

Abstract
Reversible voltammograms and a voltammetry half-wave potential versus solution pH diagram are described for a protein tyrosine radical. This work required a de novo designed tyrosine-radical protein displaying a unique combination of structural and electrochemical properties. The α3Y protein is structurally stable across a broad pH range. The redox-active tyrosine Y32 resides in a desolvated and well-structured environment. Y32 gives rise to reversible square-wave and differential pulse voltammograms at alkaline pH. The formal potential of the Y32-O•/Y32-OH redox couple is determined to 918 ± 2 mV versus the normal hydrogen electrode at pH 8.40 ± 0.01. The observation that Y32 gives rise to fully reversible voltammograms translates into an estimated lifetime of ≥30 ms for the Y32-O• state. This illustrates the range of tyrosine-radical stabilization that a structured protein can offer. Y32 gives rise to quasireversible square-wave and differential pulse voltammograms at acidic pH. These voltammograms represent the Y32 species at the upper edge of the quasirevesible range. The square-wave net potential closely approximates the formal potential of the Y32-O•/Y32-OH redox couple to 1,070 ± 1 mV versus the normal hydrogen electrode at pH 5.52 ± 0.01. The differential pulse voltammetry half-wave potential of the Y32-O•/Y32-OH redox pair is measured between pH 4.7 and 9.0. These results are described and analyzed.
Keywords: protein voltammetry, proton-coupled electron transfer
Proton-coupled electron transfer (PCET) represents a fundamental component of catalytic and long range radical-transfer processes involving tyrosine radicals (1–4). The thermodynamic and kinetic effects of coupled tyrosine oxidation/reduction and acid/base chemistry at the protein matrix are likely to play a key role in activating the aromatic residue for redox chemistry and for fine tuning its functional properties. This prediction is largely based on three sets of experimental observations that include the thermodynamic properties of aqueous tyrosine (5–7), the hydrogen-bonding properties of the kinetically well-characterized YZ and YD radicals in photosystem II (PSII) (7–10), and data derived from small-molecule model systems designed to delineate the PCET processes associated with tyrosine/phenol oxidation (4, 11–16). Tyrosine redox chemistry involves three redox couples and two pKa values. The cation Y-OH•+/Y-OH redox pair exists at pH below the pKa of the oxidized state (pKoY) whereas the tyrosinate Y-O•/Y-O- couple is observed at pH above the pKa of the reduced state (pKrY). The neutral tyrosine Y-O•/Y-OH redox couple operates in the pKoY < pH < pKrY region. With pKoY and pKrY values of -2 and 10 for aqueous tyrosine, respectively (5), Y-O•/Y-OH is predicted to be the dominant protein redox pair. Consequently, long-range electron transfer involving protein tyrosine residues is coupled to short-range proton motions between the radical species and the protein matrix. Indeed, studies on YZ and YD suggested early on that electron and proton transfers associated with tyrosine oxidation/reduction are strongly coupled events (7–9). Studies on solvated small-molecule tyrosine/phenol model systems have reinforced the key observation that local interactions critically determine the redox properties of tyrosine and the PCET mechanism by which the radical state is formed (for a recent review, see ref. 4).
The involvement of tyrosine radicals in biochemical processes has been known for decades. Yet, there is essentially a complete lack of thermodynamic data available for these redox species with published reduction potentials limited to two single-pH estimates (ref. 17 and references therein). This situation underlines the experimental challenges associated with probing the potentials of these highly oxidizing PCET molecules when they reside in a protein environment. Here we present a reduction potential vs. solution pH (Pourbaix) diagram (18) for a protein tyrosine radical. This was made possible by adopting a model protein approach guided by the following criteria. Obtaining a Pourbaix diagram for a protein tyrosine radical requires: (i) a protein system; i.e., a system that exhibits classic protein properties of cooperative structure and stability; (ii) that this protein contains a solvent-sequestered redox-active tyrosine whose potential can be measured by voltammetry across a broad pH range; (iii) that the protein voltammogram reflects a reversible electrode process from which formal tyrosine potentials can be derived. Here we report voltammetry studies of a de novo designed tyrosine radical model protein that fulfills these unique requirements.
Results and Discussion
α3Y Is a Well-Structured Protein.
