

Abstract
Colorectal cancer is the second most leading cause of cancer related deaths in the western countries. One of the forms of colorectal cancer is hereditary non-polyposis colorectal cancer (HNPCC), also known as “Lynch syndrome”. It is the most common hereditary form of cancer accounting for 5%-10% of all colon cancers. HNPCC is a dominant autosomal genetic disorder caused by germ line mutations in mismatch repair genes. Human mismatch repair genes play a crucial role in genetic stability of DNA, the inactivation of which results in an increased rate of mutation and often a loss of mismatch repair function. Recent studies have shown that certain mismatch repair genes are involved in the regulation of key cellular processes including apoptosis. Thus, differential expression of mismatch repair genes particularly the contributions of MLH1 and MSH2 play important roles in therapeutic resistance to certain cytotoxic drugs such as cisplatin that is used normally as chemoprevention. An understanding of the role of mismatch repair genes in molecular signaling mechanism of apoptosis and its involvement in HNPCC needs attention for further work into this important area of cancer research, and this review article is intended to accomplish that goal of linkage of apoptosis with HNPCC. The current review was not intended to provide a comprehensive enumeration of the entire body of literature in the area of HNPCC or mismatch repair system or apoptosis; it is rather intended to focus primarily on the current state of knowledge of the role of mismatch repair proteins in molecular signaling mechanism of apoptosis as it relates to understanding of HNPCC.
Keywords: Colorectal cancer, Hereditary non-polyposis colorectal cancer, Apoptosis, Molecular signaling mechanisms, DNA mismatch repair proteins
INTRODUCTION
DNA mismatch repair (MMR) system consists of several genes that play a crucial role in correcting DNA errors arising during DNA replication of the cell division. Besides their established role in DNA repair, MMR genes are also involved in programmed cell death or apoptosis, for example, apoptosis induced by DNA damage. Two major protein complexes namely MutL and MutS are derived following formation of unique combination of MMR proteins including MLH1, MSH2 and MSH6. These MMR protein complexes (MutL and MutS) work together in a concerted manner to form a DNA repair machinery that are responsible for majority of DNA repair whether it is base-base mismatch or insertion/deletion mutation. This machinery repairs DNA not only during cell division but also during DNA damages induced by a number of environmental factors including treatment with several types of cytotoxic drugs used as anti-cancer agents[1].
Mismatch repair genes are highly conserved from prokaryotic to eukaryotic cells. The first indication of mismatch repair was obtained from Streptococcus pnemoniae and then in Escherichia coli[2]. Loss of mismatch repair genes is associated with increased risk of cancer, destabilization of the genome and an increased rate of mutation particularly in microsatellite sequences[3]. Inherited mutations in mismatch repair genes especially MLH1 and MSH2 are associated with hereditary non-polyposis colorectal cancer (HNPCC)[4]. It has been reported that somatic mutations and epigenetic silencing of MLH1 promoter gene are observed in sporadic cancer[5]. Several studies have reported that MMR system is also involved in mediating the activation of cell cycle check points and apoptosis in response to various anti-cancer drugs that act on DNA[6,7]. Thus, cells that have deficiency in one of the mismatch repair genes would be resistant to apoptosis than cells that are proficient in mismatch repair genes[8].
Apoptosis can occur through two different pathways; extrinsic pathway or intrinsic pathway. The extrinsic pathway is activated via ligation of death receptors on cell surface membrane leading to activation of caspase 8, followed by caspase 3. This pathway bypasses mitochondria. The intrinsic pathway, on the other hand, involves depolarization of mitochondrial membrane leading to the release of cytochrome C from mitochondrial intermembrane space. Intrinsic pathway is activated via apoptotic signals produced within the cell due to developmental cues or cell stress. Proteins such as cytochrome c released from mitochondria bind to apoptotic protease activating factor 1 (Apaf1) and caspase 9. This results in activation of caspase 3, and commitment to cell death. This pathway is regulated by the B-cell lymphoma 2 family of proteins. Accumulation of Bcl-2-associated X protein or Bcl-2 homologous antagonist killer on the mitochondrial outer membrane results in a conformational change allowing for membrane insertion and pore formation. A basic description of apoptosis and apoptotic pathways is provided here before providing its link to HNPCC and DNA mismatch match repair system. Relatively detailed description of apoptotic mechanisms in relation to carcinogenesis has been reported elsewhere[9].
APOPTOSIS
Apoptosis or programmed cell death plays an important role in tissue development and homeostasis[9]. Apoptosis was first described in 1927 by Currie et al[10]. In apoptosis, cells undergo a series of biochemical and morphological changes including cell shrinkage, chromatin condensation, cell membrane blebbing, formation of apoptotic bodies, and finally ending with engulfment of apoptotic bodies by macrophages or neighboring cells[11]. A detailed description of morphological changes and activation of cellular signaling pathways that occur during apoptosis has been published in an earlier report[9]. This report also provides an in-depth analysis of intracellular signaling molecules that trigger apoptotic events and that can be exploited for chemoprevention to carcinogenesis. Apoptosis can be triggered by various stimuli from outside or inside the cell, for example, DNA damage due to defect in DNA repair mechanism, treatment with cytotoxic drugs, or by deployment of death signals[12].
