsh S, Deadman ME, Cody A, Martin A, Mansson M, Schweda EK, et al. Identification of a lipopolysaccharide α-2,3-sialyltransferase from Haemophilus influenzae. Mol Microbiol. 2001;39:341–350. doi: 10.1046/j.1365-2958.2001.02204.x. doi:10.1046/j.1365-2958.2001.02204.x. [DOI] [PubMed] [Google Scholar]
- Ishibashi Y, Imai S, Inouye Y, Okano T, Taniguchi A. Effects of carbocisteine on sialyl-Lewis x expression in an airway carcinoma cell line stimulated with tumor necrosis factor-α. Eur J Pharmacol. 2006;530:223–228. doi: 10.1016/j.ejphar.2005.11.017. doi:10.1016/j.ejphar.2005.11.017. [DOI] [PubMed] [Google Scholar]
- Ishibashi Y, Inouye Y, Okano T, Taniguchi A. Regulation of sialyl-Lewis x epitope expression by TNF-alpha and EGF in an airway carcinoma cell line. Glycoconj J. 2005;22:53–62. doi: 10.1007/s10719-005-0292-7. doi:10.1007/s10719-005-0292-7. [DOI] [PubMed] [Google Scholar]
- Jackson RJ, Hall DF, Kerr PJ. Myxoma virus encodes an α2,3-sialyltransferase that enhances virulence. J Virol. 1999;73:2376–2384. doi: 10.1128/jvi.73.3.2376-2384.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeken J. Congenital disorders of glycosylation. Ann N Y Acad Sci. 2010;1214:190–198. doi: 10.1111/j.1749-6632.2010.05840.x. doi:10.1111/j.1749-6632.2010.05840.x. [DOI] [PubMed] [Google Scholar]
- Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science. 2006;313:670–673. doi: 10.1126/science.1129594. doi:10.1126/science.1129594. [DOI] [PubMed] [Google Scholar]
- Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem. 1985;54:631–664. doi: 10.1146/annurev.bi.54.070185.003215. doi:10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- Kreisman LS, Cobb BA. Glycoantigens induce human peripheral Tr1 cell differentiation with gut-homing specialization. J Biol Chem. 2011;286:8810–8818. doi: 10.1074/jbc.M110.206011. doi:10.1074/jbc.M110.206011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Lairson LL, Rich JR, Lameignere E, Wakarchuk WW, Withers SG, Strynadka NC. Structural and kinetic analysis of substrate binding to the sialyltransferase Cst-II from Campylobacter jejuni. J Biol Chem. 2011;286:35922–35932. doi: 10.1074/jbc.M111.261172. doi:10.1074/jbc.M111.261172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leir SH, Parry S, Palmai-Pallag T, Evans J, Morris HR, Dell A, Harris A. Mucin glycosylation and sulphation in airway epithelial cells is not influenced by cystic fibrosis transmembrane conductance regulator expression. Am J Respir Cell Mol Biol. 2005;32:453–461. doi: 10.1165/rcmb.2004-0306OC. doi:10.1165/rcmb.2004-0306OC. [DOI] [PubMed] [Google Scholar]
- Lewis AL, Desa N, Hansen EE, Knirel YA, Gordon JI, Gagneux P, Nizet V, Varki A. Innovations in host and microbial sialic acid biosynthesis revealed by phylogenomic prediction of nonulosonic acid structure. Proc Natl Acad Sci USA. 2009;106:13552–13557. doi: 10.1073/pnas.0902431106. doi:10.1073/pnas.0902431106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden S, Semino-Mora C, Liu H, Rick J, Dubois A. Role of mucin Lewis status in resistance to Helicobacter pylori infection in pediatric patients. Helicobacter. 2010;15:251–258. doi: 10.1111/j.1523-5378.2010.00765.x. doi:10.1111/j.1523-5378.2010.00765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu FT, Rabinovich GA. Galectins: Regulators of acute and chronic inflammation. Ann N Y Acad Sci. 2010;1183:158–182. doi: 10.1111/j.1749-6632.2009.05131.x. doi:10.1111/j.1749-6632.2009.05131.x. [DOI] [PubMed] [Google Scholar]
