

Abstract
The epithelial Na+ channel (ENaC) in the aldosterone-sensitive distal nephron (ASDN) is under negative-feedback regulation by the renin–angiotensin–aldosterone system in protection of sodium balance and blood pressure. We test here whether aldosterone is necessary and sufficient for ENaC expression and activity in the ASDN. Surprisingly, ENaC expression and activity are robust in adrenalectomized (Adx) mice. Exogenous mineralocorticoid increases ENaC activity equally well in control and Adx mice. Plasma [AVP] is significantly elevated in Adx vs. control mice. Vasopressin (AVP) stimulates ENaC. Inhibition of the V2 AVP receptor represses ENaC activity in Adx mice. The absence of aldosterone combined with elevated AVP release compromises normal feedback regulation of ENaC in Adx mice in response to changes in sodium intake. These results demonstrate that aldosterone is sufficient but not necessary for ENaC activity in the ASDN. Aldosterone-independent stimulation by AVP shifts the role of ENaC in the ASDN from protecting Na+ balance to promoting water reabsorption. This stimulation of ENaC likely contributes to the hyponatremia of adrenal insufficiency.
Keywords: epithelial transport, hypertension, sodium excretion, diabetes insipidus, sodium wasting
Renal sodium excretion is fine-tuned in the aldosterone-sensitive distal nephron (ASDN). Here, the activity of the epithelial Na+ channel (ENaC) is limiting for sodium reabsorption (reviewed in refs. 1 and 2). ENaC serves as the apical entry pathway for electrogenic Na+ reabsorption through principal cells. Normal ENaC function is required for proper sodium balance and, thus, normal blood pressure. Gain-of-function mutations in ENaC cause inappropriate renal sodium retention and consequent increases in mean arterial pressure (2, 3). Inhibition of ENaC corrects the renal and blood pressure phenotypes resulting from such mutations. Loss-of-function mutations in ENaC, in contrast, cause renal sodium wasting and corresponding decreases in blood pressure (2, 4).
The activity of ENaC is under negative-feedback regulation by the renin–angiotensin–aldosterone system (RAAS; ref. 1). The mineralocorticoid, aldosterone, is the final hormone in this cascade. This antinatriuretic factor is essential for proper Na+ balance (5, 6). Decreases in blood pressure evoke via renin–AngII signaling secretion of aldosterone from the adrenal gland. Aldosterone through the mineralocorticoid receptor (MR) stimulates ENaC in the ASDN to minimize renal sodium excretion in protection of Na+ balance and vascular volume (2, 4). Pathological increases in aldosterone elevate blood pressure by promoting inappropriate renal sodium retention (7, 8). Inhibition of ENaC ameliorates inappropriate renal sodium retention. In contrast, pathological decreases in aldosterone result in sodium wasting arising from inappropriate increases in renal sodium excretion (4, 8, 9). MR agonism and antagonism increase and decrease ENaC activity, respectively (10–12). There is strong support for a tight positive relation between the levels and actions of aldosterone and ENaC activity, sodium balance, and blood pressure.
Key aspects of these relations, however, remain obscure. For instance, whereas the temporal coupling between changes in blood pressure and sodium excretion is tight, pressure-induced changes in circulating aldosterone are comparatively slow. Moreover, residual but significant ENaC activity is present in the ASDN of MR knockout mice (13), and, in some instances, ENaC activity is high in the absence of significant changes in aldosterone (12). Findings such as these suggest that, although aldosterone is capable of increasing ENaC activity, its absence is less effective at decreasing it.
Several hormones and paracrine factors, in addition to aldosterone, modulate the activity of ENaC. For instance, vasopressin (AVP) decreases renal sodium excretion by increasing the activity of ENaC and sodium reabsorption in the ASDN in parallel with aldosterone (14–16). Such observations suggest that aldosterone serves as one of many factors modulating ENaC activity, rather than functioning as a requisite master regulator of the channel.
Here we ask whether aldosterone is an absolute requirement for ENaC activity, testing the necessity and sufficiency of this hormone for channel expression and activity in the ASDN. We find that ENaC is expressed and active in the absence of aldosterone. Adrenal insufficiency elevates plasma AVP concentration. AVP stimulates ENaC in adrenalectomized (Adx) mice through a posttranslational mechanism via V2 receptors. Thus, although aldosterone is sufficient to stimulate ENaC activity in the ASDN, it is not necessary for activity, and ENaC activity in the ASDN can be high in the absence of this and other corticosteroids. These findings provide important insights about the role of ENaC and its regulation in pathological states of hyponatremia, such as that during adrenal insufficiency.
Results
ENaC Is Expressed and Active in the ASDN of Adx Mice.