APOPTOTIC PATHWAYS
In mammals, there are two main apoptotic pathways, extrinsic pathway (death receptor mediated pathway) and intrinsic pathway (mitochondrial mediated pathway). As shown in Figure 1, the extrinsic pathway is mediated by cell surface death receptors. The death ligands bind and ligate with death receptors such as Fas, tumor necrosis factor receptor, or tumour necrosis factor-related apoptosis-inducing ligand receptors. This results in recruitment of adaptor protein Fas-associated death domain and caspase-8 forming a death inducing signaling complex (DISC). The auto activation of caspase-8 causes the activation of other caspases (caspase-3, -6, -7) in the caspase cascade process[13] that ultimately lead to cellular destruction. Caspases are aspartate-specific cysteine proteases and members of the interleukin-1β converting enzyme family[13]. So far, 14 mammalian caspases have been identified. Caspases are synthesized as inactive zymogen, which upon proteolytic cleavage become active.
Figure 1.
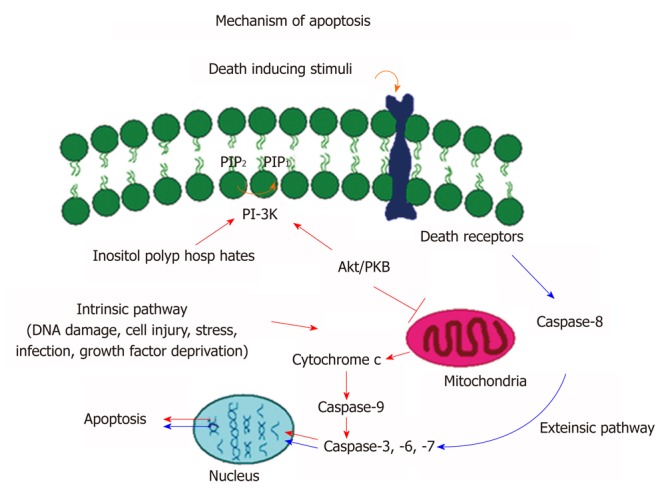
Molecular signaling mechanisms of apoptosis. Schematic representation of extrinsic and intrinsic apoptotic pathways that lead to activation of caspase cascade and programmed cell death.
The intrinsic pathway is mediated by different apoptotic stimuli. Most intrinsic signals induce depolarization of mitochondrial membrane and the release of cytochrome C into the cytoplasm. The release of cytochrome C initiates a series of biochemical events including activation of caspase cascade and thus cellular destruction. One of the most important events is that the released cytosolic cytochrome C binds to Apaf-1 and procaspase-9 that result in the formation of an intracellular DISC-like complex, apotosome. Activation of procaspase-9 leads to proteolytic processing and activation of procaspase-3, -6, and 7 resulting in an activation of caspase cascade and cell death[14]. The progression of apoptosis is highly regulated by a series of signaling pathways including those that involve in caspase cascade and PI3 kinase/AKT/PKB pathways. The caspase-cascade plays an important role in stimulation and transduction of apoptotic signals. The activation of the caspases is considered as a hallmark of apoptosis[14].
COLORECTAL CANCER
A number of gastrointestinal cancers specifically associated with various regions of the intestinal tract starting from esophageal to cancers of anus have been identified. Among gastrointestinal cancers, colorectal cancer is by far the most studied intestinal cancer type. In general, cancer is a disease caused by defective genes that transforms a normal cell into a cancerous cell in such a way that is unable to control cell growth, and thus continue to proliferate in an irregular fashion. Cancerous cells are no longer responsive to apoptotic signals, thus escaping programmed death process (apoptosis). Colorectal cancer is one of the leading causes of deaths in the world. Colorectal cancer is the second most common cause of cancer related deaths in Western countries including the United States. Colorectal cancer was reported to be responsible for 9% of new cancer cases and 10% of cancer deaths in 2010 in the United States alone[15,16].
Colorectal cancer can develop as a disease if there is any genetic disorder; the most common cause is chromosomal instability[17]. There are two major types of colorectal cancers that are primarily regulated genetically; sporadic colon cancer caused by sporadic mutation and hereditary colon cancer caused by hereditary mutation. In sporadic case, the gene mutation is induced by exposure to different carcinogens. Sporadic cancer happens by chance and no family history can be tracked. In hereditary case, the gene mutations are found in the germ lines and the defect can pass from the parents to the children that result in an accumulation of cancer in the family; for example, HNPCC[17].
An important event to prevent cells from forming clonal growth that would lead to carcinogenesis is apoptosis[18]. During carcinogenesis, apoptotic process is deregulated and thus cells tend to escape natural death process to overcome any cellular damage. In other words, cells cannot perform its normal growth function if there is a mutation in certain cancer-related genes[19]. Mismatch repair genes may represent such a scenario. Among mismatch repair genes, MLH1 and MSH2 are most studied genes that have been linked to cause abnormalities in apoptotic process. Therefore, in this review article, our efforts will be focused to provide a possible linkage of these two mismatch repair genes in HNPCC and their manifestation of apoptosis. Furthermore, understanding of apoptotic mechanisms is of utmost importance because defect causes failure in treatment with anti-cancer agents, and may also cause a number of other human diseases[20].