- Lowe JB. Glycan-dependent leukocyte adhesion and recruitment in inflammation. Curr Opin Cell Biol. 2003;15:531–538. doi: 10.1016/j.ceb.2003.08.002. doi:10.1016/j.ceb.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Mackiewicz A, Rose-John S, Schooltink H, Laciak M, Gorny A, Heinrich PC. Soluble human interleukin-6-receptor modulates interleukin-6-dependent N-glycosylation of α1-protease inhibitor secreted by HepG2 cells. FEBS Lett. 1992;306:257–261. doi: 10.1016/0014-5793(92)81012-b. doi:10.1016/0014-5793(92)81012-B. [DOI] [PubMed] [Google Scholar]
- Magalhaes A, Reis CA. Helicobacter pylori adhesion to gastric epithelial cells is mediated by glycan receptors. Braz J Med Biol Res. 2010;43:611–618. doi: 10.1590/s0100-879x2010007500049. doi:10.1590/S0100-879X2010007500049. [DOI] [PubMed] [Google Scholar]
- Mahdavi J, Sonden B, Hurtig M, Olfat FO, Forsberg L, Roche N, Angstrom J, Larsson T, Teneberg S, Karlsson KA, et al. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science. 2002;297:573–578. doi: 10.1126/science.1069076. doi:10.1126/science.1069076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann AC, Record CO, Self CH, Turner GA. Monosaccharide composition of haptoglobin in liver diseases and alcohol abuse: Large changes in glycosylation associated with alcoholic liver disease. Clin Chim Acta. 1994;227:69–78. doi: 10.1016/0009-8981(94)90136-8. doi:10.1016/0009-8981(94)90136-8. [DOI] [PubMed] [Google Scholar]
- Marek KW, Vijay IK, Marth JD. A recessive deletion in the GlcNAc-1-phosphotransferase gene results in peri-implantation embryonic lethality. Glycobiology. 1999;9:1263–1271. doi: 10.1093/glycob/9.11.1263. doi:10.1093/glycob/9.11.1263. [DOI] [PubMed] [Google Scholar]
- Markine-Goriaynoff N, Gillet L, Karlsen OA, Haarr L, Minner F, Pastoret PP, Fukuda M, Vanderplasschen A. The core 2 β-1,6-N-acetylglucosaminyltransferase-M encoded by bovine herpesvirus 4 is not essential for virus replication despite contributing to post-translational modifications of structural proteins. J Gen Virol. 2004;85:355–367. doi: 10.1099/vir.0.19715-0. doi:10.1099/vir.0.19715-0. [DOI] [PubMed] [Google Scholar]
- Martino AT, Mueller C, Braag S, Cruz PE, Campbell-Thompson M, Jin S, Flotte TR. N-glycosylation augmentation of the cystic fibrosis epithelium improves Pseudomonas aeruginosa clearance. Am J Respir Cell Mol Biol. 2011;44:824–830. doi: 10.1165/rcmb.2009-0285OC. doi:10.1165/rcmb.2009-0285OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzwa M, Ota H, Hayama M, Zhang MX, Sano K, Honda T, Ueno I, Akamatsu T, Nakayama J. Helicobacter pylori infection up-regulates gland mucous cell-type mucins in gastric pyloric mucosa. Helicobacter. 2003;8:594–600. doi: 10.1111/j.1523-5378.2003.00185.x. doi:10.1111/j.1523-5378.2003.00185.x. [DOI] [PubMed] [Google Scholar]
- Mizuguchi S, Inoue K, Iwata T, Nishida T, Izumi N, Tsukioka T, Nishiyama N, Uenishi T, Suehiro S. High serum concentrations of Sialyl Lewisx predict multilevel N2 disease in non-small-cell lung cancer. Ann Surg Oncol. 2006;13:1010–1018. doi: 10.1245/ASO.2006.05.018. doi:10.1245/ASO.2006.05.018. [DOI] [PubMed] [Google Scholar]
