

Abstract
Pseudomonas aeruginosa M18, a rhizosphere-isolated bacterial strain showing strong antifungal activity, can produce secondary metabolites such as phenazine-1-carboxylic acid and pyoluteorin (Plt). The LysR-type transcriptional regulator PltR activates the Plt biosynthesis operon pltLABCDEFG, the expression of which is induced by Plt. Here, we identified and characterized the non-conserved pltL promoter (pltLp) specifically activated by PltR and its upstream neighboring lys box from the complicated pltR–pltL intergenic sequence. The 22 bp palindromic lys box, which consists of two 9 bp complementary inverted repeats interrupted by 4 bp, was found to contain the conserved, GC-rich LysR-binding motif (T-N11-A). Evidence obtained in vivo from mutational and lacZ report analyses and in vitro from electrophoretic mobility shift assays reveals that the PltR protein directly bound to the pltLp region as the indispensable binding motif “lys box”, thereby transcriptionally activating the pltLp-driven plt operon expression. Plt, as a potential non-essential coinducer of PltR, specifically induced the pltLp expression and thus strengthened its biosynthetic plt operon expression.
Introduction
Pyoluteorin (Plt) is a Pseudomonas-produced antifungal compound with a resorcinol ring linked to a bichlorinated pyrrole moiety produced by a polyketide synthase–non-ribosomal peptide synthetase (PKS/NRPS) hybrid biosynthetic pathway [1]. The Plt biosynthetic gene cluster, which is approximately 32 kb long, has been cloned and characterized in Pseudomonas fluorescens Pf-5 [2], [3] and P. aeruginosa M18 [4], [5], and identified through complete genome sequencing in P. aeruginosa LESB58 [6]. The plt locus shows a high level of conservation in gene organization and size among these three Pseudomonas strains. P. aeruginosa M18 shares 99% nucleotide sequence identity in the plt gene locus with LESB58 [6], [7], but shows a certain degree of difference in nucleotide sequence, especially for the non-coding sequence from Pf-5 [8]. Intriguingly, strain LESB58 cannot produce Plt [9] because of a frameshift mutation in the pltB gene [6]. The plt gene cluster is composed of two pairs of divergently transcribed operons: one pair is the pltLABCDEFG and pltRM operons responsible for Plt biosynthesis and its activation, the other pair is the pltHIJKNO and pltZ operons involved in Plt export and its repression. However, the plt gene cluster has not yet been experimentally dissected for the pltR and pltL intergenic non-coding region (485 bp for Pf-5 and 652 bp for M18) containing a number of promoters and cis-acting elements in two opposite directions [2]–[5].
Plt biosynthesis is regulated by the Gac/Rsm regulatory cascade, a ubiquitous global regulatory pathway that consists of the two-component signal transduction system GacS/GacA, the sRNAs RsmY and RsmZ, and the RNA-binding repressor protein RsmA repressing access to the ribosome binding site (RBS) [10]–[13]. Our previous studies have also shown that Plt biosynthesis is strongly repressed by three quorum sensing (QS) systems, namely, LasI/LasR, RhlI/RhlR, and PQS/PqsR, in P. aeruginosa M18 [14]–[16]. Aside from these global regulatory systems, the pathway-specific regulator PltR, a typical LysR-type transcriptional regulator (LTTR), is responsible for the transcriptional activation of the pltLABCDEFG biosynthetic operon [3], [17]. Expression of the plt operon is induced by Plt [4], [18]. In addition, the TetR-type regulator PltZ negatively regulates the divergently transcribed pltHIJKNO ABC (ATP-binding cassette) export operon and, thus, indirectly downregulates Plt production [4], [5]. However, the molecular mechanism underlying the transcriptional activation and autoinduction of the plt biosynthetic operon, for example, PltR's target DNA motif, Plt's receptor protein, and the effect of Plt on PltR–DNA interaction, remains unknown.
The LTTR family represents one of the largest classes of bacterial transcriptional regulatory proteins [19], [20]. Typically, the conservative LTTR protein comprises a helix-turn-helix (HTH) DNA-binding motif at the N-terminus and a cofactor-binding motif at the C-terminus. LTTRs are mostly characterized as transcriptional activators of a single divergently transcribed gene or operon and negative autoregulators [19]. With the continuous identification of large numbers of LTTRs, some of them have been extended to function as global transcriptional activators or repressors of unlinked genes or operons involved in metabolism, QS, virulence, and so on. These LTTRs include PqsR, a response regulator in the PQS (Pseudomonas quinoline signal)-mediated QS system [14], [21]. Aside from repressing Plt biosynthesis, PqsR can also activate the phenazine-1-carboxylic acid (PCA) biosynthesis in P. aeruginosa M18 [14]. Two LTTRs with opposite effects on the plt operon, PltR and PqsR, likely differ in molecular regulatory mechanisms. The mechanisms of both remain uncharacterized.
Autoinduction of microbiological metabolism, including the QS signal and antibiotic biosynthesis, is widespread in the microbial community. The N-acyl homoserine lactones (AHLs) in Gram-negative bacteria typically induce the expression of their own LuxI-type biosynthetic genes in collaboration with the LuxR-type response regulators [22]. Aside from the autoinduction of Plt biosynthesis involved in the present study, an increasing number of secondary metabolites produced by Pseudomonas spp., including 2,4-diacetylphloroglucinol [23], PCA [24], and pyoverdine [25], have been shown to induce their own biosynthesis in a concentration-dependent manner.
The current study identifies three promoters in the pltL direction, including the non-conserved pltL promoter (pltLp) specifically activated by PltR, and one pltR promoter in the pltR direction within the intergenic region between the divergently transcribed pltL and pltR genes. The LTTR PltR directly binds to the pltLp region at the indispensable and intact 22 bp palindromic lys box and, thus, specifically activating plt operon expression. Plt, as a potential and non-essential cofactor of PltR, specifically induces the pltLp and thus strengthens the plt operon expression.
Results
Identification of the promoter in the intergenic region between divergently transcribed pltL and pltR genes
In the Plt biosynthetic structural, regulatory, and transport gene cluster of P. aeruginosa M18 (Genbank accession number AY394844), the divergently transcribed pltL and pltR genes were separated by a large and complex intergenic region (652 bp) that likely contains a number of promoters and regulatory elements. Generally, putative promoters predicted by the NNPP software (P>0.8) need further experimental confirmation through lacZ reporter fusion analysis. The NNPP analysis showed five putative promoters in both the pltR and pltL directions. These predicted promoter fragments were respectively amplified and fused with the promoterless lacZ gene in the plasmid pME6522 (Fig. 1A and 1C). β-Galactosidase activity expressed from these putative promoter–lacZ fusion plasmids were assayed in E. coli DH5α and P. aeruginosa M18. As shown in Fig. 1B, among the R1 to R4–lacZ fusion plasmids that carried five putative promoters in the pltR direction (the R3 fragment contains two putative promoters), only the R4–lacZ fusion expressed a significantly higher β-galactosidase activity in both DH5α and M18. The R1 to R3–lacZ fusions did not display noticeable differences in β-galactosidase expression from the empty plasmid pME6522 as the control. The result indicates that the R4 fragment, spanning from−116 bp to +10 bp relative to the putative TSS (transcriptional start site, +1) of pltR, harbors an actual pltR promoter that also shows a certain level of basal expression activity in E. coli DH5α.
Figure 1. Detection of promoter activity in the intergenic region between divergently transcribed pltR and pltLABCDEFG genes using the lacZ reporter gene.