HEREDITARY NON-POLYPOSIS COLORECTAL CANCER
HNPCC, also known as “Lynch Syndrome”, is the most common form of hereditary colorectal cancer accounting for 5%-10% of all colon cancers. HNPCC is a dominant autosomal genetic disorder (affected person has one copy of the mutated gene) caused by germ line mutations in mismatch repair genes[21]. Patients with HNPCC show microsatellite instability due to mutations in DNA mismatch repair genes[22]. Microsatellites are repeated sequences of DNA usually 1 to 10 nucleotides long throughout the genome. Four genes are known to be responsible for HNPCC. These are MLH1, MSH2, MSH6, and PMS2. In early 1990s, the identification of genetic basis for HNPCC began with the localization of both MLH1 and MSH2 gene[23]. HNPCC has 80%-90% mutations in MLH1 and MSH2 genes. MSH6 gene accounts for 10% of HNPCC whereas PMS2 gene accounts only for 5% of HNPCC cases[23]. In HNPCC, a simple insertion/deletion of a single nucleotide leads to frame shift mutation and thus truncated protein product formation. The frame shift mutation accounts for the majority of the mutations that have been identified in HNPCC[24]. HNPCC is also characterized by development of extra colonic tumor formation. The average age of diagnosis of HNPCC is approximately 45 years. HNPCC is subdivided into Lynch syndrome I (colorectal cancer only) and Lynch syndrome II (colorectal cancer and extra colonic tumors), the extra colonic tumors that are associated with HNPCC are cancers of endometrium, stomach, small bowel, urinary tract, and ovaries[25].
DIAGNOSIS OF HNPCC PATIENTS
Several diagnostic criteria have been developed to help identify HNPCC[26]. Amsterdam Criteria I developed in 1991 was the first international criteria used for the diagnosis of HNPCC. Amsterdam Criteria I require three observations, colorectal cancer in three or more relatives, in at least two generations, and one or more relatives diagnosed before the age of fifty years[26]. Amsterdam Criteria I led to the development of Amsterdam Criteria II to include the extracolonic malignancies[26].
Microsatellite instability (MSI) screening is used as an added criterion to establish defective DNA mismatch repair system. Microsatellites are short, tandemly repeated DNA sequences. MSI is a change in length of microsatellite allele due to insertion/deletion of repeating units during DNA replication[27]. This was used as a primary method for screening HNPCC[28,29] after its discovery in proximal colon tumors[30]. In 1997, the National Cancer Institute Workshop proposed a panel of five markers for microsatellite that could be used to detect MSI. This panel is called Bethesda panel, and includes two mononucleotide (BAT-25 and BAT-26) and three-di nucleotide (D5S346, D2S123, and D17S250) repeats. Samples are classified as MSI-high, if two or more of the five markers show instability, whereas those with one unstable marker are classified as MSI-low. Samples with no alteration are considered as MSI-stable[30]. In 2002, the National Cancer Institute workshop recommended a second panel of mononucleotide markers such as BAT-40 for detection of microsatellite instability high (MSI-H) because mononucleotide appears to be more sensitive to detect MSI-H than dinucleotide markers. Since both MSI and HNPCC are caused by mismatch repair defects, MSI can be used as primary method for screening population at risk for HNPCC. There is limitation for using MSI because MSI is not specific for HNPCC but it could exist in 10% to 15% of sporadic colorectal cancers. Hypermethylation of MLH1 promoter causes MSI in sporadic cancer. It is recommended that the patient who meets the Bethesda guidelines would be tested for MSI followed by immunohistochemistry (IHC) for the MSI-H tumors[27].
Use of IHC for MLH1, MSH2 proteins and MSI is a yet another good criterion to some extent because they are complimentary to each other in identifying HNPCC patients. IHC helps identify the mutated gene, and may detect MMR deficient case that can be missed by MSI testing[31]. MSH2 forms a dimer with MSH6 while MLH1 forms a dimer with PMS2. So a mutation in MSH2 or MLH1 will result in the loss of MSH2/MSH6 or MLH1/PMS2 staining by using IHC method. However; the reverse is not true.
DNA MISMATCH REPAIR SYSTEM
DNA Mismatch repair system consists of several genes that encode nuclear proteins responsible for maintaining genetic stability by repairing base-to-base mismatches and insertion/deletion loops that arise during S phase of the DNA replication. In eukaryotic cells, MMR repair systems include MutS and MutL proteins complexes. The genetic stability is normally dependent upon the ability of these MMR protein complexes to recognize DNA damages and repair them; failure to which causes genetic instability[32,33]. Eukaryotic DNA mismatch repair is initiated when MutS alpha (MSH2-MSH6) or MutS beta (MSH2-MSH3) recognizes and binds to mismatched DNA. The binding of these heterodimers to the mismatches recruits MutL including MutL alpha (MLH1-PMS2) or MutL beta (MLH1-PMS1)[7]. Human MutL alpha has an ATPase activity that regulates the termination of mismatch-provoked excision[7]. The formation of MutS:MutL mismatch DNA complex leads to strand discrimination and removal of the errors that are made. MMR process includes other factors that help correct these errors. These include proliferating cell nuclear antigen (PCNA), replication factor C (RF-C), exonuclease1 (Exo1), and DNA polymerase. PCNA interacts with both MSH2 and MLH1 and is thought to play a role in the initiation and DNA re-synthesis steps of the mismatch repair[7]. When there is deficiency in mismatch repair system, the replication errors in the genome are not repaired, As a result, mutations accumulate throughout the genome[34]. Thus mutations in mismatch repair genes cause predisposition to cancer.
Germ line mutation of MMR genes specially MLH1 and MSH2 were identified in the families with colorectal cancer; 70%-80% population have mutations in these two genes[35,36]. Therefore, cells that have mutation(s) in either one of these genes show mutator phenotypes and display MSI[7]. MLH1 and MSH2 are nuclear proteins with 756 and 934 amino acids that respectively encode proteins of approximately 80 KDa and 100 KDa. The MLH1 gene is located on chromosome 3p21 with nineteen exons, while MSH2 gene is on chromosome 2p16 with sixteen exons[37].