- Mizuguchi S, Nishiyama N, Iwata T, Nishida T, Izumi N, Tsukioka T, Inoue K, Kameyama M, Suehiro S. Clinical value of serum cytokeratin 19 fragment and sialyl-Lewis x in non-small cell lung cancer. Ann Thorac Surg. 2007;83:216–221. doi: 10.1016/j.athoracsur.2006.08.042. doi:10.1016/j.athoracsur.2006.08.042. [DOI] [PubMed] [Google Scholar]
- Mizuguchi S, Nishiyama N, Iwata T, Nishida T, Izumi N, Tsukioka T, Inoue K, Uenishi T, Wakasa K, Suehiro S. Serum Sialyl Lewis x and cytokeratin 19 fragment as predictive factors for recurrence in patients with stage I non-small cell lung cancer. Lung Cancer. 2007;58:369–375. doi: 10.1016/j.lungcan.2007.07.002. doi:10.1016/j.lungcan.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Mkhikian H, Grigorian A, Li CF, Chen HL, Newton B, Zhou RW, Beeton C, Torossian S, Tatarian GG, Lee SU, et al. Genetics and the environment converge to dysregulate N-glycosylation in multiple sclerosis. Nat Commun. 2011;2:334. doi: 10.1038/ncomms1333. doi:10.1038/ncomms1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran AP, Gupta A, Joshi L. Sweet-talk: Role of host glycosylation in bacterial pathogenesis of the gastrointestinal tract. Gut. 2011;60:1412–1425. doi: 10.1136/gut.2010.212704. doi:10.1136/gut.2010.212704. [DOI] [PubMed] [Google Scholar]
- Morgan R, Gao G, Pawling J, Dennis JW, Demetriou M, Li B. N-acetylglucosaminyltransferase V (Mgat5)-mediated N-glycosylation negatively regulates Th1 cytokine production by T cells. J Immunol. 2004;173:7200–7208. doi: 10.4049/jimmunol.173.12.7200. [DOI] [PubMed] [Google Scholar]
- Nanthakumar NN, Dai D, Newburg DS, Walker WA. The role of indigenous microflora in the development of murine intestinal fucosyl- and sialyltransferases. FASEB J. 2003;17:44–46. doi: 10.1096/fj.02-0031fje. [DOI] [PubMed] [Google Scholar]
- Novak J, Moldoveanu Z, Julian BA, Raska M, Wyatt RJ, Suzuki Y, Tomino Y, Gharavi AG, Mestecky J, Suzuki H. Aberrant glycosylation of IgA1 and anti-glycan antibodies in IgA nephropathy: Role of mucosal immune system. Adv Otorhinolaryngol. 2011;72:60–63. doi: 10.1159/000324607. [DOI] [PubMed] [Google Scholar]
- Opat AS, van VC, Gleeson PA. Trafficking and localisation of resident Golgi glycosylation enzymes. Biochimie. 2001;83:763–773. doi: 10.1016/s0300-9084(01)01312-8. doi:10.1016/S0300-9084(01)01312-8. [DOI] [PubMed] [Google Scholar]
- O'Reilly MK, Tian H, Paulson JC. CD22 is a recycling receptor that can shuttle cargo between the cell surface and endosomal compartments of B cells. J Immunol. 2011;186:1554–1563. doi: 10.4049/jimmunol.1003005. doi:10.4049/jimmunol.1003005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacchiarotta T, Hensbergen PJ, Wuhrer M, van NC, Nevedomskaya E, Derks RJ, Schoenmaker B, Koeleman CA, van DJ, Deelder AM, et al. Fibrinogen alpha chain O-glycopeptides as possible markers of urinary tract infection. J Proteomics. 2012;75:1067–1073. doi: 10.1016/j.jprot.2011.10.021. [DOI] [PubMed] [Google Scholar]
- Peyrard M, Seroussi E, Sandberg-Nordqvist AC, Xie YG, Han FY, Fransson I, Collins J, Dunham I, Kost-Alimova M, Imreh S, et al. The human LARGE gene from 22q12.3-q13.1 is a new, distinct member of the glycosyltransferase gene family. Proc Natl Acad Sci USA. 1999;96:598–603. doi: 10.1073/pnas.96.2.598. doi:10.1073/pnas.96.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piller F, Piller V, Fox RI, Fukuda M. Human T-lymphocyte activation is associated with changes in O-glycan biosynthesis. J Biol Chem. 1988;263:15146–15150. [PubMed] [Google Scholar]