(A and B) Promoter detection in the pltR direction. (A) Four fragments, R1 to R4, containing five putative promoters in the pltR direction (predicted by the NNPP promoter online prediction with a score cutoff 0.80) were fused with the promoterless lacZ gene on the plasmid pME6522. The R3 fragment includes two putative promoters. The putative +1 site indicates the putative TSS of the fifth predicted promoter. (B) β-Galactosidase expression (Miller Units) from above these lacZ fusion plasmids was assayed in both E. coli DH5α and P. aeruginosa M18 after 15 h of growth in KMB. The R4 fragment was detected to include a true promoter (shown by an asterisk). (C and D) Promoter detection in the pltLABCDEFG operon direction. (C) Five fragments, L1 to L5, which contain five putative promoters in the pltL direction, were respectively fused into the upstream of the promoterless lacZ gene on pME6522. Putative +1 indicates the putative TSS of the third predicted promoter. (D) β-Galactosidase expression from the above lacZ fusion recombined plasmids was measured in both DH5α and M18. The L1 fragment (marked with a triangle) exhibited a certain degree of promoter activity in DH5α, but not in M18. The other four fragments, L2 to L5, did not show any evident promoter activity. (E and F) Detection of promoter or its elements by prolongation analysis within the DNA fragment spanning from +10 to the pltL ATG, which was not predicted to carry promoters. (E) Four prolonging fragments based on L3, namely, L3–1 to L3–4, were respectively fused with the lacZ gene in pME6522. (F) The resulting lacZ fusion plasmids were assayed for β-galactosidase expressions in both DH5α and M18. The region covering +66 to +124 bp (relative to the putative +1 site) contained either a stronger promoter or its elements (marked with an asterisk). The L3–3–lacZ fusion expression significantly decreased, compared with that in the L3–2–lacZ fusion expression in E. coli DH5α, which implies that the promoter located at +66 to +124bp is not likely pltLp activated by PltR. Moreover, the region from +124 to +205 bp (marked with a triangle) possibly contains another promoter under the transcriptional activation of PltR. (G and H) Distinguishing pltLp activated by PltR from other promoters by deletion analysis. (G) Four fragments (L6 and its shortened derivative fragments, namely, L6–1 to L6–3), four continuously prolonged fragments based on L6–3 (L6-3-1 to L6-3-4), and the L6–4 fragment, were respectively cloned into pME6522. The L6 fragment was not predicted (score cutoff 0.60) to contain putative promoter regions. (H) The β-galactosidase expression from these recombinant lacZ reporter plasmids was analyzed in E. coli DH5α, P. aeruginosa M18, and the pltR mutant M18pltR. An intact promoter contained in the region from +66 to +124 (marked with an asterisk) was not controlled by PltR. The pltLp activated by PltR was located from +124 to +205 bp (marked with a triangle), relative to the putative +1 site.
Similarly, five predicted promoter fragments (L1 to L5) in the pltL direction were respectively fused with the lacZ gene of pME6522 (Fig. 1C). Two of the predicted promoters extended into the pltL ORF. In the M18 strain, all five putative promoters did not show any significant expression compared with the pME6522 control (Fig. 1D). However, the L1–lacZ fusion showed a relatively higher expression (approximately 160 Miller units) in E. coli DH5α, but was almost entirely inhibited in P. aeruginosa M18. This occurrence is probably due to the presence of negative specific repressors in M18. The 164 bp L1 fragment was partially overlapped with the pltR promoter (R4) and showed similar levels of promoter activity, implying that this region likely contains a bidirectional promoter.
With the highest score of 0.94 in the NNPP promoter prediction, the putative promoter contained in the L3 fragment was supposed to be the most probable pltL promoter. However, no promoter activity was detected in the L3 fragment by the above lacZ fusion analysis (Fig. 1D). For this, four fragments that continuously extended from the right side of L3, namely, L3–1 to L3–4, were respectively cloned into the plasmid pME6522 to construct the corresponding lacZ fusion reporter plasmids (Fig. 1E). The results of the β-galactosidase assay in the M18 strain showed that the L3–2–lacZ fusion displayed stronger expression (approximately 900 Miller units) in the presence of the fragment ranging from +66 bp to +124 bp relative to the putative +1 site (Fig. 1F), implying that this 59 bp fragment likely contains strong promoters or additional activating cis-regulatory motifs. In addition, the L3–2–lacZ fusion also expressed approximately 70 Miller units of β-galactosidase activity in E. coli DH5α. The L3–3–lacZ fusion carrying an additional extended sequence showed similar expression levels of β-galactosidase as the L3–2–lacZ fusion in the M18 strain. However, L3–3–lacZ fusion expression was inhibited to the level of the control plasmid in DH5α (Fig. 1E). These results suggest that the promoter activity expressed by the fragment ranging from +66 bp to +124 bp is probably independent of the transcriptional activator PltR and inhibited by the presence of one inhibitory motif within the region from +124 to +205 bp (relative to the putative +1). Therefore, the search for the PltR-activated pltLp in the +124 to +205 bp region was continued.
Based on the L3–1 to L3–4–lacZ fusion analysis results (Fig. 1E), the 235 bp L6 fragment (+19 bp to +253 bp) (Fig. 1G), which was not predicted by NNPP (even above a minimum promoter score of 0.6) to carry any promoters, could be speculated to actually harbor strong promoters. As expected, the L6–lacZ fusion showed stronger expression activity in the M18 strain (Fig. 1H). In addition, the expression of L6–lacZ fusion was almost entirely inhibited in the pltR mutant M18pltR (Fig. 1H), suggesting that one promoter contained in the L6 fragment is dependent on PltR. To narrow down the key promoter region, three lacZ fusion plasmids, which respectively contains three truncated fragments L6–1, L6–2 and L6–3, were constructed (Fig. 1G). These three lacZ fusions all expressed relatively higher levels of β-galactosidase activity in E. coli DH5α, P. aeruginosa M18, and M18pltR (Fig. 1H). Therefore, the L6–3 fragment ranging from +66 to +124 bp contains an intact PltR-independent promoter that may be a non-pltLp promoter.
Based on the differential expression between the L6–lacZ and L6–1–lacZ fusions in both E. coli DH5α and P. aeruginosa M18pltR (Fig. 1H), as well as the differential expression between the L3–2–lacZ and the L3–3–lacZ fusions in DH5α (Fig. 1F), the fragment from +124 bp to +205 bp contains a potential promoter activated by PltR. Another set of lacZ fusion plasmids, which contains four continually prolonged fragments L6-3-1 to L6-3-4 based on L6–3, were constructed and assayed for β-galactosidase expression in strains DH5α, M18, and M18pltR (Figs. 1F and 1H). Interestingly, the relatively higher level of L6–3–lacZ fusion expression was almost entirely inhibited by a short sequence prolongation (+124 bp to +143 bp) in all three strains. This inhibited expression in the L6-3-1 to L6-3-3–lacZ fusion was reversed to the greatest extent only in M18 when the L6-3-4 fragment extended to +205 (i.e., L6-3-4) (Fig. 1H). In addition, the L6–4–lacZ fusion expression was almost entirely abolished in both DH5α and M18pltR (Fig. 1H). These results strongly suggest that the pltLp activated by PltR is located from +124 to +205 bp. Moreover, this region was also shown to carry an inhibitory cis-element (+124 bp to +143 bp) for the upstream promoter contained in the L6–3 fragment covering from +66 bp to +124 bp relative to the putative pltL +1 site.
Mapping the TSSs of pltL and pltR
To better define the promoter region and cis-elements responsible for the regulation of plt operon expression, the TSS (+1) of the pltL and pltR genes were mapped using 5′RACE (5′Rapid amplification of cDNA end). The results are shown in Fig. 2. The actual +1 site of pltR, which is identical to the putative +1 site, was located at 63 bp upstream from the pltR translational start codon ATG (Fig. 2). The actual pltR promoter identified by 5′RACE was basically consistent with the putative pltR promoter (predicted by NNPP) contained in the R4 fragment (Fig. 1A).
Figure 2. Mapping the TSSs and promoters of pltR and pltL genes through 5′RACE and the above lacZ reporter analysis (Fig. 1).

PR, the pltR promoter; PL, the pltLp which needs to be specifically activated by PltR; PX, a non-PltR controlled promoter closely neighboring the upstream of PL; PY, another potential promoter silent in P. aeruginosa M18, but active in E. coli DH5α. The TSS (+1) of the pltR transcript was located at 63 bp upstream of the pltR translational start codon ATG, which is identical to the predicted +1 site. The TSS of the plt operon was located at 46 bp upstream of the pltL ATG. Closely linked to the pltLp with the Px promoter, a palindromic lys box, including two 9 bp inverted complementary sequences separated by 4 bases, is boxed and highlighted by two head-to-head arrows.