The synthetic α3Y model protein used here is a member of a family of de novo designed radical proteins (2, 17, 19–21). This family is based on a 67-residue scaffold containing three interacting α-helices linked by two glycine loops (Fig. S1). The radical site (position 32) is located in the middle of the central helix and occupied by a tyrosine (to form the α3Y protein), a tryptophan (α3W), or a cysteine (α3C). C32 in α3C has been used as a chemical link for phenol ligation (22). α3Y, α3W, and α3C exhibit main characteristics associated with well-folded native proteins. They form thermodynamically stable, highly helical structures across a broad pH range. Folding occurs in a reversible, cooperative manner and is driven by the hydrophobic effect. Their tertiary structures are well defined. Importantly, residue 32 resides in a desolvated and highly structured environment. This latter property uniquely separates α3Y from all previously described tyrosine/phenol radical model systems in which the radical species typically resides in a highly solvated and dynamic environment (4, 23).
α3Y Voltammetry Control Studies.
Cyclic voltammetry (CV) (24) data cannot be obtained for α3Y due to the poor electrochemical response when using this relatively insensitive technique [see ref. 17 for a comparison in the CV response of a small-molecule tyrosine system and α3Y]. Differential pulse voltammetry (DPV) (24, 25) has better sensitivity than CV and generates a well-defined Faradaic response from α3Y at both acidic and alkaline conditions. Site-directed mutagenesis unambiguously assigned the observed current to Y32 and showed that the protein scaffold itself is redox inert to at least 1.3 V vs. NHE (17, 22). Examples of raw and background-corrected differential pulse voltammograms, details regarding data processing, and error analyses are provided in SI Text. To prove that α3Y is an electrochemically reversible system we employ the similarly sensitivity technique square-wave voltammetry (SWV) (24, 26, 27). In SWV, the applied potential is stepped progressively in fixed increments, and at each increment, a forward (in this study, oxidative) potential pulse is applied followed by a reverse (reductive) pulse. This generates a forward (Ifor), a reverse (Irev), and a net (Inet = Ifor - Irev) voltammogram. The time between sampling Ifor and Irev is set by the pulse width (tp), which is determined by the square-wave frequency (f = 1/2tp). The forward and reverse square-wave voltammograms resemble a cyclic voltammogram and can be used to assess the reversibility of the electrode process (26, 27). For a reversible electrode process, the peak potential of the net current (Enet) equals the voltammetry half-wave potential (E1/2). Typically, the half-wave potential closely approximates the formal potential (E0’). For a diffusion-controlled reversible redox system, E1/2 equals E0’ when the ratio of the diffusion coefficients of the reduced and oxidized species equals unity (24).
DPV conducted with a glassy carbon (GC) working electrode generates α3Y voltammograms with good signal-to-noise ratio (S/N) (17) (Fig. S2). In contrast, SWV in combination with a GC electrode generates α3Y voltammograms with significant lower S/N and this approach was not perused beyond a preliminary assessment. A series of control measurements were conducted to refine a pyrolytic graphite “edge” (PGE) electrode system for a SWV analysis of the α3Y electrode process. Fig. S3 summarizes experiments conducted to optimize sample conditions (Fig. S3 A and B) and to determine whether α3Y gives rise to diffusion-controlled or surface-confined electrode kinetics (Fig. S3D). Fig. S4 displays typical raw and background-corrected α3Y square-wave voltammograms collected at optimized sample conditions and provides details regarding data processing and analyses. Finally, Fig. S5 illustrates the reproducibility observed between data replicates and independent SWV measurements. The main conclusions from the data displayed in Figs. S2–S5 are summarized below and we refer to the SI Text for further details. (i) SWV samples were optimized at 20–100 μM α3Y in 20 mM sodium acetate, 20 mM potassium phosphate, 20 mM sodium borate (APB buffer), and 80–140 mM KCl. The S/N of the α3Y Faradaic current decreases above and below the 20–100 μM protein concentration range. The α3Y peak potential is independent of the KCl concentration above 10 mM KCl at alkaline pH (pH 8.4) and above 60 mM KCl at acidic pH (pH 5.5). The S/N of the α3Y voltammogram declines as the KCl concentration rises and accurate determination of peak amplitudes and potentials becomes increasingly difficult above 140 mM KCl. Based on these results, subsequent SWV studies were conducted at 80 μM α3Y dissolved in a 20 mM APB, 80 mM KCl buffer. (ii) The SWV net potential (Enet) and the DPV half-wave potential (E1/2) are displayed as a function of the α3Y concentration in Fig. S3 B and C, respectively. The SWV and DPV measurements were conducted using a PGE and GC electrode, respectively, and the α3Y concentration series was collected at both acidic and alkaline pH. A very weak to no correlation (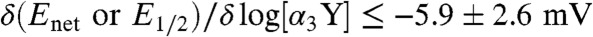 per decade) is observed for both electrode systems. We conclude that there are no distorting protein/working electrode surface interactions (such as self-inhibition, e.g., ref. 28) present at any conditions used in this report. (iii) Voltammograms were recorded at large SW pulse amplitudes to investigate the kinetic characteristics of α3Y on the PGE electrode (27, 29) (Fig. S3D). The observed response is consistent with diffusion-controlled electrode kinetics at both acidic and alkaline pH. (iv) The reproducibility in E1/2 obtained with the DPV/GC system is ± 3 mV for data replicates (Fig. S2 A and D–F) and ± 4 mV for independent measurements (Fig. S2I). The reproducibility in Enet observed for the SWV/PGE system is ± 3 mV for data replicates (Fig. S5 A–F) and ± 3 mV for independent measurements (Fig. S5 G–I). After correcting for small differences in pH, we find no significant difference (5 ± 5 mV) in the α3Y DPV E1/2 value obtained using a GC electrode (Figs. S2 and S3C) relative to a PGE electrode (Fig. S3A). This concludes the description of control experiments done in preparation for a SWV analysis of the α3Y electrode process and obtaining a Pourbaix diagram for the tyrosine radical protein.