- Poschet JF, Boucher JC, Tatterson L, Skidmore J, Van Dyke RW, Deretic V. Molecular basis for defective glycosylation and Pseudomonas pathogenesis in cystic fibrosis lung. Proc Natl Acad Sci USA. 2001;98:13972–13977. doi: 10.1073/pnas.241182598. doi:10.1073/pnas.241182598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priatel JJ, Chui D, Hiraoka N, Simmons CJ, Richardson KB, Page DM, Fukuda M, Varki NM, Marth JD. The ST3Gal-I sialyltransferase controls CD8+ T lymphocyte homeostasis by modulating O-glycan biosynthesis. Immunity. 2000;12:273–283. doi: 10.1016/s1074-7613(00)80180-6. doi:10.1016/S1074-7613(00)80180-6. [DOI] [PubMed] [Google Scholar]
- Rodrigues ML, Dobroff AS, Couceiro JN, Alviano CS, Schauer R, Travassos LR. Sialylglycoconjugates and sialyltransferase activity in the fungus Cryptococcus neoformans. Glycoconj J. 2002;19:165–173. doi: 10.1023/A:1024245606607. doi:10.1023/A:1024245606607. [DOI] [PubMed] [Google Scholar]
- Roitt IM, Cooke A. The role of autoantigen in autoimmunity. Immunol Lett. 1987;16:259–263. doi: 10.1016/0165-2478(87)90155-6. doi:10.1016/0165-2478(87)90155-6. [DOI] [PubMed] [Google Scholar]
- Roitt IM, Dwek RA, Parekh RB, Rademacher TW, Alavi A, Axford J, Bodman K, Bond A, Cooke A, Hay FC, et al. Changes in carbohydrate structure of IgG in rheumatoid arthritis. Recenti Prog Med. 1988;79:314–317. [PubMed] [Google Scholar]
- Roitt IM, Hutchings PR, Dawe KI, Sumar N, Bodman KB, Cooke A. The forces driving autoimmune disease. J Autoimmun. 1992;5(Suppl A):11–26. doi: 10.1016/0896-8411(92)90015-i. doi:10.1016/0896-8411(92)90015-I. [DOI] [PubMed] [Google Scholar]
- Rojek JM, Campbell KP, Oldstone MB, Kunz S. Old World arenavirus infection interferes with the expression of functional α-dystroglycan in the host cell. Mol Biol Cell. 2007;18:4493–4507. doi: 10.1091/mbc.E07-04-0374. doi:10.1091/mbc.E07-04-0374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen SD. Ligands for L-selectin: Homing, inflammation, and beyond. Annu Rev Immunol. 2004;22:129–156. doi: 10.1146/annurev.immunol.21.090501.080131. doi:10.1146/annurev.immunol.21.090501.080131. [DOI] [PubMed] [Google Scholar]
- Rudd PM, Elliott T, Cresswell P, Wilson IA, Dwek RA. Glycosylation and the immune system. Science. 2001;291:2370–2376. doi: 10.1126/science.291.5512.2370. doi:10.1126/science.291.5512.2370. [DOI] [PubMed] [Google Scholar]
- Rudd PM, Wormald MR, Stanfield RL, Huang M, Mattsson N, Speir JA, DiGennaro JA, Fetrow JS, Dwek RA, Wilson IA. Roles for glycosylation of cell surface receptors involved in cellular immune recognition. J Mol Biol. 1999;293:351–366. doi: 10.1006/jmbi.1999.3104. doi:10.1006/jmbi.1999.3104. [DOI] [PubMed] [Google Scholar]
- Ryan SO, Bonomo JA, Zhao F, Cobb BA. MHCII glycosylation modulates Bacteroides fragilis carbohydrate antigen presentation. J Exp Med. 2011;208:1041–1053. doi: 10.1084/jem.20100508. doi:10.1084/jem.20100508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldova R, Royle L, Radcliffe CM, Abd Hamid UM, Evans R, Arnold JN, Banks RE, Hutson R, Harvey DJ, Antrobus R, et al. Ovarian cancer is associated with changes in glycosylation in both acute-phase proteins and IgG. Glycobiology. 2007;17:1344–1356. doi: 10.1093/glycob/cwm100. doi:10.1093/glycob/cwm100. [DOI] [PubMed] [Google Scholar]