Relative to pltR, the composition of the promoters and regulatory elements in the pltL direction were more complex and diverse. The 5′RACE result shows that the pltL TSS is located 46 bp upstream from the pltL ATG site (Fig. 2). The L6 fragment (Fig. 1G), which was not predicted by NNPP to carry any promoter, actually contains two promoters, namely, the pltLp activated by PltR and its neighboring upstream promoter (Px). The putative −10 and −35 regions in these promoters differ substantially from each other and are also markedly different from the canonical −10 (TTGACA) and −35 (TATAAT) sequences. As positively activated genes or operons generally have poor −35 and/or −10 regions their transcription mostly relies on the assistance of an activator protein to stabilize promoter-RNAP interaction. The fact that PltR regulates the pltL promoter but not the Px promoter is consistent with the low level of conservation between the −10 and −35 regions of the two promoters. The Px promoter was located at the −143 bp to −86 bp relative to the pltL TSS (Fig. 2), which corresponds to the L6–3 fragment (Fig. 1G). However, several attempts to experimentally identify the TSS of the Px promoter failed. Remarkably, a perfect palindromic motif composed of two 9 bp reverse complementary sequences separated by 4 bp was sandwiched exactly between the pltL and Px promoters (Fig. 2). The preceding results show that the presence of half of this palindromic motif could abolish the expression of the Px promoter (Figs. 1G and 1H, L6-3-1). Furthermore, the 22-bp interrupted palindromic motif perfectly matched the conservative LTTR binding site (T-N11-A) [19]. Therefore, this motif, designated as the lys box hereafter, was supposed to function as the target site of the LTTR protein PltR, which was verified by the following experiments.
The pltLp promoter region (−84 bp to +1) was directly bound and activated by PltR
Among the two neighboring promoters (pltLp and Px) in the pltL direction, Px was not controlled by PltR (Figs. 1G and 1F). The next focus was on characterizing the PltR-activated pltLp. To confirm the activation function of PltR on the experimentally positioned pltLp, the pltLp–lacZ transcriptional fusion plasmid (p6522–pltLp) containing the pltLp and its upstream palindromic lys box (−84 bp to +1), was constructed and transformed into E. coli DH5α, P. aeruginosa M18, the pltR mutant M18pltR and its complementary strain M18pltR/pBBR–pltR. The β-galactosidase activity was measured in KMB to compare pltLp–lacZ fusion expression between the presence and absence of pltR. As shown in Fig. 3A, the expression of the pltLp–lacZ fusion was completely inhibited in M18pltR when compared with that (approximately 200 Miller units) in the wild-type strain M18. With the introduction of the pltR overexpression plasmid pBBR–pltR, the pltLp–lacZ expression in the pltR mutant was restored and even significantly increased, compared with that in the M18 strain. The result clearly demonstrates that the 85 bp promoter region comprising its neighboring lys box needed to be activated by PltR (Fig. 3A). In addition, no evident expression in the pltLp–lacZ fusion was detected in E. coli DH5α (Fig. 3A). The pltLp–lacZ expression was not restored in the DH5α strain harboring the double plasmids, pBBR–pltR, and p6522–pltLp (data not shown), indicating that the pltLp expression requires the participation of other activation factors aside from the indispensable activator PltR.
Figure 3. PltR transcriptionally activated pltLABCDEFG operon expression via direct binding to the pltLp region from −84 bp to +1.
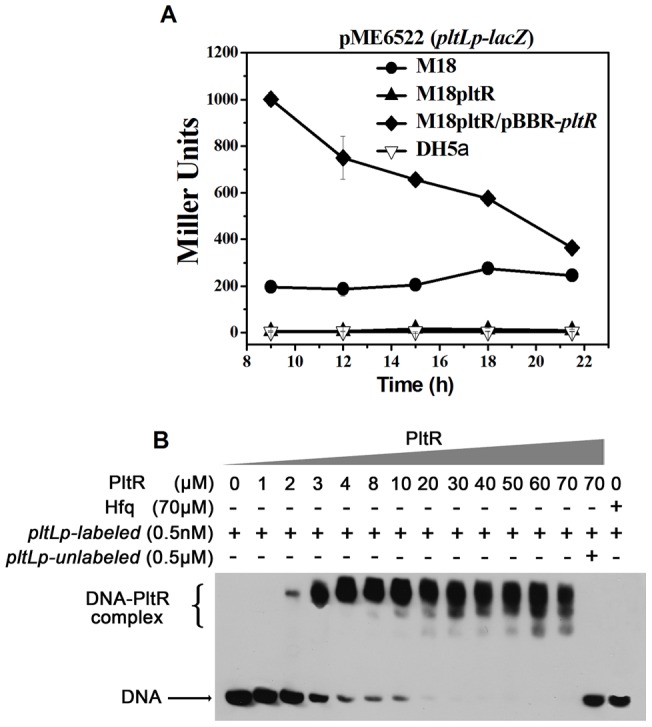
(A) Transcriptional activation of pltLp by PltR. The β-galactosidase expression (Miller Units) from pltLp–lacZ transcriptional fusion (p6522–pltLp) was assayed in M18, M18pltR, M18pltR/pBBR–pltR, and DH5α. The pltLp–lacZ expression was entirely inhibited in the pltR mutant M18pltR, which, in turn, was reversed and even enormously enhanced by the exogenous pltR overexpression from the plasmid pBBR–pltR. (B) Direct binding of PltR to pltLp, assessed by EMSA. 0.5 nM biotin-labelled pltLp promoter fragment (−84 bp to +1) were respectively incubated with increasing amounts of His–PltR protein (0 to 70 μM, Lane 1 to 13). For competition reactions, 0.5 μM unlabeled pltLp fragment was included in the binding reaction in molar as shown in Lane 14. The Hfq protein was used as a negative control (Lane 15).
EMSA was further designed to detect whether PltR directly binds to the pltLp region. The pltLp fragment (−84 bp to +1) was chosen to assess its binding affinity with PltR. As presented in Fig. 3B (Lanes 1 to 13), when 0.5 nM biotin-labeled pltLp fragments were incubated with increasing amounts of PltR protein (0 to 70 μM) in a 10 μl reaction system, the PltR protein showed specific binding activity with pltLp. Accompanied by the increasing concentration of PltR, the lagged bands representing the PltR-pltLp complex gradually strengthened while the biotin-labled free DNA probes significantly declined and was almost entirely bound by 20 μM PltR. The Hfq protein, as negative control, showed no binding activity with pltLp. A 1000-fold molar excess of unlabeled pltLp probes entirely displaced the biotin-labeled probes from PltR. Based on these data, it can be concluded that PltR directly binds the pltLp, thereby activating plt operon expression. This observation deserves further investigation to define the key sequence responsible for the binding of PltR to pltLp.
The 22 bp palindromic lys box is indispensable to the binding and activation of the pltLp by PltR
A putative secondary structure folded from pltLp and its upstream palindromic box (−84 bp to +1) by Mfold 3.2 is shown in Fig. 4A. The 22 bp lys box (−74 bp to −53 bp) constituting a typical T-N11-A LTTR binding site was supposed to form into a stem loop structure for binding and activation by PltR. Another two stem loops located from −47 bp to −4 bp were assumed as the cis-elements (−10 and −35 sequences) for RNA polymerase. Two pltLp–lacZ derivative plasmids, p6522–pltLp-2 and p6522–pltLp-3, which respectively carry a deletion of half or the entire lys box, were constructed to assess the effect of the lys box on pltLp activity (Fig. 4B). As shown in Fig. 4C, half and entire deletion of the lys box induced complete inhibition of the pltLp–lacZ fusion expression to approximately the expression level of the empty plasmid pME6522. Without the presence of the complete lys box, the pltLp could not be initiated. This confirms that the lys box is indispensable for the initiation and activation of the pltLp.
Figure 4. The 22 bp lys box (−74 to −53 bp) neighboring upstream of pltLp was indispensable to the transcriptional activation of PltR on the plt operon and the direct binding of PltR to pltLp.

(A) The putative DNA secondary structure (by MEME) of the pltLp region (−84 bp to +1). (B) The pltLp fragment (pltLp, −84 bp to +1) and two truncated fragments with a half or entire deletion of the palindrome motif (pltLp-2 and pltLp-3) were respectively fused with the lacZ reporter gene in pME6522. The palindrome lys box is shown with two thick arrows. (C) β-Galactosidase expression (Miller Units) from three lacZ fusion plasmids (p6522–pltLp, pltLp-2, pltLp-3) was assayed in the M18 strain. A half or entire deletion of the palindrome motif resulted in complete inhibition of the pltLp–lacZ expression in the M18 strain. (D) Direct binding of PltR to pltLp was eliminated by a half or entire deletion in the palindrome lys box. 0.5 nM pltLp and two truncated fragments (pltLp-2 and pltLp-3) were respectively incubated with increasing amounts of His–PltR protein.