ROLE OF DNA MISMATCH REPAIR PROTEINS IN HNPCC
The DNA mismatch repair system is considered as sensory system that can scan DNA, and when there is any insertion, deletion, or mispaired nucleotide, this system is able to detect and remove the errors[38]. DNA mismatch repair system can be divided into four phases: first recognition of mismatch base by MutS, second recruitment of MutL, third removal of incorrect base, fourth re-synthesis of corrected DNA by DNA polymerase enzyme using the parent strand as a template[39]. DNA mismatch repair was first discovered in bacteria where it was identified. Inactivation of this system increases mutation rates and fails to repair the DNA replication error[39]. In early 1990s, the importance of this system was further appreciated with the identification of genetic basis for HNPCC[23]. HNPCC is a caused by inherited germ line mutations in one of the mismatch repair genes especially MLH1 and MSH2[31,40]. About 90% of HNPCC cases display MSI. MSI has been uniquely linked with mismatch repair defects, so MSI can be used as a marker for mutator phenotype to diagnose patients at high risk for HNPCC[40].
MECHANISMS OF DNA MISMATCH REPAIR
The human mismatch repair proteins are responsible for recognition and correcting errors that are made during DNA replication. A simplified version of how DNA mismatch repair system functions to correct DNA repair during replication is shown in Figure 2. The function of MutS-α (MSH2/MSH6) is to repair base-base mismatches. The function of MutS-β (MSH2/MSH3) is to repair insertion/deletion loop that arise during replication. MSH proteins have ATPase activity, one adenosine triphosphate (ATP) binding site present at each molecule[41]. In the presence of adenosine diphosphate (ADP) MutS protein will bind tightly to the mismatches on the DNA strand, while in the presence of ATP the MutS will act as sliding clamp[38]. MutS protein will move along to the DNA to identify which strand needs to be repaired. It has been proposed that the feature of the newly synthesized strand is a single strand nick for example the gaps between Okazaki fragments in the lagging strand[38]. So MMR will excise the strand containing a nick, which needs exonuclease activity. After the binding of MutS to the mismatches, MutS:ATP complex will recruit MutL proteins. MutL binds to the complex and interact with MutS at the site of the mismatches. MutL protein will transfer DNA polymerase, PCNA, and recruit exonuclease I for excision up to kilobase of DNA[38]. Excision of the mismatches can be either 5’ to 3’ or the opposite. 5’ to 3’ requires MutS, Exo1, and replication protein A. Whereas 3’ to 5’ excision requires MutL RFC and PCNA. After the mismatches have been removed, the re-synthesis step starts by the involvement of DNA polymerase[41].
Figure 2.
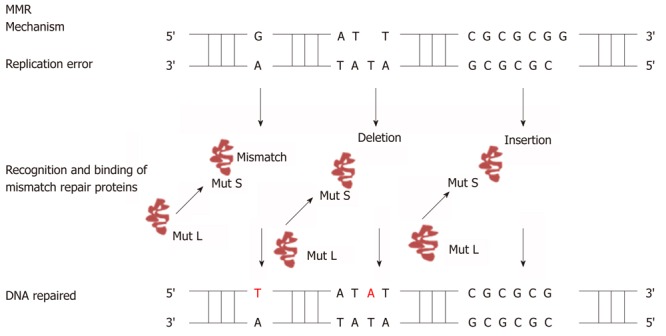
Mechanism of DNA mismatch repair. Figure shows how DNA mismatch repair system (MutS and MutL) detects DNA mismatches as well as insertion/deletion mutations during DNA replication and repairs it with the help of the DNA mismatch repair system.
There are two proposed models for the signaling of the downstream mismatch repair processes after the mismatch recognition; stationary (Trans) model and moving (Cis) model. The moving model includes translocation and molecular switch models[42]. In stationary model, the binding of MSH proteins to the DNA strand considered as protein -protein interaction, this cause DNA to bend and bring the two distant sites together. In moving model, MSH proteins will bind to the mismatch on the DNA and then this protein will move away from the site to look for the strand discrimination signal. Translocation model suggests that hydrolysis of ATP drives unidirectional movement of MSH proteins, and this will result in the creation of α-loop[7]. Molecular switch model hypothesized that the binding of MSH proteins to the mismatch will trigger an ADP to ATP exchange that will support the bi-directional sliding of MSH proteins away from the mismatch. After the movement of the MSH proteins from the site another MSH proteins will occupy the empty site. When MSH proteins reach the strand break, the excision step begins[7].
ROLE OF MISMATCH REPAIR PROTEINS IN APOPTOSIS
As mentioned before, DNA mismatch repair system has been implicated in correction of base/base mismatches and insertion/deletion loops (Figure 2) that arise during DNA replication[43]. The inactivation or defects in MMR, usually MSH2 or MLH1, is associated with HNPCC and responsible for microsatellite instability[44]. A recent study shows that MMR system plays an important role in apoptotic machinery, activation of cell cycle check points[45], and in cytotoxicity induced by certain types of DNA damaging drugs. A simplified relationship of DNA mismatch repair system with apoptosis is shown in Figure 3. However, the exact mechanism by which the mismatch repair proteins mediate apoptosis is not yet understood[46]. It has been shown that loss of DNA mismatch repair causes resistance to certain types of DNA damaging agents because MMR deficient cells display defects in G2/M cell cycle arrest when treated with these agents[47,48]. MMR have been linked to resistance to a number of chemotherapeutic drugs such as 6-thioguanine and DNA methylating agents. Loss of MMR proteins also result in low level resistance to cisplatin. Cisplatin works by binding with DNA and creating DNA adducts that lead to intrastrand or interstrand cross-links which disrupt the structure of DNA helix. Proficient MMR system is important to recognize the damaged DNA created by cisplatin. The complex (DNA and cisplatin) interferes with the normal activity of MMR and prevents the repair process. Therefore the inability to complete the repair of damaged DNA caused by this drug leads to apoptosis whereas in MMR deficient system, cells continue to proliferate and cause resistance to the drug[49].