We tested the necessity of adrenal steroids, including mineralocorticoids, to the expression and activity of ENaC in principal cells by assaying directly the activity of this channel with patch-clamp electrophysiology in split-open ASDN isolated from Adx mice. As expected, adrenalectomy significantly decreased plasma corticosterone levels to the lower limit of quantification, and it significantly increased plasma [K+], and decreased plasma osmolality and body weight (Fig. S1). Surprisingly, ENaC expression and activity were robust in ASDN from Adx mice. Fig. 1 (see also Table 1) shows typical single-channel current traces from cell-attached patches formed on the apical membranes of principal cells from control and Adx mice (Fig. 1A), as well as corresponding summary graphs of the open probability (Po; Fig. 1B), number of active channels (N; Fig. 1C), and activity (NPo; Fig. 1D) for ENaC in these patches. The Po of ENaC was not different between control and Adx mice; however, N was significantly greater in Adx mice, with ENaC in this latter group having elevated activity.
Fig. 1.

Mineralocorticoid is not necessary for ENaC activity in the ASDN. (A) Representative gap-free current traces from cell-attached patches made on the apical membrane of principal cells in split-open murine ASDN from control (Upper) and Adx (Lower) mice. These seals contain at least two ENaC. The closed state (c) is denoted with a dashed line. Inward current is downward. The holding potential for these patches was −Vp = −60 mV. (B–D) Summary graphs of Po (B), N (C), and NPo (D) for ENaC in control (gray) and Adx (black) mice. Data are from experiments identical to that in A. *Significantly greater compared with control.
Table 1.
ENaC activity in control and Adx mice
| Drinking water | Treatment† | NPo | N | Po | f‡ |
| Control | |||||
| H2O | — | 0.78 ± 0.17* | 2.4 ± 0.30* | 0.28 ± 0.03* | 0.46 (36/79) |
| 1% saline | — | 0.25 ± 0.06 | 1.5 ± 0.19 | 0.15 ± 0.03 | 0.39 (20/51) |
| H2O | DOCA | 1.4 ± 0.22*,** | 3.0 ± 0.40 | 0.44 ± 0.04*,** | 0.60 (29/48) |
| 1% saline | DOCA | 0.76 ± 0.15** | 2.7 ± 0.35** | 0.22 ± 0.02** | 0.56 (33/59) |
| H2O | AVP | 1.78 ± 0.17** | 3.8 ± 0.42** | 0.44 ± 0.03** | 0.75 (30/40)** |
| 1% saline | Tolvaptan | 0.13 ± 0.04 | 1.4 ± 0.15 | 0.08 ± 0.02 | 0.31 (19/62) |
| Adx | |||||
| H2O | — | 1.4 ± 0.59* | 4.1 ± 0.90*,+ | 0.23 ± 0.02 | 0.44 (10/23) |
| 1% saline | — | 0.53 ± 0.11+ | 2.0 ± 0.20 | 0.22 ± 0.03 | 0.50 (26/52) |
| H2O | DOCA | 1.6 ± 0.21* | 3.8 ± 0.40* | 0.36 ± 0.05** | 0.65 (35/54) |
| 1% saline | DOCA | 0.76 ± 0.10 | 2.2 ± 0.19 | 0.31 ± 0.03** | 0.65 (32/49) |
| 1% saline | Tolvaptan | 0.17 ± 0.04* | 1.7 ± 0.16 | 0.09 ± 0.01* | 0.34 (33/96) |
All groups were maintained with regular chow containing 0.32% [Na+].
*Significant increase/decrease compared with 1% saline drinking water. **Significantly greater compared with no treatment. +Significantly greater compared with control mice under identical conditions.
†Injected with 2.4 mg of DOCA (in 150 μL of olive oil) for 3 consecutive days or treated with 30 mg/kg Tolvaptan added to drinking water for 2 d before patch-clamp analysis or isolated ASDN treated with 1 μM AVP for at least 30 min before patch-clamp analysis.
‡f, frequency (patches with at least one active channel/total number of viable seals for that condition) compared with a z test.
The results of immunofluorescence studies of ENaC expression in the ASDN of control and Adx mice, as shown in Fig. 2 and Fig. S2, are consistent with these electrophysiology findings. All three ENaC subunits are clearly expressed in AQP2-positive cells of the ASDN in both control and Adx mice. This finding is in agreement with what has been reported for the expression of ENaC subunits during MR antagonism (17) and in Adx rats (18, 19).
Fig. 2.

ENaC is expressed in the ASDN of Adx mice. Representative (n ≥ 3) fluorescence micrographs of ASDN from control (Upper) and Adx (Lower) mice maintained with tap water probed with anti-ENaC (left; red) and anti-AQP2 (second from left; green) antibodies and corresponding merged (third from left) and bright-field images (right). Nuclear staining (blue) with DAPI is included in merged images. Staining with anti–γ-ENaC and anti–β-ENaC antibodies are shown here for control and Adx mice, respectively. Complete images with all three ENaC antibodies for both conditions are shown in Fig. S2.
Aldosterone Is Sufficient to Increase ENaC Activity.