Correspondingly, the biotin-labeled pltLp, pltLp-2, and pltLp-3 DNA probes (0.5 nM) were respectively utilized to carry out the EMSA analysis with increasing PltR concentrations (0 to 60 μM). The results are shown in Fig. 4D. The PltR protein displayed stronger binding affinity with the pltLp fragment with the intact lys box. As expected, half or whole deletion of the lys box completely abolished the binding of pltLp with PltR. No evident binding complexes were observed in the EMSA between the biotin-labeled pltLp-2 or pltLp-3 probe and the PltR protein (Fig. 4D, Lanes 4 to 9 and Lanes 10 to 15). The results suggest that PltR directly binds to the pltLp region at the indispensable and intact palindromic lys box and thus activate the pltLp expression.
Effects of point mutations in the lys box on the expression and PltR binding of pltLp
To determine the effects of the lys box mutations on plt operon expression and PltR–pltLp binding, four pltLp derivative fragments carrying a serial of point mutations in the lys box were amplified and used to construct lacZ fusion plasmids and carry out an EMSA analysis. To open the putative stem-loop structure formed by the lys box (Fig. 4A), 2 bp (M2), 4 bp (M4), and 6 bp (M6) substitutions were designed in one of two 9 bp reversely complementary sequences in the lys box (Fig. 5A). The other 4 bp replacement was introduced to the M4 mutant to be complementary to the originally replaced 4 bp, producing the M4-M pltLp mutant, which could regenerate a 22 bp stem-loop structure with different base arrangements from the wild-type pltLp. The β-galactosidase activity expressed from the pltLp–lacZ fusion wild-type plasmid (p6522–pltLp) and its four derived mutant plasmids were assayed in P. aeruginosa M18. As depicted in Fig. 5B, the pltLp–lacZ fusion expression was significantly reduced, even entirely abolished, by the four lys-box mutants (Fig. 5B). The β-galactosidase expression of pltLp–lacZ, pltLp-M2–lacZ, pltLp-M4–lacZ, to pltLp-M6–lacZ fusion, showed a gradual and significant decreasing trend with the gradual opening of the stem-loop structure, which suggests that the secondary structure of the lys box plays a key role in the binding and activation of the pltLp by PltR. In addition, the inhibited β-galactosidase expression from the pltLp-M4–lacZ fusion was not restored; it continued to decrease to the expression level of the empty plasmid pME6522 by a complementary 4 bp substitution carried by the M4-M mutant. This finding implies that the sequence specificity of the lys box is also essential for the binding and activation of the pltLp by PltR.
Figure 5. Effects of lys box mutations on pltLp expression and the interaction between PltR and pltLp.

(A) Construction map of the mutagenized pltLp with lys box mutations. Three pltLp mutant fragments, pltLp-M2, -M4, and -M6, respectively carried 2 bp, 4 bp, and 6 bp replacements in the lys box. The pltLp mutant M4-M was constructed by introducing a 4 bp complementary replacement into the pltLp-M4 mutant. The above four pltLp mutagenized fragments were respectively cloned into pME6522. (B) β-galactosidase expression (Miller Units) from the above lacZ fusion plasmids was measured in the M18 strain. The pltLp expression gradually and significantly decreased, even entirely inhibited, with the opening of the lys box formed stem loop (Fig. 4A) by base substitutions. (C) The binding of PltR to pltLp was significantly weakened and even eliminated by the lys box mutations. pltLp and its derivative mutated fragments (0.5 nM) were respectively incubated with increasing amounts of PltR protein. The concentration of PltR was 0, 10, and 60 μM, respectively.
Biotin-labeled DNA probes corresponding to the wild-type and mutagenized fragments of pltLp were subjected to an EMSA to assess the influence of point mutations in the lys box on the PltR–pltLp binding interaction. Subsequently, 0.5 nM biotin-labeled DNA probes were co-incubated with 0, 10, and 60 μM PltR. As shown in Fig. 5C, a 2 bp (or 4 bp, 6 bp) substitution and an 8 bp complementary substitution in the lys box resulted in significant decrease in the pltLp binding activity by PltR. The decrease was clearly reflected by an enormous reduction in the intensity of the retarded PltR–pltLp-M2 (or M4, M6, M4-M) complex bands, compared with those of the PltR–pltLp–wt complex bands. With the number of substituted bases increasing from 2 bp to 6 bp, the stem-loop structure formed by the lys box was opened step by step and thus the binding between the DNA probes and PltR gradually declined. Although the stem-loop structure was reshaped by a 4 bp complementary substitution in pltLp–M4-M, its binding with PltR was not restored to levels exceeding that of pltLp-M4 with PltR. This experiment confirms that the secondary structure and the primary sequence of the lys box are crucial for the PltR-activated expression of the plt operon.
Plt specifically induce the pltL promoter and thus enhance the plt operon expression
The autoinduction phenomenon, wherein Plt enhance the expression of its own biosynthetic operon pltLABCDEFG, has been reported in P. fluorescens Pf-5 [18] and P. aeruginosa M18 [4]. To assess whether the pltLp is specifically induced by Plt, two lacZ fusion plasmids, p6522–pltLp and p6522–L6-1, which respectively carry the pltLp and its upstream Px promoter, were transformed into M18pltB mutant not producing Plt. β-Galactosidase activity was assayed in KMB media amended without or with 10 µg ml−1 exogenous Plt. As depicted in Figs. 6A and 6B, the pltLp–lacZ fusion expression was significantly enhanced by the addition of exogenous Plt, whereas the Px–lacZ fusion expression did not reveal any noticeable difference between the KMB with and without 10 µg ml−1 exogenous Plt. The data clearly suggests that the PltR-activated pltLp was specifically induced by the secondary metabolite Plt. Moreover, the qRT-PCR result also shows that the transcript accumulation of pltA, which is the second gene in the pltLABCDEFG biosynthetic structural operon, in the M18pltB strain was strongly increased in KMB with 10 µg ml−1 exogenous Plt compared with that in KMB without the addition of Plt (Fig. 6C).
Figure 6. Exogenous Plt specifically induced the pltLp promoter expression and thus enhanced the plt operon transcription.
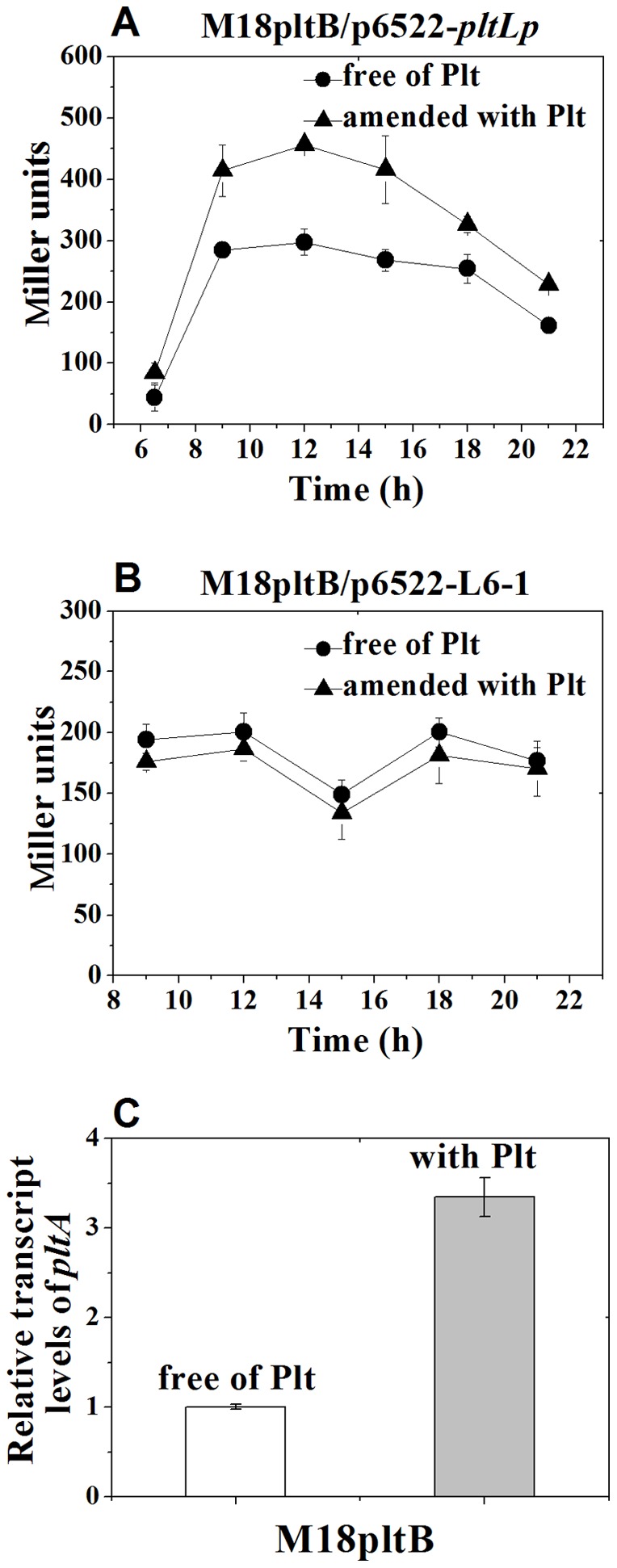
(A and B) β-galactosidase expression (Miller Units) from p6522–pltLp and p6522–L6-1, which respectively carry pltLp (−84 bp to +1) and its upstream Px promoter (+66 bp to +124 bp), was assayed in the KMB without or with 10 μg mL−1 exogenous Plt. The addition of 10 μg ml−1 exogenous Plt significantly enhanced the pltLp–lacZ expression (A), but did not influence PX–lacZ fusion expression (B) in the Plt-defective mutant M18pltB. (C) qRT-PCR analysis of the pltA transcript level in the strain M18pltB grown in the KMB without or with 10 μg ml−1 exogenous Plt. The pltA transcript level was significantly enhanced by the addition of exogenous Plt.