Figure 3.
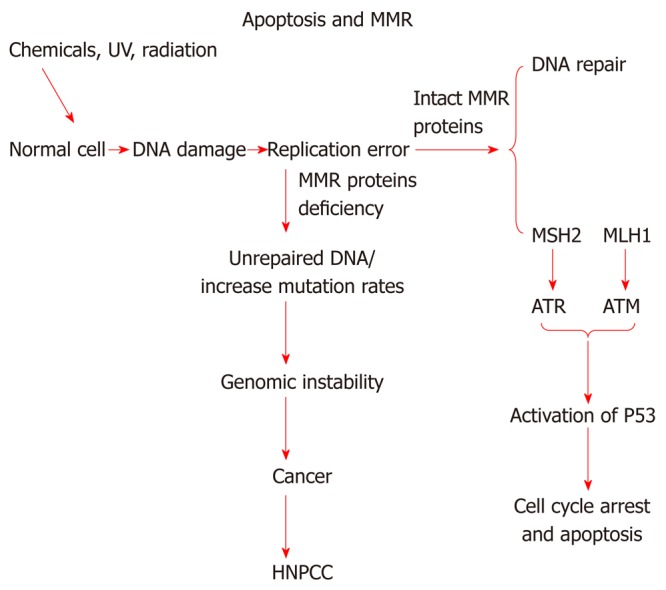
Relationship of mismatch repair proteins with apoptosis. This diagram depicts possible relationship between DNA mismatch repair proteins and HNPCC, and its possible link with apoptosis. MMR: DNA mismatch repair; ATR: Ataxia telangiectasia and Rad3-related protein; ATM: Ataxia telangiectasia mutated; HNPCC: Hereditary non-polyposis colorectal cancer.
There are two models that described the role of mismatch repair system in DNA damaging signaling[7]. The first model is direct signaling model; this model propose that MSH-MLH complex identify DNA adduct, and this will recruit ataxia telangiectasia and Rad3-related protein (ATR) or ataxia telangiectasia mutated (ATM) to the adduct site as a result activation of downstream damage signaling. The second model named as futile DNA repair cycle[7]. This model suggests that DNA adducts will trigger the strand specific MMR which targets the newly replicated DNA. The adduct in the template strand cannot be removed, and this will provoke new cycle of MMR. If futile repair cycle persists this will activate ATM/ATR to promote cell cycle arrest and apoptosis[7].
In 1999, Zhang et al[50] found that over expression of MSH2 or MLH1 induced apoptosis[50]. Over expression of MSH2 was toxic to the cells, and develop severe nuclear abnormalities that caused cells to undergo apoptosis. They explained that the over expression of MLH1 or MSH2 in cells causes apoptosis because of the increased levels of these two proteins may alter or sequester other proteins such as PCNA that have a role in cell cycle progression or induction of apoptosis. Therefore, over expression of one of these two proteins might sequester PCNA from its role in DNA synthesis. As a result, apoptosis may be induced. Later, Chen et al[51] (2004), have shown that the mismatch repair protein MLH1 acts as a substrate for caspase-3 which proteolyzed MLH1 in cancer cells that are treated with anti-cancer drugs that inhibit topoisomerase II. The cells, in turn, undergo apoptosis.
Human MLH1 is cleaved by caspase-3 at Asp418 residue to produce a proapoptotic carboxyl- terminal product[51] which partially redistributes from the nucleus to the cytoplasm. Losses of mismatch repair proteins (MLH1 or MSH2) cause resistant to cisplatin. Aebi et al[52] (1996)found that human colon cancer cell line HCT116 that is deficient in hMLH1 protein was 2 fold resistant to cisplatin when compared to cells that express hMLH1. Similar results were found when hMSH2 deficient cell lines were compared with hMSH2 proficient cell lines against resistance to cisplatin[52] indicating that both MLH1 and MSH2 proteins play a role in apoptotic cell signaling. Wu et al[53] (2008), has shown that human MLH1 protein is involved in the cellular response to psoralen interstrand crosslinks (ICLs). It has been found that MLH1 deficient cells are more resistant to psoralen ICLs than cells that have deficiency in MSH2[53]. In cells that have deficiency in MLH1 protein, apoptosis was not induced by psoralen ICLs, as well as CHK2 checkpoint homolog phosphorylation was undetectable when they compare with MLH1 proficient cells. This indicate that MLH1 is involved in signaling psoralen ICL- induced check point activation[53].
Thus, cell lines that are proficient or deficient in expression of MLH1 or MSH2 proteins can serve excellent model systems to study the role of these proteins in signaling mechanisms of apoptosis in relation to their involvement in HNPCC. In our laboratory, we have used such a model cell lines (SW480, HTC116 and LoVo) to accomplish just that. Figure 4 shows that, in a Western blotting experiment using specific monoclonal antibodies to MLH1 and MSH2 protein on the same blot, colorectal adenocarcinoma cell lines SW480 expresses both full length MLH1 (80 KDa) and MSH2 (100 KDa) proteins. HCT116 expresses only full length MSH2 and LoVo expresses only full length MLH1. Similar data has also been reported earlier[54] in a collaborative publication with Dr. Bruce Boman’s group at Helen F Graham Cancer Center, Newark DE. These cell lines were then used to induce apoptosis by treatment with 100 μmol/L etoposide, a known cancer treatment drug used to induce apoptosis. It is apparent that the cell lines that are deficient in one of the mismatch repair proteins (HCT116 for MLH1 and LoVo for MSH2) are resistant to apoptosis (diminished red staining) than the cell line (SW480) which expresses both these proteins. This clearly links the role of these mismatch proteins to apoptosis. Further studies in evaluating the molecular signaling mechanism of apoptosis using these cell lines are in progress.