- Sato S, St-Pierre C, Bhaumik P, Nieminen J. Galectins in innate immunity: Dual functions of host soluble β-galactoside-binding lectins as damage-associated molecular patterns (DAMPs) and as receptors for pathogen-associated molecular patterns (PAMPs) Immunol Rev. 2009;230:172–187. doi: 10.1111/j.1600-065X.2009.00790.x. doi:10.1111/j.1600-065X.2009.00790.x. [DOI] [PubMed] [Google Scholar]
- Schachter H. The joys of HexNAc. The synthesis and function of N- and O-glycan branches. Glycoconj J. 2000;17:465–483. doi: 10.1023/a:1011010206774. doi:10.1023/A:1011010206774. [DOI] [PubMed] [Google Scholar]
- Schauer R. Sialic acids as regulators of molecular and cellular interactions. Curr Opin Struct Biol. 2009;19:507–514. doi: 10.1016/j.sbi.2009.06.003. doi:10.1016/j.sbi.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer R, Srinivasan GV, Wipfler D, Kniep B, Schwartz-Albiez R. O-Acetylated sialic acids and their role in immune defense. Adv Exp Med Biol. 2011;705:525–548. doi: 10.1007/978-1-4419-7877-6_28. doi:10.1007/978-1-4419-7877-6_28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz BL, Sloane AJ, Robinson LJ, Prasad SS, Lindner RA, Robinson M, Bye PT, Nielson DW, Harry JL, Packer NH, et al. Glycosylation of sputum mucins is altered in cystic fibrosis patients. Glycobiology. 2007;17:698–712. doi: 10.1093/glycob/cwm036. doi:10.1093/glycob/cwm036. [DOI] [PubMed] [Google Scholar]
- Severi E, Hood DW, Thomas GH. Sialic acid utilization by bacterial pathogens. Microbiology. 2007;153:2817–2822. doi: 10.1099/mic.0.2007/009480-0. doi:10.1099/mic.0.2007/009480-0. [DOI] [PubMed] [Google Scholar]
- Shafi R, Iyer SP, Ellies LG, O'Donnell N, Marek KW, Chui D, Hart GW, Marth JD. The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc Natl Acad Sci USA. 2000;97:5735–5739. doi: 10.1073/pnas.100471497. doi:10.1073/pnas.100471497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley P, Guidos CJ. Regulation of Notch signaling during T- and B-cell development by O-fucose glycans. Immunol Rev. 2009;230:201–215. doi: 10.1111/j.1600-065X.2009.00791.x. doi:10.1111/j.1600-065X.2009.00791.x. [DOI] [PubMed] [Google Scholar]
- Stanley P, Okajima T. Roles of glycosylation in Notch signaling. Curr Top Dev Biol. 2010;92:131–164. doi: 10.1016/S0070-2153(10)92004-8. doi:10.1016/S0070-2153(10)92004-8. [DOI] [PubMed] [Google Scholar]
- Stanley P, Schachter H, Taniguchi N. N-Glycans. 2009. pp. 101–114. In: Varki A, Cummings RD, Esko JD, et al., editors. Essentials of Glycobiology, 2nd edition. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press. [PubMed] [Google Scholar]