per decade) is observed for both electrode systems. We conclude that there are no distorting protein/working electrode surface interactions (such as self-inhibition, e.g., ref. 28) present at any conditions used in this report. (iii) Voltammograms were recorded at large SW pulse amplitudes to investigate the kinetic characteristics of α3Y on the PGE electrode (27, 29) (Fig. S3D). The observed response is consistent with diffusion-controlled electrode kinetics at both acidic and alkaline pH. (iv) The reproducibility in E1/2 obtained with the DPV/GC system is ± 3 mV for data replicates (Fig. S2 A and D–F) and ± 4 mV for independent measurements (Fig. S2I). The reproducibility in Enet observed for the SWV/PGE system is ± 3 mV for data replicates (Fig. S5 A–F) and ± 3 mV for independent measurements (Fig. S5 G–I). After correcting for small differences in pH, we find no significant difference (5 ± 5 mV) in the α3Y DPV E1/2 value obtained using a GC electrode (Figs. S2 and S3C) relative to a PGE electrode (Fig. S3A). This concludes the description of control experiments done in preparation for a SWV analysis of the α3Y electrode process and obtaining a Pourbaix diagram for the tyrosine radical protein.
α3Y Gives Rise to Reversible Voltammograms.
Voltammetry measurements on α3Y are expected to follow an EC mechanism where “E” represents the electrode reactions (electron and proton transfers associated with the electrode-driven oxidation/reduction of Y32) and “C” represents coupled chemical reactions (i.e., side reactions of the formed Y32-O• radical). The kinetic and thermodynamic properties of both the E and C processes will influence the observed voltammogram (26, 27, 30–32). In SWV the time between the oxidative and the reductive pulse is set by the SW frequency, which has a practical range of 8–2,000 Hz (tp 62.5 ms–250 μs). This predicts that for an EC system with a very long-lived radical (i.e., t1/2 on the tens of milliseconds time scale), the SW frequency can be set to outcompete the follow-up chemical reaction(s). This would generate voltammograms with properties that only reflect the E process. Remarkably, the protein environment stabilizes the Y32-O• radical into the required time frame such that voltammograms representing a fully reversible electrode process could be obtained. This is shown below.
α3Y square-wave voltammograms were recorded using a frequency ranging from 30 Hz (tp 16.7 ms) to 960 Hz (tp 521 μs). The three top boxes in Fig. 1 display voltammograms from this data series. Background-corrected forward and reverse square-wave voltammograms collected at 190 Hz (tp 2.63 ms), 540 Hz (tp 926 μs), and 750 Hz (tp 667 μs) are shown in Fig. 1 A–C, respectively. Fig. S4 illustrates the quality of the raw voltammograms from this data series and describes methods used for background corrections and data analyses. The key results from the SW frequency series are summarized in Fig. 1 D and E. Fig. 1D shows the change in the peak potentials as a function of the SW frequency. Importantly, the net peak potential (Enet) and the peak potentials of the forward (Efor) and reverse (Erev) currents become independent of the SW frequency at 750 Hz (Fig. 1D). This is a hallmark characteristic of a fully reversible electrode process (26, 27). We further observe that Ifor/Irev declines from 1.71 ± 0.14 at 30 Hz and reaches a limiting value of 1.01 ± 0.02 for the 750 - 960 Hz range (Fig. 1E). Whereas Efor - Erev varies between -14 ± 1 and -8 ± 2 in the quasireversible range (30–540 Hz), the effects on Enet remain small with a change of 8 ± 2 mV between 30 and 120 Hz and only 4 ± 2 mV for the 120–960 Hz range (Fig. 1D). Enet equals 918 ± 2 mV vs. NHE for the 750–960 Hz range and represents the formal potential (E0’) of the Y32-O•/Y32-OH redox couple at pH 8.40 ± 0.01. This statement assumes that the diffusion coefficient of α3Y does not change as a function of the protein redox state. The diffusion coefficient of reduced α3Y was determined to be 1.47 ± 0.01 × 10-6 cm2 s-1 by pulsed field gradient NMR experiments (33) (Fig. S6). This value is typical for a small soluble protein and is not likely to change significantly as Y32 loses an electron and a proton.