Fig. 3 (see also Table 1) shows the summary graph of Po for ENaC in control (gray bars) and Adx (black bars) mice with (hatched bars) and without (filled bars) mineralocorticoid supplementation for 3 d. Mineralocorticoid increased ENaC Po in both control and Adx mice with a similar relative effective. A mineralocorticoid-dependent increase in ENaC activity is consistent with previous findings from our laboratory (14, 20, 21) and those of others (10). As expected, exogenous mineralocorticoid significantly decreased PK in Adx mice from 6.1 ± 0.8 (n = 5) to 3.8 ± 0.4 mM (n = 6), which is near that (4.1 ± 0.3 mM; n = 15) in control mice (data not shown in a figure).
Fig. 3.

ENaC in Adx mice responds to exogenous mineralocorticoid. Summary graph shows Po for ENaC in control (gray) and Adx (black) mice in the absence (filled bars) and presence (hatched bars) of deoxycorticosterone acetate (DOCA). Data are from experiments similar to that in Fig. 1A. *Significantly greater compared with the absence of DOCA treatment.
ENaC in Adx Mice Is Capable of Responding to Changes in Sodium Intake via Changes in N but Not Po.
As shown in Fig. S3, support of Adx mice with 1% saline compared with tap water offered some protection, as expected (6, 9, 22–26), against the volume depletion and hyponatremia of their hypoadrenal, sodium- and water-wasting state.
To test whether a functional adrenal gland—and, thus, the ability to have dynamic mineralocorticoid signaling—is an absolute requirement for dietary sodium-dependent regulation of ENaC, we next compared the activity of ENaC in ASDN isolated from control (gray bars) and Adx (black bars) mice maintained with tap water (filled bars) and with 1% saline drinking solution (striped bars). As shown in Fig. 4 (see also Table 1), an increase in sodium intake significantly decreases ENaC Po (Fig. 4A), N (Fig. 4B), and activity (Fig. 4C) in control mice; restated, a decrease in sodium intake causes a corresponding increase in ENaC activity. This change in sodium intake, in contrast, is without effect on Po in Adx mice. Channel number and activity, however, do significantly increase in Adx mice in response to a decrease in sodium intake. Although changed in both groups, ENaC activity remains significantly greater in Adx compared with control mice in the presence of 1% saline drinking solution.
Fig. 4.

Feedback regulation of ENaC is compromised in Adx mice. (A–C) Summary graphs of Po (A), N (B), and NPo (C) for ENaC in control (gray) and Adx (black) mice drinking tap water (solid bars) and 1% saline solution (striped bars). Data are from experiments similar to that in Fig. 1A. *Significantly greater vs. 1% saline drinking water. **Significantly greater compared with control under identical conditions. (D) Fractional ENaC activity (NPo drinking 1% saline solution/NPo drinking tap water) for control (gray) and Adx (black) mice in the absence and presence of DOCA.
Feedback Regulation of ENaC Is Compromised in Adx Mice.
To better understand the effects of exogenous mineralocorticoid and changes in dietary sodium intake on ENaC activity in Adx compared with control mice, we plotted summarized NPo as a function of both parameters (Fig. S4) and as fractional ENaC activity in the presence and absence of exogenous mineralocorticoid (Fig. 4D). The latter—which is activity when maintained with 1% saline drinking solution divided by activity in the presence of drinking tap water—reflects how capable signaling pathways are at adjusting ENaC activity to counter changes in Na+ balance: Elevated fractional ENaC activity denotes a loss of responsiveness to changes in sodium balance (21). Because changes in sodium intake do not change Po in mice with compromised adrenal function, ENaC is less responsive to this perturbation in Adx mice. Exogenous mineralocorticoid clamps ENaC activity high in both groups, disrupting normal feedback regulation to the channel in response to changes in sodium intake, which is shown as elevations in fractional ENaC activity [in the presence of deoxycorticosterone acetate (DOCA)].
Adrenal Insufficiency Increases Plasma [AVP].
The above results demonstrate that some regulatory factor stimulates ENaC in the absence of adrenal steroids in Adx mice. We tested first whether AngII could function in this regard, and results were negative. The finding that plasma [AVP], as shown in Fig. 5, is significantly increased in Adx compared with control mice—maintained with normal chow and tap water—identifies this hormone as a potential candidate mediating this effect. This observation that loss of adrenal gland function increases plasma [AVP] is consistent with the findings of others (22, 27–29).
Fig. 5.

Plasma AVP concentration is increased in Adx mice. Summary graph of plasma [AVP] in control (gray; n = 20) vs. Adx (black; n = 13) mice maintained with tap water. *Significantly increased vs. control.
AVP Increases ENaC Activity.
To test whether AVP can serve as a stimulator of ENaC activity in the absence of adrenal gland function, we assessed the actions of this neurohormone on channel activity as shown in Fig. 6 (see also Table 1). As can be seen clearly in the summary graphs of Po (Fig. 6A), N (Fig. 6B), and NPo (Fig. 6C), AVP significantly increases ENaC activity by increasing the Po and number of functional channels in the membrane (N and f). This finding is in agreement with those made earlier by us and others (14–16).
Fig. 6.