- Stone EL, Ismail MN, Lee SH, Luu Y, Ramirez K, Haslam SM, Ho SB, Dell A, Fukuda M, Marth JD. Glycosyltransferase function in core 2-type protein O glycosylation. Mol Cell Biol. 2009;29:3770–3782. doi: 10.1128/MCB.00204-09. doi:10.1128/MCB.00204-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sujino K, Jackson RJ, Chan NW, Tsuji S, Palcic MM. A novel viral α2,3-sialyltransferase (v-ST3Gal I): Transfer of sialic acid to fucosylated acceptors. Glycobiology. 2000;10:313–320. doi: 10.1093/glycob/10.3.313. doi:10.1093/glycob/10.3.313. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. doi:10.1016/S1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- Thompson S, Dargan E, Griffiths ID, Kelly CA, Turner GA. The glycosylation of haptoglobin in rheumatoid arthritis. Clin Chim Acta. 1993;220:107–114. doi: 10.1016/0009-8981(93)90011-r. doi:10.1016/0009-8981(93)90011-R. [DOI] [PubMed] [Google Scholar]
- Thompson S, Kelly CA, Griffiths ID, Turner GA. Abnormally-fucosylated serum haptoglobins in patients with inflammatory joint disease. Clin Chim Acta. 1989;184:251–258. doi: 10.1016/0009-8981(89)90058-2. doi:10.1016/0009-8981(89)90058-2. [DOI] [PubMed] [Google Scholar]
- Thon V, Lau K, Yu H, Tran BK, Chen X. PmST2: A novel Pasteurella multocida glycolipid α2-3-sialyltransferase. Glycobiology. 2011;21:1206–1216. doi: 10.1093/glycob/cwr054. doi:10.1093/glycob/cwr054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toscano MA, Bianco GA, Ilarregui JM, Croci DO, Correale J, Hernandez JD, Zwirner NW, Poirier F, Riley EM, Baum LG, et al. Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat Immunol. 2007;8:825–834. doi: 10.1038/ni1482. doi:10.1038/ni1482. [DOI] [PubMed] [Google Scholar]
- Tricot G. New insights into role of microenvironment in multiple myeloma. Lancet. 2000;355:248–250. doi: 10.1016/S0140-6736(00)00019-2. doi:10.1016/S0140-6736(00)00019-2. [DOI] [PubMed] [Google Scholar]
- Troy FA. Polysialylation: From bacteria to brains. Glycobiology. 1992;2:5–23. doi: 10.1093/glycob/2.1.5. doi:10.1093/glycob/2.1.5. [DOI] [PubMed] [Google Scholar]
- Tsuboi S, Fukuda M. Roles of O-linked oligosaccharides in immune responses. Bioessays. 2001;23:46–53. doi: 10.1002/1521-1878(200101)23:1<46::AID-BIES1006>3.0.CO;2-3. doi:10.1002/1521-1878(200101)23:1<46::AID-BIES1006>3.3.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Turner GA, Goodarzi MT, Thompson S. Glycosylation of alpha-1-proteinase inhibitor and haptoglobin in ovarian cancer: Evidence for two different mechanisms. Glycoconj J. 1995;12:211–218. doi: 10.1007/BF00731322. doi:10.1007/BF00731322. [DOI] [PubMed] [Google Scholar]
- Tzianabos AO, Wang JY, Lee JC. Structural rationale for the modulation of abscess formation by Staphylococcus aureus capsular polysaccharides. Proc Natl Acad Sci USA. 2001;98:9365–9370. doi: 10.1073/pnas.161175598. doi:10.1073/pnas.161175598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Geijn FE, Wuhrer M, Selman MH, Willemsen SP, de Man YA, Deelder AM, Hazes JM, Dolhain RJ. Immunoglobulin G galactosylation and sialylation are associated with pregnancy-induced improvement of rheumatoid arthritis and the postpartum flare: Results from a large prospective cohort study. Arthritis Res Ther. 2009;11:R193. doi: 10.1186/ar2892. doi:10.1186/ar2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heeckeren AM, Schluchter MD, Drumm ML, Davis PB. Role of Cftr genotype in the response to chronic Pseudomonas aeruginosa lung infection in mice. Am J Physiol Lung Cell Mol Physiol. 2004;287:L944–L952. doi: 10.1152/ajplung.00387.2003. doi:10.1152/ajplung.00387.2003. [DOI] [PubMed] [Google Scholar]