Fig. 1.
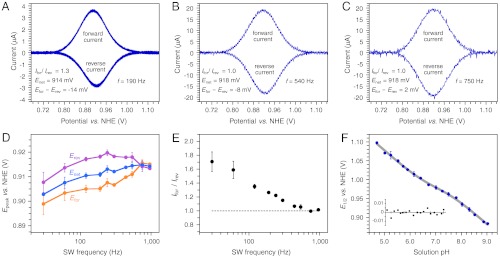
α3Y forward (oxidation) and reverse (reduction) square-wave voltammograms obtained at pH 8.40 ± 0.01 and using a SW frequency of (A) 190 Hz (tp 2.6 ms), (B) 540 Hz (tp 926 μs), and (C) 750 Hz (tp 667 μs). These voltammograms form part of a SW frequency series spanning from 30 Hz (tp 16.7 ms) to 960 Hz (tp 521 μs). (D) describes the change in the peak potential of the net (Enet), forward (Efor), and reverse (Erev) currents as a function of the SW frequency. E plots the ratio of the peak currents of the forward (Ifor) and reverse (Irev) components as a function of the SW frequency. SWV settings: 80 μM α3Y in 20 mM APB, 80 mM KCl, pH 8.40 ± 0.01; PGE working electrode, temperature 25 °C, step potential 0.15 mV, SW pulse amplitude 25 mV. (F) α3Y differential pulse half-wave potential (E1/2) of as a function of pH. The solid gray line represents a nonlinear regression curve fit to Eq. 1. The insert displays the corresponding fitting residuals. DPV settings: 200 μM α3Y in 10 mM APB, 100 mM KCl, GC working electrode, temperature 23 °C, interval time 0.1 s, step potential 1.95 mV, scan rate 19.5 mV s-1, modulation time 2 ms, modulation amplitude 25 mV.
The observation that Y32 gives rise to a fully reversible voltammogram means that the radical state is stable on the time scale of the electrochemical oxidation/reduction reactions. Theoretical treatments for EC systems where “E” represents a diffusion-controlled reversible electrode process and “C” is a coupled chemical reaction have been described for SWV (30, 31). They predict that the influence of the coupled chemical reaction on Enet approaches zero when log(2tpkEC) ≤ -1.5. This predicts that rate constants (kEC) associated with Y32-O• side reactions are ≤ 24 s-1, which, in turn, translates into a radical lifetime of ≥30 ms. The well-structured protein environment of the Y32 site clearly suppresses radical side reactions with a remarkable efficiently. Possible side reactions include intermolecular radical-radical and radical-protein reactions as well as intramolecular radical-protein reactions. For example, for freely solvated tyrosine the oxidized species rapidly dimerizes (4–8 × 108 M-1 s-1) to form Cortho-Cortho and Cortho-O dityrosine as the major and minor products, respectively. Intermolecular radical reactions are not expected to occur for the Y32 system to any significant extent. The Y32 site is located inside the protein, and the global stability of α3Y predicts that only 0.2% of the protein population is unfolded at any given time (17, 19). The absence of intermolecular side reactions is consistent with the insensitivity in the SWV Enet (190 Hz; Fig. S3B) and DPV E1/2 (Fig. S3C) potentials to the protein concentration. As a comparison, aqueous N-acetyl-tyrosinamide (NAYA) displays a δE1/2/δ log[NAYA] dependence of -30 ± 3 mV per decade (Fig. S3C) reflecting rapid intermolecular radical reactions. Intramolecular radical-protein reactions could occur in α3Y. 1H-1H nuclear Overhauser effect spectroscopy (NOESY) spectra show that the Y32 site is comprised of numerous aliphatic C-H groups (17, 19). This suggests that hydrogen-atom abstraction from C-H bonds may represent the dominant route for chemical reduction of Y32-O•. These reactions typically exhibit low rate constants (34). They are also predicted to be pH independent, which becomes important for the interpretation of the Pourbaix diagram derived for α3Y (vide infra).