Discussion
In this study, we identified three promoters in the pltL direction, including the pltL promoter (pltLp) that is specifically activated by PltR and its upstream closely neighboring promoter (Px) that is not controlled by PltR, and one promoter (pltR promoter) in the pltR direction from the complex intergenic region between the divergently transcribed pltR and pltLABCDEFG operons (Fig. 2). PltR, a pathway-specific transcriptional activator of the plt operon, was shown to directly bind to the pltLp region at the indispensable lys box, a 22 bp palindromic sequence closely linking two neighboring promoters pltLp and Px. Plt, as a potential and non-essential cofactor of PltR, specifically induces the expression of the pltLp promoter and thus enhance Plt's own biosynthetic operon expression and biosynthesis. These are summarized in a proposed model describing transcriptional activation and autoinduction of Plt biosynthesis in P. aeruginosa M18 (Fig. 7A). In addition to the pathway-specific regulation, the plt biosynthetic operon is also subject to extensive global regulation from the Las, Rhl, and PQS QS systems [14]–[16] and the Gac/Rsm signal transduction system [12], [13].
Figure 7. Proposed model for the transcriptional activation of PltR and autoinduction of pyoluteorin on Plt biosynthetic operon in P. aeruginosa M18.

(A) Thick arrows indicate activation, autoinduction, and positive regulation; thin arrows indicate biogenesis. PL and PR denote the promoters of pltL and pltR. PX and PY are two uncharacterized promoters. The TSS is shown with +1. The LysR-type regulator PltR transcriptionally activates the plt operon expression by directly binding to the pltLp region at the indispensable palindromic lys box. PltR also functions as a candidate receptor of Plt. Plt, as a potential and nonessential cofactor of PltR, specifically induces the expression of the pltL promoter and thus the plt operon. (B) The PltR protein comprise two domains: the N-terminal LysR-family HTH (helix-turn-helix) DNA-binding domain and the C-terminal co-inducer binding domain (predicted by Pfam 25.0). aa, amino acids.
LTTRs are known to bind mostly at the palindromic lys box neighboring or overlapping the upstream region of the target promoter. The lys box typically consists of the T-N11-A sequence [19]. Similarly, as a typical LTTR protein, PltR contains a DNA-binding HTH motif for the potential binding of pltLp and a co-inducer binding motif as the candidate binding site of Plt (Fig. 7B). The target lys box of PltR, a 22 bp interrupted (by 4 bp) palindromic sequence, was localized from −53 bp to −84 bp upstream of the pltL gene TSS. Moreover, it also carries the classic T-N11-A sequence. Interestingly, this 22 bp palindromic sequence was shown (by BLAST analysis) to be the only homologous sequence commonly shared by two pltR–pltL intergenic regions from P. aeruginosa M18 and P. fluorescens Pf-5 (data not shown). This phenomenon suggests that the common mechanisms involved in PltR activation and Plt autoinduction may exist between both these strains.
Studies on transcriptional activation by several members of the LTTR family have revealed many mechanistic details, and the “sliding dimer” model, a general model for inducer-dependent activation, has been proposed [19], [26]. According to this model, two binding sites, designated RBS (regulatory binding site) and ABS (activation binding site), was respectively occupied by two dimers and thus induced to form a sharp bending. However, the pltLp activation by PltR may not comply with the “sliding dimer” model because of the presence of the Px promoter closely neighboring upstream of the pltLp. Intriguingly, the gradual opening of the lys box formed stem-loop through a 2 bp to 6 bp substitution gave rise to a significant decrease and even complete inhibition in both the pltLp activity and the PltR–pltLp binding. This decrease was not restored by the reformation of similar stem-loops via a complementary mutation of a 4 bp replacement (Figs. 4A and 5). Thus, not only secondary structure but also primary sequence of the target DNA may be critical for the specific binding of PltR–pltLp.
Autoinduction is widespread in gene regulation involved in bacterial secondary metabolism, such as QS signal and antibiotic biosynthesis [9], [18], [23], [25]. In contrast to that the transcriptional activator PltR mediated the induction of plt operon expression by Plt, the TetR-type transcriptional repressor PhlF was confirmed to mediate the induction of phl operon expression by 2,4-DAPG (diacetylphloroglucinol) in P. fluorescens CHA0 [23]. PhlF represses phlA transcription by binding to the phlA operator in P. fluorescens F113 [27], which in turn can be derepressed potentially through the dissociation of PhlF from the phlA operator by 2,4-DAPG [23]. Phloroglucinol, a precursor of 2,4-DAPG, was reported to mediate cross-talk between Plt and 2,4-DAPG biosynthetic pathways in P. fluorescens Pf-5 [9]. In the present study, Plt may directly bind to its candidate receptor PltR, thereby promoting the binding of PltR with the pltLp at the lys box and transcriptional activation efficiency of PltR. However, further research is needed to confirm the direct binding of Plt to PltR and to assess whether the Plt binding causes the binding enhancement and allosteric transition of the PltR–DNA complex.
Two promoters, pltLp and Px (Fig. 2), which were not identified by the NNPP prediction program, might not carry the conserved promoter sequence. The concurrent presence of three promoters in the pltL direction implies that these promoters may be recognized by different sigma factors in response to different signals. An rpoS mutant or multiple copies of rpoD reportedly stimulates the overproduction of Plt and DAPG in P. fluorescens Pf-5 and CHA0 [28], [29] and Plt overproduction in P. aeruginosa M18 [30], [31]. Further research is needed to identify the TSS and function of Px, define the relationship between the pltLp and Px, and assess the possibility of the occurrence of sRNA within the pltR–pltL intergenic region. In addition, the result that the LacZ expression deriving from p6522-L1 was started in E. coli DH5α, whereas was entirely inhibited in P. aeruginosa M18 (Figs. 1C and 1D) suggests that a potential promoter (PY) in the pltL direction is far from the neighboring pltLp and Px promoters. The PY promoter was shown to partly overlap with the pltR promoter (PR) (Figs. 1A and 1C); they displayed similar expression in E. coli DH5α(Figs. 1B and 1D). However, whether the PY and PR promoters constitute a bidirectional promoter remains unknown.
In summary, pltLp and its upstream neighboring lys box have been identified and characterized for PltR binding from the complicated pltR–pltL intergenic region. Plt, a potential and non-essential cofactor of PltR, specifically induces the pltLp expression and thus strengthens its own plt biosynthetic operon. Further work is needed to assess the possibility for the direct binding of Plt by PltR and ascertain whether the Plt binding causes the enhancement and conformation transition of PltR-pltLp binding and thereby promotes the transcriptional activation efficiency of PltR. In addition, we will focus on elaborating the complicated regulatory mechanisms hidden within the 652 bp pltR–pltL intergenic sequence, including the promoters, cis-elements, and potential sRNAs involved in the regulation of Plt biosynthesis in P. aeruginosa M18.