Figure 4.
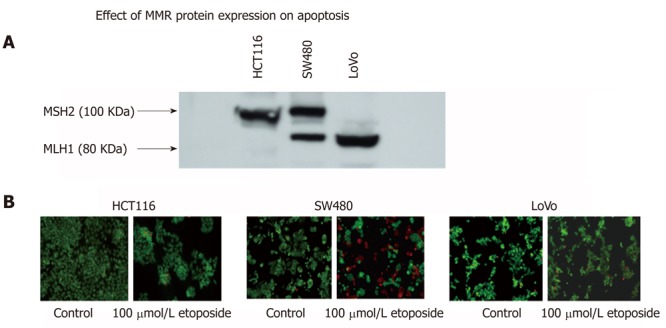
Possible link of the expression of mismatch repair proteins MLH1 and MSH2 with apoptosis. A differential expression of MSH2 and MLH1 mismatch repair proteins in SW480, HCT116 and LoVo colorectal carcinoma cell lines (A). These cells also show variation in etoposide induced apoptosis (B) suggesting a possible link of mismatch repair proteins and apoptosis.
CONCLUSION
Indeed, colorectal cancer biology especially hereditary carcinogenesis such as HNPCC is a challenging area of research to gain insight into molecular events that leads to this disease. We have described the genetic basis of HNPCC and the role of mismatch repair proteins in onset of the syndrome. While a lot has been learnt about the mechanism of DNA mismatch repair system and how it regulates HNPCC, a lot more still need to be learnt about this syndrome. Many fundamental questions still remain unanswered; besides mismatch repair genes, what other genes are involved in the pathogenesis of this disease? Are there adequate methods available to screen population using genetic markers that may predispose HNPCC? How crucial are signaling molecules in mediating actions of DNA mismatch repair genes to HNPCC and apoptosis? So far we have only learnt a linkage of HNPCC to apoptotic pathway. It is not yet clear how one can exploit apoptotic mechanism to therapeutic intervention of HNPCC? A better understanding of the apoptotic signaling pathways that link mismatch repair genes such as MLH1 and MSH2 to HNPCC are attractive approaches to answering many of these and other burning questions. Clearly, this is an exciting time for HNPCC research and discovery as more efforts with improved methodologies especially those that dissect out apoptotic signaling pathways linking this disorder become available.
ACKNOWLEDGMENTS
Samar Hassen is grateful to the Graduate Institute of Technology, University of Arkansas at Little Rock for a research assistantship. We thank Dr. Rakhee Agarwal for help with Figure 1. Authors are also thankful to Dr. Bruce Boman, Helen F Graham Cancer Center, Newark, DE) for helpful discussions on HNPCC.
Footnotes
Supported by NSF-EPSCoR P3 Center and NASA-EOSCoR Research Infrastructure Development Funds to Ali N
Peer reviewer: Mitsunori Yamakawa, Professor, Department of Pathological Diagnostics, Faculty of Medicine, Yamagata University, 2-2-2 Iida-Nishi, Yamagata 990-9585, Japan
S- Editor Wu X L- Editor A E- Editor Zhang DN
References
- 1.Fink D, Aebi S, Howell SB. The role of DNA mismatch repair in drug resistance. Clin Cancer Res. 1998;4:1–6. [PubMed] [Google Scholar]
- 2.Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 3.Wu J, Gu L, Wang H, Geacintov NE, Li GM. Mismatch repair processing of carcinogen-DNA adducts triggers apoptosis. Mol Cell Biol. 1999;19:8292–8301. doi: 10.1128/mcb.19.12.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peltomäki P. Deficient DNA mismatch repair: a common etiologic factor for colon cancer. Hum Mol Genet. 2001;10:735–740. doi: 10.1093/hmg/10.7.735. [DOI] [PubMed] [Google Scholar]
- 5.O'Brien V, Brown R. Signalling cell cycle arrest and cell death through the MMR System. Carcinogenesis. 2006;27:682–692. doi: 10.1093/carcin/bgi298. [DOI] [PubMed] [Google Scholar]
- 6.Stojic L, Brun R, Jiricny J. Mismatch repair and DNA damage signalling. DNA Repair ( Amst) 2004;3:1091–1101. doi: 10.1016/j.dnarep.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 7.Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18:85–98. doi: 10.1038/cr.2007.115. [DOI] [PubMed] [Google Scholar]
- 8.Papouli E, Cejka P, Jiricny J. Dependence of the cytotoxicity of DNA-damaging agents on the mismatch repair status of human cells. Cancer Res. 2004;64:3391–3394. doi: 10.1158/0008-5472.CAN-04-0513. [DOI] [PubMed] [Google Scholar]
- 9.Ali N, MacLeod S, Hine RJ, Chowdhury P. Cellular Signaling Mechanisms in Pancreatic Apoptosis. In: Chen GG, Lai PBS, editors. Apoptosis in Carcinogenesis and Chemotherapy. New York: Springer; 2009. pp. 295–325. [Google Scholar]
- 10.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gewies A. Introduction to Apoptosis. ApoReview. 2003:1–26. [Google Scholar]
- 12.Fadeel B, Orrenius S. Apoptosis: a basic biological phenomenon with wide-ranging implications in human disease. J Intern Med. 2005;258:479–517. doi: 10.1111/j.1365-2796.2005.01570.x. [DOI] [PubMed] [Google Scholar]
- 13.Jin Z, El-Deiry WS. Overview of cell death signaling pathways. Cancer Biol Ther. 2005;4:139–163. doi: 10.4161/cbt.4.2.1508. [DOI] [PubMed] [Google Scholar]