Collecting SW voltammograms in the pH 5.5 region was more challenging for two reasons. The Faradaic current decreases at lower pH (e.g., compare Fig. 1 A and B to Fig. 2 A and B) whereas the Y32 potential increases to above +1.0 V vs. NHE. This makes background currents (arising from the aqueous solvent and the electrode itself) more pronounced. Nonetheless, voltammograms of satisfactory S/N were collected at 190 and 540 Hz. Background-corrected forward and reverse voltammograms obtained at pH 5.52 ± 0.01 are shown in Fig. 2. Examples of raw data triplicates of the net, forward, and reverse components are shown in Fig. S5 D–F, respectively. The properties of the low-pH voltammograms exhibit the same overall frequency dependence as the high-pH data. Ifor/Irev drops from 1.44 ± 0.03 (190 Hz) to 1.15 ± 0.01 (540 Hz), Efor - Erev decreases from -11 ± 1 mV (190 Hz) to -3 ± 1 mV (540 Hz), and a minor change from 1,066 ± 2 mV (190 Hz) to 1,070 ± 1 mV (540 Hz) is observed for Enet. These characteristics strongly suggest that the 540 Hz voltammogram represents the Y32 system at the upper edge of the quasireversible range and that Enet (pH 5.52; 540 Hz) deviates from E0’ (pH 5.52) by a few mV at most (27). We therefore conclude that 1,070 ± 1 mV vs. NHE closely approximates the formal potential of the Y32-O•/Y32-OH redox couple at pH 5.52 ± 0.01.
Fig. 2.

α3Y forward (oxidation) and reverse (reduction) square-wave voltammograms obtained at 5.52 ± 0.01 and using a SW frequency of (A) 190 Hz (tp 2.6 ms) and (B) 540 Hz (tp 926 μs). SWV settings: 80 μM α3Y in 20 mM APB, 80 mM KCl, pH 5.52 ± 0.01; PGE working electrode, temperature 25 °C, step potential 0.15 mV, SW pulse amplitude 25 mV.
Pourbaix Diagram of α3Y.
Fig. 1F shows the DPV E1/2 potential of α3Y as a function of the solution pH. The α3Y potential displays a nonlinear relationship across the investigated 4.7–9.0 pH range. The SWV analysis described above provides an explanation to the observed pH dependence. The formal potential of the Y32-O•/Y32-OH redox couple is 918 ± 2 mV at pH 8.40 ± 0.01. From the E1/2 vs. pH plot in Fig. 1F we extract a potential of 916 ± 3 mV at pH 8.40. There is no significant difference between the formal potential of the Y32-O•/Y32-OH redox couple as determined by SWV and the E1/2 value derived by DPV. Consequently, the α3Y SW voltammogram (pH 8.40; ≥750 Hz) and the DP voltammogram (pH 8.40) are both derived from a fully reversible electrode process.
The DPV E1/2 (pH 5.52) value is 22 ± 3 mV lower relative to the 1,070 ± 1 mV (pH 5.52; 540 Hz) value determined by SWV. Tyrosine oxidation is a PCET event, that is, the kinetic and thermodynamic properties of the electron transfer depend on the position of one (or several protons) at any given time (35). It is well established that the hydrogen-bonding properties of the phenol OH group and the characteristics of the base accepting the phenolic proton upon oxidation strongly influence the oxidation mechanism and associated PCET rate constants. The small 22 mV shift in the DPV E1/2 potential relative to the SWV Enet value is likely to arise from coupled protonic reactions lowering the Y32/electrode electron-transfer rate into the quasireversible regime for the DPV experiment. The alternative explanation is that kEC increases as the pH decreases and makes the system quasireversible. We favor the former explanation because the effects of coupled protonic reaction are expected to be highly sensitive to the pH (e.g., by pH-induced changes in the hydrogen-bonding properties of Y32) whereas kEC is not (vide supra).