- Varki A. Since there are PAMPs and DAMPs, there must be SAMPs? Glycan “self-associated molecular patterns” dampen innate immunity, but pathogens can mimic them. Glycobiology. 2011;21:1121–1124. doi: 10.1093/glycob/cwr087. doi:10.1093/glycob/cwr087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecchi C, Montosi G, Pietrangelo A. Huh-7: A human “hemochromatotic” cell line. Hepatology. 2010;51:654–659. doi: 10.1002/hep.23410. doi:10.1002/hep.23410. [DOI] [PubMed] [Google Scholar]
- Vimr ER, Kalivoda KA, Deszo EL, Steenbergen SM. Diversity of microbial sialic acid metabolism. Microbiol Mol Biol Rev. 2004;68:132–153. doi: 10.1128/MMBR.68.1.132-153.2004. doi:10.1128/MMBR.68.1.132-153.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Balog CI, Stavenhagen K, Koeleman CA, Scherer HU, Selman MH, Deelder AM, Huizinga TW, Toes RE, Wuhrer M. Fc-glycosylation of IgG1 is modulated by B-cell stimuli. Mol Cell Proteomics. 2011;10:M110. doi: 10.1074/mcp.M110.004655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Tan J, Sutton-Smith M, Ditto D, Panico M, Campbell RM, Varki NM, Long JM, Jaeken J, Levinson SR, et al. Modeling human congenital disorder of glycosylation type IIa in the mouse: Conservation of asparagine-linked glycan-dependent functions in mammalian physiology and insights into disease pathogenesis. Glycobiology. 2001;11:1051–1070. doi: 10.1093/glycob/11.12.1051. doi:10.1093/glycob/11.12.1051. [DOI] [PubMed] [Google Scholar]
- Wesley A, Forstner J, Qureshi R, Mantle M, Forstner G. Human intestinal mucin in cystic fibrosis. Pediatr Res. 1983;17:65–69. doi: 10.1203/00006450-198301000-00013. doi:10.1203/00006450-198301000-00013. [DOI] [PubMed] [Google Scholar]
- Wieland CW, Koppel EA, den DJ, Florquin S, McKenzie AN, van KY, van der Poll T, Geijtenbeek TB. Mice lacking SIGNR1 have stronger T helper 1 responses to Mycobacterium tuberculosis. Microbes Infect. 2007;9:134–141. doi: 10.1016/j.micinf.2006.10.018. doi:10.1016/j.micinf.2006.10.018. [DOI] [PubMed] [Google Scholar]
- Xia B, Royall JA, Damera G, Sachdev GP, Cummings RD. Altered O-glycosylation and sulfation of airway mucins associated with cystic fibrosis. Glycobiology. 2005;15:747–775. doi: 10.1093/glycob/cwi061. doi:10.1093/glycob/cwi061. [DOI] [PubMed] [Google Scholar]
- Yamauchi J, Kawai Y, Yamada M, Uchikawa R, Tegoshi T, Arizono N. Altered expression of goblet cell- and mucin glycosylation-related genes in the intestinal epithelium during infection with the nematode Nippostrongylus brasiliensis in rat. APMIS. 2006;114:270–278. doi: 10.1111/j.1600-0463.2006.apm_353.x. doi:10.1111/j.1600-0463.2006.apm_353.x. [DOI] [PubMed] [Google Scholar]
- Ye Z, Marth JD. N-Glycan branching requirement in neuronal and postnatal viability. Glycobiology. 2004;14:547–558. doi: 10.1093/glycob/cwh069. doi:10.1093/glycob/cwh069. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Sato Y, Isaji T, Fukuda T, Matsumoto A, Miyoshi E, Gu J, Taniguchi N. Branched N-glycans regulate the biological functions of integrins and cadherins. FEBS J. 2008;275:1939–1948. doi: 10.1111/j.1742-4658.2008.06346.x. doi:10.1111/j.1742-4658.2008.06346.x. [DOI] [PubMed] [Google Scholar]
- Zhu X, Borchers C, Bienstock RJ, Tomer KB. Mass spectrometric characterization of the glycosylation pattern of HIV-gp120 expressed in CHO cells. Biochemistry. 2000;39:11194–11204. doi: 10.1021/bi000432m. doi:10.1021/bi000432m. [DOI] [PubMed] [Google Scholar]