- 14.Wang J, Lenardo MJ. Roles of caspases in apoptosis, development, and cytokine maturation revealed by homozygous gene deficiencies. J Cell Sci. 2000;113(Pt 5):753–757. doi: 10.1242/jcs.113.5.753. [DOI] [PubMed] [Google Scholar]
- 15.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 16.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 17.Raevaara TE, Korhonen MK, Lohi H, Hampel H, Lynch E, Lönnqvist KE, Holinski-Feder E, Sutter C, McKinnon W, Duraisamy S, et al. Functional significance and clinical phenotype of nontruncating mismatch repair variants of MLH1. Gastroenterology. 2005;129:537–549. doi: 10.1016/j.gastro.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 18.Meier P, Finch A, Evan G. Apoptosis in development. Nature. 2000;407:796–801. doi: 10.1038/35037734. [DOI] [PubMed] [Google Scholar]
- 19.Scott W. Lowe and Athina W.Lin. Apoptosis in cancer. Carcinogenesis. 2000;21:485–495. doi: 10.1093/carcin/21.3.485. [DOI] [PubMed] [Google Scholar]
- 20.Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–348. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- 21.Niessen RC, Berends MJ, Wu Y, Sijmons RH, Hollema H, Ligtenberg MJ, de Walle HE, de Vries EG, Karrenbeld A, Buys CH, et al. Identification of mismatch repair gene mutations in young patients with colorectal cancer and in patients with multiple tumours associated with hereditary non-polyposis colorectal cancer. Gut. 2006;55:1781–1788. doi: 10.1136/gut.2005.090159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamasaki Y, Matsushima M, Tanaka H, Tajiri S, Fukuda R, Ozawa H, Takagi A, Hirabayashi K, Sadahiro S. Patient with eight metachronous gastrointestinal cancers thought to be hereditary nonpolyposis colorectal cancer (HNPCC) Intern Med. 2010;49:209–213. doi: 10.2169/internalmedicine.49.2316. [DOI] [PubMed] [Google Scholar]
- 23.Learn PA, Kahlenberg MS. Hereditary colorectal cancer syndromes and the role of the surgical oncologist. Surg Oncol Clin N Am. 2009;18:121–44, ix. doi: 10.1016/j.soc.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 24.Apessos A, Mihalatos M, Danielidis I, Kallimanis G, Agnantis NJ, Triantafillidis JK, Fountzilas G, Kosmidis PA, Razis E, Georgoulias VA, et al. hMSH2 is the most commonly mutated MMR gene in a cohort of Greek HNPCC patients. Br J Cancer. 2005;92:396–404. doi: 10.1038/sj.bjc.6602260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peltomäki P. Role of DNA mismatch repair defects in the pathogenesis of human cancer. J Clin Oncol. 2003;21:1174–1179. doi: 10.1200/JCO.2003.04.060. [DOI] [PubMed] [Google Scholar]
- 26.Zhang L. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part II. The utility of microsatellite instability testing. J Mol Diagn. 2008;10:301–307. doi: 10.2353/jmoldx.2008.080062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perucho M. Correspondence re: C.R. Boland et al., A National Cancer Institute workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res., 58: 5248-5257, 1998. Cancer Res. 1999;59:249–256. [PubMed] [Google Scholar]
- 28.Shia J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part I. The utility of immunohistochemistry. J Mol Diagn. 2008;10:293–300. doi: 10.2353/jmoldx.2008.080031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gu L, Hong Y, McCulloch S, Watanabe H, Li GM. ATP-dependent interaction of human mismatch repair proteins and dual role of PCNA in mismatch repair. Nucleic Acids Res. 1998;26:1173–1178. doi: 10.1093/nar/26.5.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Veigl ML, Kasturi L, Olechnowicz J, Ma AH, Lutterbaugh JD, Periyasamy S, Li GM, Drummond J, Modrich PL, Sedwick WD, et al. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc Natl Acad Sci USA. 1998;95:8698–8702. doi: 10.1073/pnas.95.15.8698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paraf F, Gilquin M, Longy M, Gilbert B, Gorry P, Petit B, Labrousse F. MLH1 and MSH2 protein immunohistochemistry is useful for detection of hereditary non-polyposis colorectal cancer in young patients. Histopathology. 2001;39:250–258. doi: 10.1046/j.1365-2559.2001.01203.x. [DOI] [PubMed] [Google Scholar]
- 32.Bellacosa A. Functional interactions and signaling properties of mammalian DNA mismatch repair proteins. Cell Death Differ. 2001;8:1076–1092. doi: 10.1038/sj.cdd.4400948. [DOI] [PubMed] [Google Scholar]
- 33.Obmolova G, Ban C, Hsieh P, Yang W. Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature. 2000;407:703–710. doi: 10.1038/35037509. [DOI] [PubMed] [Google Scholar]