Based on the discussion presented above, the α3Y Pourbaix diagram is modeled by (36):
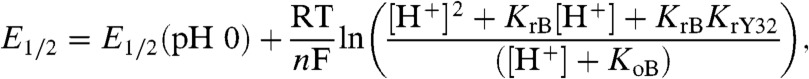 |
[1] |
where E1/2(pH 0) is the half-wave potential at pH 0 (V); KrY32 is the acid dissociation constant of Y32 in its reduced state (pKapp = 11.3, see ref. 17); KrB and KoB represent apparent acid dissociation constants (Kapp) of a protein residue B when Y32 is in its reduced and oxidized state, respectively; n is the number of electron equivalents required to convert one mol of oxidant to one mol of reductant; T is the temperature (296 K); R is the gas constant (8.314 J mol-1 K-1); and F is the Faraday constant (96.485 kJ V-1 mol-1). Eq. 1 was fitted to the α3Y Pourbaix diagram with the following results: E1/2(pH 0) = 1.37 ± 0.02 V, ln(10)RT/nF = 59 ± 5 mV, pKrB = 6.7 ± 0.2, and pKoB = 7.4 ± 0.2. The fitted line is displayed in gray in Fig. 1F and the fitting residuals are shown in the Inset. In this model, Y32 is thermodynamically coupled to a residue B in the protein matrix. Residue B is protonated at low pH and unavailable to participate in the protonic reactions associated with the oxidation of Y32. As the pH increases and approaches the pKrB range, residue B deprotonates and now becomes available to participate in the PCET reactions associated with Y32 oxidation. The redox-induced shift in the pKa of residue B gives rise to the nonlinearity of the Pourbaix diagram describing the pH dependence of the Y32-O•/Y32-OH redox couple.
Conclusions
This report describes three important experimental results. (i) Square-wave and differential pulse voltammograms representing a reversible electrode process have been obtained for a protein tyrosine radical. This represents a unique experimental demonstration of obtaining a formal potential for a protein Y-O•/Y-OH redox pair. (ii) The observation of reversible voltammograms translates into an estimated lifetime for the tyrosine radical of ≥30 ms. This is a remarkably long lifetime for a phenol-based radical and illustrates that the protein environment can stabilize the Y32-O• state by inhibiting deleterious radical side reactions. (iii) The first Pourbaix diagram for a protein tyrosine radical is described and analyzed. These results represent a significant advance that is long overdue considering that these important protein redox species were identified as early as 1977 (37). The historically prohibitive challenges associated with obtaining thermodynamic information for protein tyrosine radicals stem from the fact that electrochemical measurements must be performed at the very upper edge of the biological redox scale. Moreover, voltammetry on phenols typically follows an EC mechanism where the electrochemical reduction of the radical state is outcompeted by the coupled chemical reaction. This gives rise to irreversible voltammograms from which E1/2 potentials cannot be derived without independent information on the rate of the chemical follow-up reaction. Finally, the voltammetry measurements must be conducted in a pH-stable protein background and be sensitive to interactions at the radical site to generate a meaningful Pourbaix diagram for this PCET cofactor. These experimental barriers have been successfully overcome by the α3Y system.
The recently published high-resolution crystal structure of PSII provides a striking example as to why it is vital to understand the details associated with tyrosine-mediated PCET processes in nature. Seventeen years ago, Gerald T. Babcock and co-workers proposed a model for photosynthetic water oxidation in which PCET at the level of YZ provides the key mechanism by which protons formed during the water-splitting process are transported from the active site to the thylakoid lumen (38, 39). This hypothesis broke significantly with the contemporary metal-centered view of photosynthetic water oxidation as it proposed a mechanistically essential PCET function for the tyrosine rather than a traditional ET role in which YZ forms a simple link in an electron-transfer chain. The hypothesis of directional proton transfer driven by the YZ redox cycle gave rise to two core structural predictions: That YZ is in hydrogen-bonding contact with substrate molecules ligated to the metal cluster and that there is a proton exit channel from the tyrosine via its hydrogen-bonding partner D1-H190 to the thylakoid lumen. The recent 1.9 Å resolution crystal structure of PSII shows features remarkably consistent with these predictions (10). In light of the new PSII structure, a straightforward refinement of the 1995 hypothesis is to propose a “global” reprotonation of the YZ radical upon reduction with a proton from the extended hydrogen-bonding network connecting the metal/substrate/YZ molecules. This would allow directional PCET at the YZ site as originally proposed but remove the requirement of direct hydrogen-atom transfer from metal-ligated substrate to the YZ radical. Overall, the intriguing structural properties of the PSII active site will undoubtedly spark an even more intense interest in the amazing chemistry performed by protein tyrosine radicals.
Materials and Methods
Differential pulse and square-wave voltammetry measurements were performed using an Autolab PGSTAT12 potentiostat equipped with a three-electrode glass cell containing a GC or a PGE working electrode. The working electrode was carefully polished between each measurement. All samples were prepared using ultra-pure chemicals and the measurements made under an argon atmosphere. NMR spectra were collected using a 500 MHz Bruker Advance III spectrometer fitted with a cryo probe. Details regarding electrode preparations, voltammetry setup and measurements, data processing, and data analyses can be found in the SI Text.
Supplementary Material
ACKNOWLEDGMENTS.
Funding was provided by National Institutes of Health (NIH) Grant GM079190 and by NIH predoctoral fellowship GM096756 to M.C.M.R.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1112057109/-/DCSupplemental.