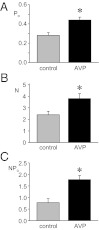
AVP increases ENaC activity. Summary graphs show Po (A), N (B), and NPo (C) for ENaC in control mice maintained with normal chow and tap water in the absence (gray) and presence of 1 μM AVP (black). Data are from experiments similar to that in Fig. 1A. *Significantly greater vs. the absence of AVP.
AVP via V2 Receptors Maintains ENaC Activity High in Adx Mice.
To test whether AVP stimulates ENaC in Adx mice, the expression and activity of ENaC in ASDN from control and Adx mice in the absence and presence of treatment with the V2 antagonist Tolvaptan was compared. As shown in the summary graph of NPo in Fig. 7A (see also Table 1), V2 antagonism significantly decreased the activity of ENaC in Adx mice to levels that were not different from that in control animals. Although decreasing ENaC activity, Tolvaptan as shown in Fig. 7B (see also Fig. S5) had no overt effect on the expression of ENaC subunits in AQP2-positive cells of the ASDN of Adx mice. This finding excludes decreases in expression as the cause of decreased ENaC activity in Adx mice with V2 receptor blockade. Such findings are consistent with aldosterone-independent activation of ENaC by AVP involving a posttranslational mechanism.
Fig. 7.

AVP stimulates ENaC through a posttranslational mechanism in Adx mice. (A) Summary graph of ENaC activity in control (gray) and Adx (black) mice maintained with 1% saline drinking solution in the absence (filled bars) and presence (hatched bars) of 30 mg/kg V2 receptor inhibitor Tolvaptan. Data are from experiments similar to that in Fig. 1A. *Significant decrease vs. the absence of Tolvaptan. (B) Representative (n ≥ 7) fluorescence micrographs of ASDN from Adx mice treated with Tolvaptan probed with anti-ENaC (Left Upper; red) and anti-AQP2 (Left Lower; green) antibodies and corresponding merged (Right Upper) and bright field images (Right Lower). Nuclear staining (blue) with DAPI is included in merged images. Staining with the anti–α-ENaC antibody is shown here. A complete image with all three ENaC antibodies is shown in Fig. S5.
Discussion
The expression and activity of ENaC are surprisingly robust in the absence of adrenal steroids in Adx mice. Adrenalectomy increases plasma [AVP]. An increase in AVP via V2 receptors maintains ENaC activity high via a posttranslational mechanism in the ASDN of Adx mice, resulting in elevated activity at all levels of sodium intake tested in the present study. Addition of exogenous mineralocorticoid increases the activity of ENaC equally well in control and Adx mice, independent of sodium intake. These findings demonstrate that aldosterone is sufficient, but not necessary, for ENaC activity in the ASDN and that elevations in AVP resulting from adrenal insufficiency are capable of stimulating ENaC in an adrenal steroid-independent manner. A consequence of elevated AVP and loss of regulation by adrenal steroids is that ENaC is no longer under normal feedback control in response to changes in sodium balance in Adx mice.
All studies investigating the actions of aldosterone on (amiloride-sensitive) renal sodium excretion, transport, and the activity of ENaC in the ASDN are in agreement that increases in aldosterone are sufficient to increase ENaC activity (11, 12, 30, 31). Conclusions from the current results are consistent with aldosterone being sufficient to increase ENaC activity.
We report here that aldosterone, although sufficient, is not necessary for ENaC activity in the ASDN. The results in support of this finding are the observations that ENaC expression and activity are robust in Adx mice, although these mice lack significant levels of adrenal gland function and plasma corticosteroids and are clearly in a hypoadrenal, sodium- and water-wasting state. In the only other study directly investigating ENaC activity with patch-clamp analysis in Adx animals, no significant ENaC activity was observed in cortical collecting tubules from Adx rats maintained with a normal sodium diet (12). This experimental condition, in general, is similar to that used in the present studies. There are potentially important differences, however. For instance, rodents were maintained with some glucocorticoid replacement in the earlier study but not in the present study. As discussed below, glucocorticoids can influence the circulating concentration of AVP. Interestingly, a high K+ diet increased ENaC activity in Adx and control rats in the absence of significant changes in plasma aldosterone levels in the earlier study (12). It is not clear yet whether these discrepancies represent true incongruence or whether they reflect rather slight variations in experimental conditions or differences in sensitivity.
Our rationale is that we consistently find significant ENaC activity, albeit at lower levels compared with a sodium deficient diet, in ASDN isolated from rats and mice maintained with regular-sodium (0.32% [Na+]) and high-sodium (2% [Na+]) diets (14, 20, 21). Aldosterone should be low (extremely so in the latter case) with these feeding regimens. In contrast, these others find no significant ENaC activity in cortical collecting tubules isolated from rodents maintained with normal and high-sodium diets (11, 30). We interpret our results as showing that the activity of ENaC in the ASDN is high in the presence of aldosterone and low, but significant, in the absence of this hormone in normal animals.