Materials and Methods
Bacterial strains, plasmids, oligonucleotides, and culture conditions
The bacterial strains, plasmids, and primers used in the present study are listed in Table 1 and Table S1. Escherichia coli was cultured in Luria–Bertani (LB) broth at 37°C [32]. P. aeruginosa M18 and its derivatives were grown in King's medium B (KMB) at 28°C [33]. When needed, 0.5 mM IPTG and 20 μg ml−1 X-Gal were used for blue/white screening, and 4 mg ml−1 ortho-nitrophenyl-β-d-galactopyranoside (ONPG) in 100 mM phosphate buffer (pH 7.0) were used for β-galactosidase assays. Antibiotics were added in the following final concentrations (μg ml−1): for Pseudomonas, spectinomycin (Sp) 100, tetracycline (Tc) 120, gentamicin (Gm) 45, kanamycin (Km) 50 and chloramphenicol (Cm) 200 for Pseudomonas; Tc 15, Gm 10, Km 50 and Cm 40 for E. coli.
Table 1. Bacterial strains and plasmids used in this study.
| Strains/plasmids | Genotype and relevant characteristics | Reference |
| E. coli | ||
| DH5α | supE44ΔlacU169(Φ80lacZΔM15)hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | |
| BL21 (DE3) | E.coli B, F- , ompT, hsdSB(rB-mB-), gal, dcm (DE3) | |
| P. aeruginosa | ||
| M18WT | Wild type | Huang et al. [5] |
| M18pltR | pltR::Gmr, Spr Gmr | Yan et al. [15] |
| M18pltB | pltB::Gmr, Spr Gmr | Huang et al. [4] |
| Plasmids | ||
| pME6522 | pVS1-p15A E.coli – Pseudomonas shuttle vector for transcriptional lacZ fusions and promoter probing, Tcr | Blumer et al. [38] |
| p6522-R1 to R4 | Four fragments, R1 to R4, which respectively harbor five putative promoters in the pltR direction (R3 includes two putative promoters), were fused with the upstream of promoter-less lacZ gene on pME6522, Tcr | This study |
| p6522-L1 to L5 | Five fragments, L1 to L5, which contain five putative promoters in the pltL direction, were respectively fused into the upstream of lacZ gene in pME6522, Tcr | This study |
| p6522-L3-1 to L3-4 | Four prolonging fragments based on L3, L3-1 to L3-4, was respectively fused with the lacZ gene in pME6522, Tcr | This study |
| P6522-L6 | The L6 fragment (covering 235 bp upstream the pltL translational start codon) in the pltL direction, which was not predicted to contain promoter, was fused into the upstream of lacZ gene in pME6522, Tcr | This study |
| P6522-L6-1 to L6-3 | Three shortened derivative fragments based on L6, L6–1 to L6–3, and the L6–4 fragment, were respectively cloned into pME6522, Tcr | This study |
| p6522-L6-3-1 to L6-3-4 | Five continuously prolonged fragments based on L6–3, L6-3-1 to L6-3-4, were respectively cloned into pME6522, Tcr | This study |
| p6522-pltLp | The 85 bp pltL promoter fragment was cloned into pME6522, Tcr | This study |
| p6522-pltLp-2 to pltLp-3 | Two mutagenized pltL promoter fragments (64 bp) with a half or entire deletion of palindromic lys box were respectively cloned into pME6522, Tcr | This study |
| p6522-pltLp-M2 to pltLp-M6 | Three mutant fragments of pltLp promoter, pltLp-M2, -M4, and -M6, which respectively carry 2-bp, 4-bp, and 6-bp replacement in the lys box, were cloned into pME6522, Tcr | This study |
| P6522-pltLp-M4-M | The pltLp-M4-M fragment, which was generated by introducing a 4-bp complementary replacement in the lys box into the pltLp-M4 mutant, was cloned into pME6522; Tcr | This study |
| pBBR1MCS | Cloning vector, broad host range, IncP IncQ, Cmr | Kovach et al. [39] |
| pBBR-pltR | A 1198 bp BamHI-HindIII fragment containing the 1032 bp entire ORF of pltR and its upstream promoter/operator region was cloned into pBBR1MCS, Cmr | This study |
| pET28a | T7 expression vector, Kmr | Novagen |
| pET-pltR | A 1052 bp NdeI-HindIII fragment containing the 1032 bp entire ORF of pltR was cloned into pET28a, Kmr | This study |
DNA manipulation and analyses
All molecular biology methods not depicted in detail were carried out using standard procedures [32]. General molecular biological reagents or kits were used according to the manufacturers' specifications, including Taq, LA Taq (TaKaRa), and KOD plus DNA polymerase (Toyobo), restriction endonucleases, DNA ligase, DNA and protein markers, plasmid DNA (TaKaRa or BioDev) and genomic DNA extraction kits (BBI), and DNA gel recovery kits (Qiagen or Axygen). DNA was synthesized by Shanghai Sangon Biological Engineering Technology and Services Co., Ltd., and sequenced by the Invitrogen Biotechnology Corporation and the Beijing HuaDa Genomics Institute.
Neural Network Promoter Prediction (NNPP) software (v. 2.2) was used to identify putative promoter elements of the pltR–pltL intergenic region, with a minimum promoter score of 0.8 [34]. The secondary structure of the pltLp sequence was predicted by Mfold 3.2 at 28°C. The most stable structure was chosen when multiple structures were generated [35]. The probable protein domain analysis of PltR was performed using Pfam [36].
5′Rapid amplification of cDNA end (5′RACE) for identifying the transcriptional start sites (TSS) of pltR and pltLABCDEFG
5′RACE was performed using an Invitrogen 5′RACE system. Total RNAs were isolated using the NucleoSpin® RNA II kit (Macherey–Nagel). The first strand cDNAs of pltR and pltL were respectively synthesized from total RNA templates using DNA specific primers Ptsr1 and Ptsl1, and purified with a S.N.A.P column. A homopolymeric C tail was then added to the 3′-end of cDNAs using deoxynucleotidyl transferase (TdT) and dCTP. PCR amplification of the dC-tailed cDNA was carried out using the novel deoxyinosine-containing abridged anchor primer (AAP) and the nested DNA specific primers Ptsr2 or Ptsl2. The PCR products were purified and cloned into the pMD18-T vectors (TaKaRa). The TSSs were identified through the sequencing of multiple independent clones.
Construction of lacZ fusion plasmids and β-galactosidase activity assay
The reporter plasmid pME6522, which carries a promoterless E. coli lacZ gene downstream from a multiple cloning site and encodes Tc resistance, was used to probe unknown promoter activity or construct transcriptional fusion plasmid. As shown in Figs. 1A and 1C, two sets of DNA fragments containing all putative promoters (predicted by NNPP, P>0.8) at the 5′-terminals of pltR and pltL operons were respectively amplified and cloned into the EcoRI/PstI-digested pME6522 to identify actual promoter activity. Four fragments (L3–1 to L3–4), which extend continuously from the right of the L3 fragment, were cloned into pME6522 to probe for potential promoter activity and location within the L6 fragment not predicted to carry any promoters of the pltLABCDEFG operon (Fig. 1E). The L6 fragment and its eight systematically deleted fragments were inserted into the plasmid pME6522 to analyze the cis-regulatory elements in the 5′-leader of the plt operon, including promoters and the PltR's acting site (Fig. 1G).
The pltLp–lacZ transcriptional fusion plasmid (p6522–pltLp) was constructed by introducing a 85 bp DNA fragment, which ranges from −84 bp to the actual TSS (+1) and harbors the pltLp and its upstream neighboring palindromic lys box (Fig. 2A), into the plasmid pME6522, thereby generating p6522–pltLp (Fig. 3A and 4B). The pltLp fragments with deletion of half or the entire lys box were cloned into pME6522, generating two fusion plasmids, p6522–pltLp-2 and p6522–pltLp-3 (Fig. 4B). All recombined lacZ reporter plasmids in this study have been verified by sequencing with a lacZ primer.