References
- 1.Stubbe J, Nocera DG, Yee CS, Chang MCY. Radical initiation in the class I ribonucleotide reductase: Long-range proton-coupled electron transfer? Chem Rev. 2003;103:2167–2201. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- 2.Hoganson CW, Tommos C. The function and characteristics of tyrosyl radical cofactors. Biochim Biophys Acta. 2004;1655:116–122. doi: 10.1016/j.bbabio.2003.10.017. [DOI] [PubMed] [Google Scholar]
- 3.Reece SY, Hodgkiss JM, Stubbe J, Nocera DG. Proton-coupled electron transfer: The mechanistic underpinning for radical transport and catalysis in biology. Phil Trans R Soc B. 2006;1472:1351–1364. doi: 10.1098/rstb.2006.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dempsey JL, Winkler JR, Gray HB. Proton-coupled electron flow in protein redox machines. Chem Rev. 2010;110:7024–7039. doi: 10.1021/cr100182b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dixon WT, Murphy D. Determination of the acidity constants of some phenol radical cations by means of electron spin resonance. J Chem Soc Faraday Trans II. 1976;72:1221–1230. [Google Scholar]
- 6.Harriman A. Further comments on the redox properties of tryptophan and tyrosine. J Phys Chem. 1987;91:6102–6104. [Google Scholar]
- 7.Tommos C, Babcock GT. Proton and hydrogen currents in photosynthetic water oxidation. Biochim Biophys Acta. 2000;1458:199–219. doi: 10.1016/s0005-2728(00)00069-4. [DOI] [PubMed] [Google Scholar]
- 8.Hays A-MA, Vassiliev IR, Golbeck JH, Debus RJ. Role of D1-His190 in the proton-coupled oxidation of tyrosine YZ in manganese-depleted photosystem II. Biochemistry. 1999;38:11851–11865. doi: 10.1021/bi990716a. [DOI] [PubMed] [Google Scholar]
- 9.Tommos C. Electron, proton and hydrogen-atom transfer in photosynthetic water oxidation. Phil Trans R Soc B. 2002;357:1383–1394. doi: 10.1098/rstb.2002.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Umena Y, Kawakami K, Shen J-R, Kamiya N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature. 2011;473:55–60. doi: 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- 11.Sjödin M, Styring S, Åkemark B, Sun L, Hammarström L. Proton-coupled electron transfer from tyrosine in a tyrosine-ruthenium-tris-bipyridine complex: Comparison with tyrosineZ oxidation in photosystem II. J Am Chem Soc. 2000;122:3932–3936. [Google Scholar]
- 12.Mayer JM. Proton-coupled electron transfer: A reaction chemist’s view. Annu Rev Phys Chem. 2004;55:363–90. doi: 10.1146/annurev.physchem.55.091602.094446. [DOI] [PubMed] [Google Scholar]
- 13.Huynh MHV, Meyer TJ. Proton-coupled electron transfer. Chem Rev. 2007;107:5004–5064. doi: 10.1021/cr0500030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reece SY, Nocera DG. Proton-coupled electron transfer in biology: Results from synergistic studies in natural and model system. Annu Rev Biochem. 2009;78:673–699. doi: 10.1146/annurev.biochem.78.080207.092132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Costentin C, Robert M, Savéant J-M. Concerted proton-electron transfers: Electrochemical and related approaches. Acc Chem Res. 2010;43:1019–1029. doi: 10.1021/ar9002812. [DOI] [PubMed] [Google Scholar]
- 16.Warren JJ, Tronic TA, Mayer JM. Thermochemistry of proton-coupled electron transfer reagents and its implications. Chem Rev. 2010;110:6961–7001. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martínez-Rivera MC, Berry BW, Valentine KG, Westerlund K, Hay S, Tommos C. Electrochemical and structural properties of a protein system designed to generate tyrosine Pourbaix diagrams. J Am Chem Soc. 2011;133:17786–17795. doi: 10.1021/ja206876h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pourbaix M. Atlas of Electrochemical Equilibria in Aqueous Solutions. 2nd English Ed. Houston, TX: National Association of Corrosion Engineers; 1974. [Google Scholar]
- 19.Tommos C, Skalicky JJ, Pilloud DL, Wand AJ, Dutton PL. De novo proteins as models of radical enzymes. Biochemistry. 1999;38:9495–9507. doi: 10.1021/bi990609g. [DOI] [PubMed] [Google Scholar]