Nevertheless, it is accepted that in ASDN from normal animals, ENaC activity is related in a positive manner with aldosterone levels (11, 12, 14, 21). Adx mice, which are not normal (see below), represent an exception, then, where ENaC activity is high in the absence of aldosterone and other adrenal steroids. This exception demonstrates that ENaC can be active in the absence of aldosterone and, thus, that aldosterone is not necessary for the activity of this channel in the ASDN.
This interpretation is consistent with what has been reported for ENaC activity in neonatal MR-null mice (13). These mice do not survive long after the first week of life without sodium supplementation due to pathological renal sodium excretion. During this critical phase of early life, renal sodium loss cannot be compensated by nursing pups because of the low sodium and water content of mother’s milk. However, neonatal MR-null mice retain residual, but significant, ENaC activity—∼24% of normal—as extrapolated from amiloride-sensitive fractional Na+ excretion and transport across isolated, perfused collecting ducts.
Knockout of aldosterone synthase (AS) agrees with findings from MR-null mice (25, 32). AS-null mice have pronounced renal sodium and water wasting, which cannot be compensated during the critical neonatal period. Sodium and water wasting results in dehydration, failure to thrive, and death of ∼1/3 of AS-null mice in the first weeks of life. Sodium restriction exacerbates renal salt and water wasting in both AS- and MR-null mice compared with control animals, which have appropriate feedback regulation by RAAS of ENaC and other mechanism for decreasing renal Na+ excretion. These observations are reminiscent of human infants carrying inactivating MR mutations and AS deficiency, which require sodium supplementation to survive (4, 8, 9, 33).
Clearly the hormonal state, and thus phenotype, of loss-of-function of MR or AS do not completely overlap that of adrenalectomy. With MR dysfunction, the RAAS is up-regulated (13). With AS dysfunction, aldosterone is absent, but the levels of other adrenal steroids capable of mineralocorticoid action—in particular, corticosterone—are increased (9, 32, 33). In Adx mice, there is no aldosterone or other adrenal steroids and catecholamines; thus, these animals receive no input from the adrenal gland to ENaC. Moreover, akin to adrenal insufficiency, removal of the adrenal glands, as shown here and by others (27–29), causes marked increases in plasma AVP levels. Increases in AVP are not present with either MR or AS dysfunction (25).
Usually, AVP release is primarily controlled by plasma osmolality. Elevated AVP release in adrenal-insufficient states (that lack both glucocorticoids and mineralocorticoids) results from two events. There is loss of negative-feedback regulation by glucocorticoids of the hypothalamic–pituitary axis controlling AVP release. There also is strong nonosmotic stimulation of AVP release, resulting from volume depletion due to sodium and water wasting by the kidney (27, 28).
It is recognized that adrenal insufficiency and central diabetes insipidus are counterpoints when considering the equilibrium distribution of sodium and water: The pattern of sodium and water distribution in either deficiency depends in part on the activity of the remaining gland. As such, the hyponatremia of adrenal insufficiency is absent when combined with neurohypophyseal deficiency and in the Brattleboro rat, which has central diabetes insipidus (22, 27, 29, 34). Thus, the hyponatremia of adrenal insufficiency is dependent on elevated AVP release.
As we (14) and others (15, 16) have demonstrated, AVP stimulates renal Na+ reabsorption in the ASDN by increasing ENaC activity. The current results demonstrating that AVP increases ENaC activity are consistent with these earlier findings.
Akin to its regulation of aquaporin 2 water channels, AVP stimulates ENaC in principal cells via the V2 receptor (14). The current findings that AVP levels are increased in Adx mice and that inhibition of the V2 receptor decreases ENaC activity in ASDN from these mice to levels that are identical to those observed in control animals demonstrates that elevated AVP is the driving force maintaining ENaC activity high in Adx mice. Moreover, the findings that all three ENaC subunits are expressed in the ASDN of Adx mice and that V2 receptor antagonism in these animals does not overtly affect ENaC subunit expression are consistent with AVP stimulating ENaC via a posttranslational mechanism.
The current electrophysiology results enable elaboration of the mechanism by which elevated AVP levels in Adx mice increase ENaC activity. We find that ENaC Po is clamped and N is elevated in these mice resulting from the new balance between stimulation by AVP as countered by loss of stimulation by a dearth of aldosterone. Both aldosterone and AVP increase ENaC Po and N with the major, long-term effect of AVP being an increase in N (10, 11, 14, 21).
Irrespective of the exact molecular mechanism, a consequence of adrenalectomy is that ENaC is no longer regulated in a normal manner by feedback signaling in response to changes in sodium balance. Loss of feedback regulation is a result of stimulation by elevated AVP release combined with disruption of regulation by the RAAS.
In the presence of AVP, water via activated aquaporin 2 channels is free to follow the Na+ reabsorbed through activated ENaC in the ASDN. Sodium reabsorbed at the ASDN in this manner facilitates water reabsorption by supporting the axial corticomedullary hyperosomotic gradient as established by the loop of Henle (35).