P. aeruginosa M18 and its derivative strains carrying the lacZ reporter plasmids above were inoculated from the overnight culture into 500-ml Erlenmeyer flasks containing 100 ml KMB media to a final concentration of OD600 = 0.05, and grown at 28°C with shaking at 200 rpm. β-Galactosidase activities were measured according to the method by Miller [37] and as described by Huang et al. [5].
qRT-PCR
P. aeruginosa M18 and its derivative mutant M18pltB were grown in KMB with or without 10 µg ml−1 Plt. Cells were harvested from 0.5 ml of cultures with an OD600 of 3.5 to 5 and submitted to quantify the accumulation level of pltA mRNA by quantitative reverse transcription PCR (qRT-PCR). Total RNA was isolated with NucleoSpin RNA II kit (Macherey–Nagel) and treated with RQ1 RNase-free DNase (Promega). First strand cDNA was synthesized using RevertAid™ First Strand cDNA Synthesis Kit (MBI Fermentas) and quantified using a RealMasterMix (SYBR Green) RT-PCR Kit (Tiangen). The qRT-PCR amplicons of pltA (157 bp) and the reference gene rpoD (173 bp) were synthesized from their first strand cDNA templates with the corresponding gene-specific primers. The results were normalized using the constitutively expressed gene rpoD as the internal control. Experiments were carried out in triplicate.
Overexpression and purification of PltR
An 1178 bp fragment, including the entire pltR gene (1032 bp) and its operator/promoter region (146 bp), was amplified and cloned into the E. coli–Pseudomonas shuttle vector pBBR1MCS at the BamHI and HindIII sites, thus constructing the pltR overexpression plasmid pBBR–pltR.
The entire pltR ORF (1032 bp) was amplified and inserted into the NdeI/BamHI-digested pET28a plasmid. The resulting plasmid, pET–pltR, was transformed into E. coli BL21 for overexpression and purification of the C-terminus His6–tagged PltR protein. E. coli BL21 carrying the pET28a–pltR plasmid was grown at 37°C in 1000 ml LB media, with shaking to an OD600 of 0.8, and induced with 0.5 mM IPTG. Subsequently, cell cultures were incubated at 16°C for 16 h. The cells were harvested by centrifugation and resuspended in 10 ml nickel A buffer (20 mM imidazole, 300 mM NaCl, 25 mM Tris–HCl at pH 8.0). The resuspending solution was added 1 μg ml−1 leupeptin, 1 μg ml−1 aprotinin, and 50 μM phenylmethylsulfonyl fluoride (PMSF) and shaken at 4°C for 30 min. The cells were lysed in ultrasonic cell disruptor. The protein extract was loaded onto a Ni–NTA agarose column (GE healthcare) and washed twice with 5 ml nickel buffer. The PltR protein was eluted with the elution buffer containing 500 mM imidazole. The purified PltR protein was stored in a buffer containing 20 mM Tris–HCl (pH 8.0), 200 mM NaCl, 1 mM DTT, and 1 mM EDTA at −80°C after removing the imidazole by HPLC (AKTA).
Electrophoretic mobility shift assay (EMSA) of DNA–protein interaction
DNA probes were generated by PCR with the corresponding biotin-labeled primers (Table S1). EMSA was performed using a Thermo Scientific LightShift Chemiluminescent EMSA Kit (No. 20148). A total of 0.5 nM biotin-labeled DNA probes were incubated with increasing concentrations of PltR protein in 10 ul binding buffer. The reaction mixtures were incubated for 20 min to 40 min at room temperature. Subsequently, 6 ul of the samples were separated by electrophoresis on 6% native polyacrylamide gel and electrotransferred to a positively charged nylon membrane (Ambion). The transferred PltR–DNA complexes and free DNA probes were cross-linked to the membrane for 2 min with a 320 nm UV light cross-linking instrument. Finally, the biotin-labeled bands were detected using a Thermo Scientific Chemiluminescent Nucleic Acid Detection Module.
Site-directed mutagenesis of the 22 bp palindromic lys box
Three mutants of the pltLp region (−84 bp to +1), which respectively carried 2 bp (M2: G74T, C73A), 4 bp (M4: G74T, C73A, T71G, T70C), and 6 bp replacements (M6: G74T, C73A, T71G, T70C, G68A, C67T) in the lys box (Fig. 5A), were generated by PCR amplification with the respective mutant forward primer and the same non- or biotin-labeled reverse primer (Table S1). Similarly, a 4 bp complementary replacement was introduced to the M4 mutant to construct the M4-M (G74T, C73A, T71G, T70C, and A57G, A56C, G54T, C53A) pltLp mutant (Fig. 5A). The mutant PCR products were cloned into the EcoRI/PstI-digested pME6522, producing the corresponding lacZ fusion reporter plasmids. In addition, the biotin-labeled mutant PCR products were used for the EMSA analysis.
Effect of exogenous Plt on the pltLp and plt operon expression
The pltB mutant M18pltB carrying the lacZ reporter plasmid p6522–pltLp or p6522–L6-1 was grown in KMB with or without Plt at a final concentration of 10 μg ml−1 (dissolved in methanol). β-Galactosidase activity was measured to confirm the specific induction of pltLp by Plt. Similarly, the pltA mRNA level of the M18pltB strain was assayed by qRT-PCR in KMB with or without 10 μg ml–1 Plt.
Supporting Information
Primers used in this study.
(DOC)
Acknowledgments
We thank Dr. Dieter Hass for providing the lacZ fusion reporter plasmid pME6522 in this study. We thank Dr. Geng Wu for providing the protein purification equipment.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was supported by grants from the National Natural Science Foundation of China (No. 30800009), the National High-Tech Research and Development Program (863 Program) of China (No. 2007AA02Z215), the National Key Basic Research Program (973 Program) of China (No. 2009CB118906), the Key Project of the Shanghai Committee of Science and Technology (No. 08391911900), and the Shanghai Leading Academic Discipline Project (No. B203). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Bender C, Rangaswamy V, Loper J. Polyketide production by plant-associated Pseudomonads. Annu Rev Phytopathol. 1999;37:175–196. doi: 10.1146/annurev.phyto.37.1.175. [DOI] [PubMed] [Google Scholar]
- 2.Brodhagen M, Paulsen I, Loper JE. Reciprocal regulation of pyoluteorin production with membrane transporter gene expression in Pseudomonas fluorescens Pf-5. Appl Environ Microbiol. 2005;71:6900–6909. doi: 10.1128/AEM.71.11.6900-6909.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nowak-Thompson B, Chaney N, Wing JS, Gould SJ, Loper JE. Characterization of the pyoluteorin biosynthetic gene cluster of Pseudomonas fluorescens Pf-5. J Bacteriol. 1999;181:2166–2174. doi: 10.1128/jb.181.7.2166-2174.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang X, Yan A, Zhang X, Xu Y. Identification and characterization of a putative ABC transporter PltHIJKN required for pyoluteorin production in Pseudomonas sp. M18. Gene. 2006;376:68–78. doi: 10.1016/j.gene.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 5.Huang X, Zhu D, Ge Y, Hu H, Zhang X, et al. Identification and characterization of pltZ, a gene involved in the repression of pyoluteorin biosynthesis in Pseudomonas sp. M18. FEMS Microbiol Lett. 2004;232:197–202. doi: 10.1016/S0378-1097(04)00074-6. [DOI] [PubMed] [Google Scholar]
- 6.Winstanley C, Langille MG, Fothergill JL, Kukavica-Ibrulj I, Paradis-Bleau C, et al. Newly introduced genomic prophage islands are critical determinants of in vivo competitiveness in the Liverpool Epidemic Strain of Pseudomonas aeruginosa. Genome Res. 2009;19:12–23. doi: 10.1101/gr.086082.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu DQ, Ye J, Ou HY, Wei X, Huang X, et al. Genomic analysis and temperature-dependent transcriptome profiles of the rhizosphere originating strain Pseudomonas aeruginosa M18. BMC Genomics. 2011;12:438. doi: 10.1186/1471-2164-12-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paulsen IT, Press CM, Ravel J, Kobayashi DY, Myers GS, et al. Complete genome sequence of the plant commensal Pseudomonas fluorescens Pf-5. Nat Biotechnol. 2005;23:873–878. doi: 10.1038/nbt1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kidarsa TA, Goebel NC, Zabriskie TM, Loper JE. Phloroglucinol mediates cross-talk between the pyoluteorin and 2,4-diacetylphloroglucinol biosynthetic pathways in Pseudomonas fluorescens Pf-5. Mol Microbiol. 2011;81:395–414. doi: 10.1111/j.1365-2958.2011.07697.x. [DOI] [PubMed] [Google Scholar]