- 34.Lei X, Zhu Y, Tomkinson A, Sun L. Measurement of DNA mismatch repair activity in live cells. Nucleic Acids Res. 2004;32:e100. doi: 10.1093/nar/gnh098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chao EC, Lipkin SM. Molecular models for the tissue specificity of DNA mismatch repair-deficient carcinogenesis. Nucleic Acids Res. 2006;34:840–852. doi: 10.1093/nar/gkj489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valenzuela CD, Moore HG, Huang WC, Reich EW, Yee H, Ostrer H, Pachter HL. Three synchronous primary carcinomas in a patient with HNPCC associated with a novel germline mutation in MLH1: Case report. World J Surg Oncol. 2009;7:94. doi: 10.1186/1477-7819-7-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Silva FC, Valentin MD, Ferreira Fde O, Carraro DM, Rossi BM. Mismatch repair genes in Lynch syndrome: a review. Sao Paulo Med J. 2009;127:46–51. doi: 10.1590/S1516-31802009000100010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jascur T, Boland CR. Structure and function of the components of the human DNA mismatch repair system. Int J Cancer. 2006;119:2030–2035. doi: 10.1002/ijc.22023. [DOI] [PubMed] [Google Scholar]
- 39.Jacob S, Praz F. DNA mismatch repair defects: role in colorectal carcinogenesis. Biochimie. 2002;84:27–47. doi: 10.1016/s0300-9084(01)01362-1. [DOI] [PubMed] [Google Scholar]
- 40.Heinen CD, Schmutte C, Fishel R. DNA repair and tumorigenesis: lessons from hereditary cancer syndromes. Cancer Biol Ther. 2002;1:477–485. doi: 10.4161/cbt.1.5.160. [DOI] [PubMed] [Google Scholar]
- 41.Jun SH, Kim TG, Ban C. DNA mismatch repair system. Classical and fresh roles. FEBS J. 2006;273:1609–1619. doi: 10.1111/j.1742-4658.2006.05190.x. [DOI] [PubMed] [Google Scholar]
- 42.Fishel R. Mismatch repair, molecular switches, and signal transduction. Genes Dev. 1998;12:2096–2101. doi: 10.1101/gad.12.14.2096. [DOI] [PubMed] [Google Scholar]
- 43.Jacob S, Aguado M, Fallik D, Praz F. The role of the DNA mismatch repair system in the cytotoxicity of the topoisomerase inhibitors camptothecin and etoposide to human colorectal cancer cells. Cancer Res. 2001;61:6555–6562. [PubMed] [Google Scholar]
- 44.Lin DP, Wang Y, Scherer SJ, Clark AB, Yang K, Avdievich E, Jin B, Werling U, Parris T, Kurihara N, et al. An Msh2 point mutation uncouples DNA mismatch repair and apoptosis. Cancer Res. 2004;64:517–522. doi: 10.1158/0008-5472.can-03-2957. [DOI] [PubMed] [Google Scholar]
- 45.Jiricny J, Nyström-Lahti M. Mismatch repair defects in cancer. Curr Opin Genet Dev. 2000;10:157–161. doi: 10.1016/s0959-437x(00)00066-6. [DOI] [PubMed] [Google Scholar]
- 46.Topping RP, Wilkinson JC, Scarpinato KD. Mismatch repair protein deficiency compromises cisplatin-induced apoptotic signaling. J Biol Chem. 2009;284:14029–14039. doi: 10.1074/jbc.M809303200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davis TW, Wilson-Van Patten C, Meyers M, Kunugi KA, Cuthill S, Reznikoff C, Garces C, Boland CR, Kinsella TJ, Fishel R, et al. Defective expression of the DNA mismatch repair protein, MLH1, alters G2-M cell cycle checkpoint arrest following ionizing radiation. Cancer Res. 1998;58:767–778. [PubMed] [Google Scholar]
- 48.de las Alas MM, Aebi S, Fink D, Howell SB, Los G. Loss of DNA mismatch repair: effects on the rate of mutation to drug resistance. J Natl Cancer Inst. 1997;89:1537–1541. doi: 10.1093/jnci/89.20.1537. [DOI] [PubMed] [Google Scholar]
- 49.Martin LP, Hamilton TC, Schilder RJ. Platinum resistance: the role of DNA repair pathways. Clin Cancer Res. 2008;14:1291–1295. doi: 10.1158/1078-0432.CCR-07-2238. [DOI] [PubMed] [Google Scholar]
- 50.Zhang H, Richards B, Wilson T, Lloyd M, Cranston A, Thorburn A, Fishel R, Meuth M. Apoptosis induced by overexpression of hMSH2 or hMLH1. Cancer Res. 1999;59:3021–3027. [PubMed] [Google Scholar]
- 51.Chen F, Arseven OK, Cryns VL. Proteolysis of the mismatch repair protein MLH1 by caspase-3 promotes DNA damage-induced apoptosis. J Biol Chem. 2004;279:27542–27548. doi: 10.1074/jbc.M400971200. [DOI] [PubMed] [Google Scholar]
- 52.Aebi S, Kurdi-Haidar B, Gordon R, Cenni B, Zheng H, Fink D, Christen RD, Boland CR, Koi M, Fishel R, et al. Loss of DNA mismatch repair in acquired resistance to cisplatin. Cancer Res. 1996;56:3087–3090. [PubMed] [Google Scholar]
- 53.Wu Q, Vasquez KM. Human MLH1 protein participates in genomic damage checkpoint signaling in response to DNA interstrand crosslinks, while MSH2 functions in DNA repair. PLoS Genet. 2008;4:e1000189. doi: 10.1371/journal.pgen.1000189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hassen S, Boman BM, Ali N, Parker M, Somerman C, Ali-Khan Catts ZJ, Ali AA, Fields JZ. Detection of DNA mismatch repair proteins in fresh human blood lymphocytes--towards a novel method for hereditary non-polyposis colorectal cancer (Lynch syndrome) screening. J Exp Clin Cancer Res. 2011;30:100. doi: 10.1186/1756-9966-30-100. [DOI] [PMC free article] [PubMed] [Google Scholar]