Adx mice, similar to other adrenal-insufficient states, however, have pronounced renal sodium and volume wasting. Yet, as shown here, ENaC activity is high in Adx mice because of elevations in AVP. It is unclear whether the latter is a compensatory response or an effect of adrenal insufficiency, independent of actions on vascular volume. Nevertheless, that ENaC activity is high in the face of elevated sodium excretion suggests that there are other aldosterone-dependent processes involved in control of renal sodium and volume excretion—perhaps the Na–Cl cotransporter—that are also compromised in Adx animals and that elevations in AVP are not able to stimulate these aldosterone-dependent, non-ENaC processes to compensate fully for sodium and volume loss. Regardless of whether increases in ENaC activity are compensatory or a primary effect, a consequence of adrenal insufficiency is that AVP-stimulated ENaC, in the absence of input from RAAS, becomes a slave to water reabsorption rather than a key mediator of sodium balance, exacerbating the hyponatremia of this state.
Methods
Animals.
All animal use and welfare adhered to the NIH Guide for the Care and Use of Laboratory Animals (36) following a protocol reviewed and approved by the Institutional Laboratory Animal Care and Use Committee of the University of Texas Health Science Center at San Antonio. Adult (∼25g, 6–8 wk old) male C57BL/6J control mice and those with bilateral adrenalectomy (Adx) were purchased from Jackson Laboratories. Experiments were conducted 2–3 wk after surgery. Mice were maintained with standard chow (0.32% [Na+]; TD.7912; Harlan Teklad). One week before experimentation, mice were divided into two groups: one maintained with tap water and the other with 1% saline drinking solution. For some experiments, mice were injected s.c. with 2.4 mg of DOCA dissolved in 150 μL of olive oil for three consecutive days before euthanizing. In others, mice were provided with 30 mg/kg AVP V2 receptor inhibitor Tolvaptan in drinking water for 2 d.
Isolated, Split-Open ASDN Preparation.
Isolation of the ASDN containing connecting tubule and collecting duct suitable for electrophysiology has been described (14, 20, 21). Refer to SI Methods for additional description.
Patch-Clamp Electrophysiology.
ENaC activity in principal cells of murine ASDN was quantified in cell-attached patches of the apical membrane made under voltage-clamp conditions (−Vp = −60 mV) by using standard procedures (14, 20, 21). For the current experiments, typical bath and pipette solutions were (in mM): 150 NaCl, 5 KCl, 1 CaCl2, 2 MgCl2, 5 glucose, and 10 Hepes (pH 7.4); and 140 LiCl, 2 MgCl2, and 10 Hepes (pH 7.4), respectively. For each experimental condition, ASDN from at least three different mice was assayed. Refer to SI Methods for additional description.
Immunohistochemistry.
Kidneys were prepared and sectioned for immunolabeling and subsequently imaged for immunofluorescence by using standard methods, closely following those published for the anti-ENaC antibodies used in the current studies (17). Antibodies to each of the three ENaC subunits were a gift from Mark A. Knepper (National Institutes of Health, Bethesda, MD) and have been characterized (17, 37). Kidney slices immunolabeled with anti-ENaC and anti-AQP2 antibodies were imaged with epifluorescence on an inverted microscope by using a 40× objective (1.4 NA) on a system built around a Zeiss Axiovert 200M equipped with DAPI, FITC, and CY5 excitation and emission filters. Images were collected and analyzed with Slidebook 4.2 software. Refer to SI Methods for additional description.
Analysis of Hormones and Electrolytes.
Urinary and plasma electrolyte concentrations and osmolality were determined by using standard procedures with a flame photometer and vapor pressure osmometer, respectively. Plasma corticosterone and AVP concentrations were quantified by using standard procedures with HPLC on a reversed-phase, cation exchange column (4.6 × 150 mm; 5-μm particle size; part number C-18; fAlltech) and a competitive enzyme-linked immunoassay (Arg8-Vasopressin EIA kit; Enzo Life Sciences), respectively (38). Refer to SI Methods for additional description.
Statistical Analysis and Data Presentation.
Data are reported as mean ± SEM. Unpaired data were compared with a two-sided unpaired Student t test. The criterion for significance was P ≤ 0.05. For presentation, slow baseline drifts were corrected, and current data from some patches were software-filtered at 50 Hz.
Supplementary Material
Acknowledgments
We thank Dr. M. A. Javors, J. J. Sanchez, and C. R. Herrera for assistance quantifying plasma (corticosterone) with HPLC. This work was supported by National Institutes of Health Grant R01 DK59594 (to J.D.S.) and American Heart Association Fellowship 10POST3200019 (to V.B.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1201978109/-/DCSupplemental.