- 10.Haas D, Keel C. Regulation of antibiotic production in root-colonizing Pseudomonas spp. and relevance for biological control of plant disease. Ann Rev Phytopathol. 2003;41:117–153. doi: 10.1146/annurev.phyto.41.052002.095656. [DOI] [PubMed] [Google Scholar]
- 11.Lapouge K, Schubert M, Allain FHT, Haas D. Gac/Rsm signal transduction pathway of gamma-proteobacteria: from RNA recognition to regulation of social behaviour. Mol Microbiol. 2008;67:241–253. doi: 10.1111/j.1365-2958.2007.06042.x. [DOI] [PubMed] [Google Scholar]
- 12.Zhang X, Wang S, Geng H, Ge Y, Huang X, et al. Differential regulation of rsmA gene on biosynthesis of pyoluteorin and phenazine-1-carboxylic acid in Pseudomonas sp. M18. W J Microbiol Biotechnol. 2005;21:883–889. [Google Scholar]
- 13.Ge Y, Huang X, Wang S, Zhang X, Xu Y. Phenazine-1-carboxylic acid is negatively regulated and pyoluteorin positively regulated by gacA in Pseudomonas sp. M18. FEMS Microbiol Lett. 2004;237:41–47. doi: 10.1016/j.femsle.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 14.Lu J, Huang X, Li K, Li S, Zhang M, et al. LysR family transcriptional regulator PqsR as repressor of pyoluteorin biosynthesis and activator of phenazine-1-carboxylic acid biosynthesis in Pseudomonas sp. M18. J Biotechnol. 2009;143:1–9. doi: 10.1016/j.jbiotec.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 15.Yan A, Huang X, Liu H, Dong D, Zhang D, et al. An rhl-like quorum-sensing system negatively regulates pyoluteorin production in Pseudomonas sp. M18. Microbiology. 2007;153:16–28. doi: 10.1099/mic.0.29211-0. [DOI] [PubMed] [Google Scholar]
- 16.Chen Y, Wang X, Huang X, Zhang X, Xu Y. Las-like quorum-sensing system negatively regulates both pyoluteorin and phenazine-1-carboxylic acid production in Pseudomonas sp. M18. Sci China C Life Sci. 2008;51:174–181. doi: 10.1007/s11427-008-0026-8. [DOI] [PubMed] [Google Scholar]
- 17.Yan A, Wang X, Zhang X, Xu Y. LysR family factor PltR positively regulates pyoluteorin production in a pathway-specific manner in Pseudomonas sp. M18. Sci China Ser C-Life Sci. 2007;37:325–332. doi: 10.1007/s11427-007-0054-9. [DOI] [PubMed] [Google Scholar]
- 18.Brodhagen M, Henkels MD, Loper JE. Positive autoregulation and signaling properties of pyoluteorin, an antibiotic produced by the biological control organism Pseudomonas fluorescens Pf-5. Appl Environ Microbiol. 2004;70:1758–1766. doi: 10.1128/AEM.70.3.1758-1766.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maddocks SE, Oyston PC. Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology. 2008;154:3609–3623. doi: 10.1099/mic.0.2008/022772-0. [DOI] [PubMed] [Google Scholar]
- 20.Schell MA. Molecular biology of the LysR family of transcriptional regulators. Annu Rev Microbiol. 1993;47:597–626. doi: 10.1146/annurev.mi.47.100193.003121. [DOI] [PubMed] [Google Scholar]
- 21.Xiao GP, Deziel E, He JX, Lepine F, Lesic B, et al. MvfR, a key Pseudomonas aeruginosa pathogenicity LTTR-class regulatory protein, has dual ligands. Mol Microbiol. 2006;62:1689–1699. doi: 10.1111/j.1365-2958.2006.05462.x. [DOI] [PubMed] [Google Scholar]
- 22.Venturi V. Regulation of quorum sensing in Pseudomonas. FEMS Microbiol Rev. 2006;30:274–291. doi: 10.1111/j.1574-6976.2005.00012.x. [DOI] [PubMed] [Google Scholar]
- 23.Schnider-Keel U, Seematter A, Maurhofer M, Blumer C, Duffy B, et al. Autoinduction of 2,4-diacetylphloroglucinol biosynthesis in the biocontrol agent Pseudomonas fluorescens CHA0 and repression by the bacterial metabolites salicylate and pyoluteorin. J Bacteriol. 2000;182:1215–1225. doi: 10.1128/jb.182.5.1215-1225.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Y, Du X, Lu ZJ, Wu D, Zhao Y, et al. Regulatory feedbackloop of two phz gene clusters through 5'-untranslated regions in Pseudomonas sp. M18. PLoS One. 2011;6:e19413. doi: 10.1371/journal.pone.0019413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lamont IL, Beare PA, Ochsner U, Vasil AI, Vasil ML. Siderophore-mediated signaling regulates virulence factor production in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2002;99:7072–7077. doi: 10.1073/pnas.092016999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Porrua O, Platero AI, Santero E, del Solar G, Govantes F. Complex interplay between the LysR-type regulator AtzR and its binding site mediates atzDEF activation in response to two distinct signals. Molecular Microbiology. 2010;76:331–347. doi: 10.1111/j.1365-2958.2010.07100.x. [DOI] [PubMed] [Google Scholar]
- 27.Abbas A, Morrissey JP, Marquez PC, Sheehan MM, Delany IR, et al. Characterization of interactions between the transcriptional repressor PhlF and its binding site at the phlA promoter in Pseudomonas fluorescens F113. J Bacteriol. 2002;184:3008–3016. doi: 10.1128/JB.184.11.3008-3016.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarniguet A, Kraus J, Henkels MD, Muehlchen AM, Loper JE. The sigma factor sigma s affects antibiotic production and biological control activity of Pseudomonas fluorescens Pf-5. Proc Natl Acad Sci U S A. 1995;92:12255–12259. doi: 10.1073/pnas.92.26.12255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schnider U, Keel C, Blumer C, Troxler J, Defago G, et al. Amplification of the housekeeping sigma factor in Pseudomonas fluorescens CHA0 enhances antibiotic production and improves biocontrol abilities. J Bacteriol. 1995;177:5387–5392. doi: 10.1128/jb.177.18.5387-5392.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ge Y, Pei D, Li W, Zhao Y, Xu Y. Insertional mutation of the rpoS gene contributes to alteration in biosynthesis of antifungal agents in Pseudomonas sp. M18. Biol Control. 2006;39:186–192. [Google Scholar]
- 31.Zhu D, Xu W, Geng H, Zhang X, Xu Y. Gene cloning of rpoD and its impact on biosynthesis of antibiotics in fluorescent Pseudomonas M18. Wei Sheng Wu Xue Bao. 2003;43:315–323. [PubMed] [Google Scholar]
- 32.Sambrook J, Russell DW. Cold Spring Harbor: CSH Press. 2001. Molecular cloning: a laboratory manual 3rd edn.
- 33.King EO, Ward MK, Raney DE. Two simple media for the demonstration of pyocyanin and fluorescin. J Lab Clin Med. 1954;44:301–307. [PubMed] [Google Scholar]
- 34.Reese MG. Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput Chem. 2001;26:51–56. doi: 10.1016/s0097-8485(01)00099-7. [DOI] [PubMed] [Google Scholar]
- 35.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, et al. The Pfam protein families database. Nucleic Acids Res. 2002;30:276–280. doi: 10.1093/nar/30.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor, N.Y., USA. : Cold Spring Harbor Laboratory Press. 1972.
- 38.Blumer C, Heeb S, Pessi G, Haas D. Global GacA-steered control of cyanide and exoprotease production in Pseudomonas fluorescens involves specific ribosome binding sites. Proc Natl Acad Sci U S A. 1999;96:14073–14078. doi: 10.1073/pnas.96.24.14073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kovach ME, Phillips RW, Elzer PH, Roop RM, 2nd, Peterson KM. pBBR1MCS: a broad-host-range cloning vector. Biotechniques. 1994;16:800–802. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primers used in this study.
(DOC)