- 20.Dai Q-H, et al. Structure of a de novo designed protein model of radical enzymes. J Am Chem Soc. 2002;124:10952–10953. doi: 10.1021/ja0264201. [DOI] [PubMed] [Google Scholar]
- 21.Westerlund K, Berry BW, Privett HK, Tommos C. Exploring amino-acid radical chemistry: Protein engineering and de novo design. Biochim Biophys Acta. 2005;1707:103–116. doi: 10.1016/j.bbabio.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 22.Hay S, Westerlund K, Tommos C. Moving a phenol hydroxyl group from the surface to the interior of a protein: Effects on the phenol potential and pKA. Biochemistry. 2005;44:11891–11902. doi: 10.1021/bi050901q. [DOI] [PubMed] [Google Scholar]
- 23.Sibert RS, Josowicz M, Barry BA. Control of proton and electron transfer in de novo designed, biomimetic hairpins. Am Chem Soc Chem Biol. 2010;5:1157–1168. doi: 10.1021/cb100138m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bard AJ, Faulkner LR. Electrochemical Methods: Fundamentals and Applications. 2nd Ed. New York: John Wiley & Sons, Inc.; 2001. [Google Scholar]
- 25.Parry EP, Osteryoung RA. Evaluation of analytical pulse polarography. Anal Chem. 1965;37:1634–1637. [Google Scholar]
- 26.Osteryoung J, O’Dea JJ. Square-wave voltammetry. In: Bard AJ, editor. Electroanalytical Chemistry. Vol 5. New York: Marcel Dekker; 1986. pp. 209–308. [Google Scholar]
- 27.Mirčeski V, Komorsky-Lovrić Š, Lovrić M. Square-wave voltammetry: Theory and applications. In: Scholz F, editor. Monographs in Electrochemistry. Berlin: Springer-Verlag; 2007. [Google Scholar]
- 28.Costentin C, Louault C, Robert M, Savéant J-M. The electrochemical approach to concerted proton-electron transfers in the oxidation of phenols in water. Proc Natl Acad Sci USA. 2009;106:18143–18148. doi: 10.1073/pnas.0910065106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jeuken LJC, McEvoy JP, Armstrong FA. Insights into gated electron-transfer kinetics at the electrode-protein interface: A square wave voltammetry study of the blue copper protein azurin. J Phys Chem B. 2002;106:2304–2313. [Google Scholar]
- 30.O’Dea JJ, Osteryoung J, Osteryoung RA. Theory of square wave voltammetry for kinetic systems. Anal Chem. 1981;53:695–701. [Google Scholar]
- 31.Miles AB, Compton RG. Simulation of square-wave voltammetry: EC and ECE electrode processes. J Phys Chem B. 2000;104:5331–5342. [Google Scholar]
- 32.Garay F, Milivoj Lovrić M. Square-wave voltammetry of quasi-reversible electrode processes with coupled homogeneous chemical reactions. J Electroanal Chem. 2002;518:91–102. [Google Scholar]
- 33.Zheng G, Prince WS. Simultaneous convection compensation and solvent suppression in biomolecular NMR diffusion experiments. J Biomol NMR. 2009;45:295–299. doi: 10.1007/s10858-009-9367-2. [DOI] [PubMed] [Google Scholar]
- 34.Mahoney LR, DaRooge MA. Kinetic behavior and thermochemical properties of phenoxy radical. J Am Chem Soc. 1975;97:4722–4731. [Google Scholar]
- 35.Hammes-Schiffer S, Stuchebrukhov AA. Theory of coupled electron and proton transfer reactions. Chem Rev. 2010;110:6939–6960. doi: 10.1021/cr1001436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clarke WM. Oxidation-Reduction Potentials of Organic Systems. Baltimore, MD: The Williams & Wilkins Company, Waverly Press Inc.; 1960. [Google Scholar]
- 37.Sjöberg B-M, Reichard P, Gräslund A, Ehrenberg A. Nature of free-radical in ribonucleotide reductase from escherichia-coli. J Biol Chem. 1977;252:536–541. [PubMed] [Google Scholar]
- 38.Tommos C, Tang X-S, Warncke K, Hoganson CW, Styring S, McCracken J, Diner BA, Babcock GT. Spin-density distribution, conformation, and hydrogen bonding of the redox-active tyrosine YZ in photosystem II from multiple electron magnetic-resonance spectroscopies: Implication for photosynthetic oxygen evolution. J Am Chem Soc. 1995;117:10325–10335. [Google Scholar]
- 39.Hoganson CW, Lydakis-Simantiris N, Tang X-S, Tommos C, Warncke K, Babcock GT, Diner BA, McCracken J, Styring S. A hydrogen-atom abstraction model for the function of YZ in photosynthetic oxygen evolution. Photosynth Res. 1995;46:177–184. doi: 10.1007/BF00020428. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.