References
- 1.Kellenberger S, Schild L. Epithelial sodium channel/degenerin family of ion channels: A variety of functions for a shared structure. Physiol Rev. 2002;82:735–767. doi: 10.1152/physrev.00007.2002. [DOI] [PubMed] [Google Scholar]
- 2.Rossier BC, Pradervand S, Schild L, Hummler E. Epithelial sodium channel and the control of sodium balance: Interaction between genetic and environmental factors. Annu Rev Physiol. 2002;64:877–897. doi: 10.1146/annurev.physiol.64.082101.143243. [DOI] [PubMed] [Google Scholar]
- 3.Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104:545–556. doi: 10.1016/s0092-8674(01)00241-0. [DOI] [PubMed] [Google Scholar]
- 4.Geller DS. Mineralocorticoid resistance. Clin Endocrinol (Oxf) 2005;62:513–520. doi: 10.1111/j.1365-2265.2005.02229.x. [DOI] [PubMed] [Google Scholar]
- 5.Horisberger J-D, Diezi J. Effects of mineralocorticoids on Na+ and K+ excretion in the adrenalectomized rat. Am J Physiol. 1983;245:F89–F99. doi: 10.1152/ajprenal.1983.245.1.F89. [DOI] [PubMed] [Google Scholar]
- 6.Edmonds CJ. Sodium excretion in the adrenalectomized rat after intravenous saline infusion. J Endocrinol. 1960;20:112–122. doi: 10.1677/joe.0.0200112. [DOI] [PubMed] [Google Scholar]
- 7.Gómez-Sánchez CE. Primary aldosteronism and its variants. Cardiovasc Res. 1998;37:8–13. doi: 10.1016/s0008-6363(97)00230-7. [DOI] [PubMed] [Google Scholar]
- 8.White PC. Disorders of aldosterone biosynthesis and action. N Engl J Med. 1994;331:250–258. doi: 10.1056/NEJM199407283310408. [DOI] [PubMed] [Google Scholar]
- 9.White PC. Aldosterone synthase deficiency and related disorders. Mol Cell Endocrinol. 2004;217:81–87. doi: 10.1016/j.mce.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 10.Kemendy AE, Kleyman T, Eaton DC. Aldosterone alters the open probability of amiloride-blockable sodium channels in A6 epithelia. Am J Physiol. 1992;263:C825–C837. doi: 10.1152/ajpcell.1992.263.4.C825. [DOI] [PubMed] [Google Scholar]
- 11.Pácha J, Frindt G, Antonian L, Silver RB, Palmer LG. Regulation of Na channels of the rat cortical collecting tubule by aldosterone. J Gen Physiol. 1993;102:25–42. doi: 10.1085/jgp.102.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palmer LG, Antonian L, Frindt G. Regulation of apical K and Na channels and Na/K pumps in rat cortical collecting tubule by dietary K. J Gen Physiol. 1994;104:693–710. doi: 10.1085/jgp.104.4.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berger S, et al. Mineralocorticoid receptor knockout mice: Pathophysiology of Na+ metabolism. Proc Natl Acad Sci USA. 1998;95:9424–9429. doi: 10.1073/pnas.95.16.9424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bugaj V, Pochynyuk O, Stockand JD. Activation of the epithelial Na+ channel in the collecting duct by vasopressin contributes to water reabsorption. Am J Physiol Renal Physiol. 2009;297:F1411–F1418. doi: 10.1152/ajprenal.00371.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reif MC, Troutman SL, Schafer JA. Sodium transport by rat cortical collecting tubule. Effects of vasopressin and desoxycorticosterone. J Clin Invest. 1986;77:1291–1298. doi: 10.1172/JCI112433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tomita K, Pisano JJ, Knepper MA. Control of sodium and potassium transport in the cortical collecting duct of the rat. Effects of bradykinin, vasopressin, and deoxycorticosterone. J Clin Invest. 1985;76:132–136. doi: 10.1172/JCI111935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nielsen J, Kwon TH, Frøkiaer J, Knepper MA, Nielsen S. Maintained ENaC trafficking in aldosterone-infused rats during mineralocorticoid and glucocorticoid receptor blockade. Am J Physiol Renal Physiol. 2007;292:F382–F394. doi: 10.1152/ajprenal.00212.2005. [DOI] [PubMed] [Google Scholar]
- 18.de Seigneux S, Kim SW, Hemmingsen SC, Frøkiaer J, Nielsen S. Increased expression but not targeting of ENaC in adrenalectomized rats with PAN-induced nephrotic syndrome. Am J Physiol Renal Physiol. 2006;291:F208–F217. doi: 10.1152/ajprenal.00399.2005. [DOI] [PubMed] [Google Scholar]
- 19.Loffing J, et al. Aldosterone induces rapid apical translocation of ENaC in early portion of renal collecting system: Possible role of SGK. Am J Physiol Renal Physiol. 2001;280:F675–F682. doi: 10.1152/ajprenal.2001.280.4.F675. [DOI] [PubMed] [Google Scholar]
- 20.Stockand JD, et al. Purinergic inhibition of ENaC produces aldosterone escape. J Am Soc Nephrol. 2010;21:1903–1911. doi: 10.1681/ASN.2010040377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pochynyuk O, et al. Dietary Na+ inhibits the open probability of the epithelial sodium channel in the kidney by enhancing apical P2Y2-receptor tone. FASEB J. 2010;24:2056–2065. doi: 10.1096/fj.09-151506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Foy JM, Schnieden H. The effects of endocrine gland ablation and of corticosteroid substitution on the rate of water turnover in the adrenalectomized rat. J Endocrinol. 1965;31:89–94. doi: 10.1677/joe.0.0310089. [DOI] [PubMed] [Google Scholar]
- 23.van der Hoek J, Hoorn EJ, de Jong GM, Janssens EN, de Herder WW. Severe hyponatremia with high urine sodium and osmolality. Clin Chem. 2009;55:1905–1908. doi: 10.1373/clinchem.2009.125575. [DOI] [PubMed] [Google Scholar]
- 24.Schwartz MJ, Kokko JP. Urinary concentrating defect of adrenal insufficiency. Permissive role of adrenal steroids on the hydroosmotic response across the rabbit cortical collecting tubule. J Clin Invest. 1980;66:234–242. doi: 10.1172/JCI109849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Makhanova N, Sequeira-Lopez ML, Gomez RA, Kim HS, Smithies O. Disturbed homeostasis in sodium-restricted mice heterozygous and homozygous for aldosterone synthase gene disruption. Hypertension. 2006;48:1151–1159. doi: 10.1161/01.HYP.0000249902.09036.e7. [DOI] [PubMed] [Google Scholar]
- 26.Hamelink CR, Currie PJ, Chambers JW, Castonguay TW, Coscina DV. Corticosterone-responsive and -unresponsive metabolic characteristics of adrenalectomized rats. Am J Physiol. 1994;267:R799–R804. doi: 10.1152/ajpregu.1994.267.3.R799. [DOI] [PubMed] [Google Scholar]
- 27.Ahmed AB, George BC, Gonzalez-Auvert C, Dingman JF. Increased plasma arginine vasopressin in clinical adrenocortical insufficiency and its inhibition by glucosteroids. J Clin Invest. 1967;46:111–123. doi: 10.1172/JCI105504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishikawa SE, Schrier RW. Pathophysiological roles of arginine vasopressin and aquaporin-2 in impaired water excretion. Clin Endocrinol (Oxf) 2003;58:1–17. doi: 10.1046/j.1365-2265.2003.01647.x. [DOI] [PubMed] [Google Scholar]
- 29.Friedman SM, Sreter FA, Nakashima M, Friedman CL. Pitressin or aldosterone effects in rats with adrenal and neurohypophyseal deficiency. Am J Physiol. 1962;203:702–708. doi: 10.1152/ajplegacy.1962.203.4.702. [DOI] [PubMed] [Google Scholar]
- 30.Frindt G, Masilamani S, Knepper MA, Palmer LG. Activation of epithelial Na channels during short-term Na deprivation. Am J Physiol Renal Physiol. 2001;280:F112–F118. doi: 10.1152/ajprenal.2001.280.1.F112. [DOI] [PubMed] [Google Scholar]
- 31.Frindt G, McNair T, Dahlmann A, Jacobs-Palmer E, Palmer LG. Epithelial Na channels and short-term renal response to salt deprivation. Am J Physiol Renal Physiol. 2002;283:F717–F726. doi: 10.1152/ajprenal.00379.2001. [DOI] [PubMed] [Google Scholar]
- 32.Lee G, et al. Homeostatic responses in the adrenal cortex to the absence of aldosterone in mice. Endocrinology. 2005;146:2650–2656. doi: 10.1210/en.2004-1102. [DOI] [PubMed] [Google Scholar]
- 33.Pascoe L, Curnow KM, Slutsker L, Rösler A, White PC. Mutations in the human CYP11B2 (aldosterone synthase) gene causing corticosterone methyloxidase II deficiency. Proc Natl Acad Sci USA. 1992;89:4996–5000. doi: 10.1073/pnas.89.11.4996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Friedman SM, Sreter FA, Nakashima M, Friedman CL. Adrenal cortex and neurohypophyseal deficiency in salt and water homeostasis of rats. Am J Physiol. 1962;203:697–701. doi: 10.1152/ajplegacy.1962.203.4.697. [DOI] [PubMed] [Google Scholar]
- 35.Stockand JD. Vasopressin regulation of renal sodium excretion. Kidney Int. 2010;78:849–856. doi: 10.1038/ki.2010.276. [DOI] [PubMed] [Google Scholar]
- 36.Committee for the Update of the Guide for the Care and Use of Laboratory Animals . Guide for the care and use of laboratory animals. Washington, D.C: The National Academies Press; 2011. [Google Scholar]
- 37.Masilamani S, Kim GH, Mitchell C, Wade JB, Knepper MA. Aldosterone-mediated regulation of ENaC alpha, beta, and gamma subunit proteins in rat kidney. J Clin Invest. 1999;104:R19–R23. doi: 10.1172/JCI7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roos KP, Strait KA, Raphael KL, Blount MA, Kohan DE. Collecting duct-specific knockout of adenylyl cyclase type VI causes a urinary concentration defect in mice. Am J Physiol Renal Physiol. 2012;302:F78–F84. doi: 10.1152/ajprenal.00397.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.