

Abstract
Muscarinic type 3 receptor (M3R) plays a pivotal role in the induction of glandular fluid secretions. Although M3R is often the target of autoantibodies in Sjögren's syndrome (SjS), chemical agonists for M3R are clinically used to stimulate saliva secretion in patients with SjS. Aside from its activity in promoting glandular fluid secretion, however, it is unclear whether activation of M3R is related to other biological events in SjS. This study aimed to investigate the cytoprotective effect of chemical agonist-mediated M3R activation on apoptosis induced in human salivary gland (HSG) cells. Carbachol (CCh), a muscarinic receptor-specific agonist, abrogated tumor necrosis factor α/interferon γ-induced apoptosis through pathways involving caspase 3/7, but its cytoprotective effect was decreased by a M3R antagonist, a mitogen-activated protein kinase kinase/extracellular signal-regulated kinase (ERK) inhibitor, a phosphatidylinositol 3-kinase/Akt inhibitor, or an epidermal growth factor receptor (EGFR) inhibitor. Ligation of M3R with CCh transactivated EGFR and phosphorylated ERK and Akt, the downstream targets of EGFR. Inhibition of intracellular calcium release or protein kinase C δ, both of which are involved in the cell signaling of M3R-mediated fluid secretion, did not affect CCh-induced ERK or Akt phosphorylation. CCh stimulated Src phosphorylation and binding to EGFR. A Src inhibitor attenuated the CCh/M3R-induced cytoprotective effect and EGFR transactivation cascades. Overall, these results indicated that CCh/M3R induced transactivation of EGFR through Src activation leading to ERK and Akt phosphorylation, which in turn suppressed caspase 3/7-mediated apoptotic signals in HSG cells. This study, for the first time, proposes that CCh-mediated M3R activation can promote not only fluid secretion but also survival of salivary gland cells in the inflammatory context of SjS.
Introduction
Sjögren's syndrome (SjS) is a chronic autoimmune disease characterized by lymphocytic infiltration, gland destruction, and eye and mouth dryness (Fox and Kang, 1992; Kroneld et al., 1997; Fox and Stern, 2002). Although the cause of SjS remains unclear, many studies have suggested that T and B lymphocytes that infiltrate the affected glands are involved in the pathogenesis of SjS, because of their production of tissue-destructive proinflammatory cytokines and autoantibodies, respectively (Lee et al., 2009). It was reported that levels of proinflammatory cytokines, such as tumor necrosis factor α (TNFα) and interferon γ (IFNγ), are elevated in the affected glands in SjS (Fox et al., 1994; Kolkowski et al., 1999). Those proinflammatory cytokines can induce apoptosis of salivary gland cells through caspase 3 signaling (Kamachi et al., 2002; Kulkarni et al., 2006). In contrast, it is thought that hypofunction of fluid secretion from affected glands is caused by autoantibodies against muscarinic type 3 receptor (M3R) (Li et al., 2004; Koo et al., 2008). M3R is the major muscarinic acetylcholine receptor in the salivary glands, and it plays a pivotal role in the induction of salivary fluid secretion (Baum, 1993). More specifically, acetylcholine released from the parasympathetic nerves activates M3R to induce intracellular Ca2+ influx, which initiates the cell signaling required for fluid secretion from acinar cells (Ambudkar et al., 1993; Ambudkar, 2000; Park et al., 2001). Chemical agonists for M3R, such as pilocarpine and cevimeline, are often used clinically to stimulate saliva secretion among patients with SjS (Mavragani and Moutsopoulos, 2007).
M3R, a G protein-coupled receptor (GPCR), belongs to the largest transmembrane receptor superfamily in humans and mice, and it is characterized by a seven-transmembrane α-helix structure (Pierce et al., 2002). In general, the binding of an agonistic ligand to its cognate GPCR elicits Ca2+ and/or PKC signaling cascades that induce the expression of genes required for multiple fundamental functions, including exocrine and endocrine secretion, smooth muscle and cardiac muscle contraction, pain transmission, fluid homeostasis, blood pressure regulation, and immune responses (Pierce et al., 2002). GPCRs, such as endothelin receptors and protease-activated receptor I, also activate mitogenic signaling networks, such as PKC/protein kinase D, MEK/ERK, and the PI3K/Akt cascade, which leads to the induction of a variety of biological responses, including cell proliferation, differentiation, migration, and survival (Rozengurt, 1998, 2007).
Many studies have shown the relevance of Ca2+ signaling in M3R-induced fluid secretion. However, mitogenic signaling pathways such as those involving PKC, ERK, and Akt, as well as their M3R-mediated downstream events, are still poorly understood in the context of salivary gland cells. Only recently has a muscarinic receptor agonist, namely, carbachol (CCh), been reported to stimulate in vitro ERK phosphorylation in human immortalized salivary gland cells or rat submandibular acinar cells (Soltoff and Hedden, 2010). Because SjS is a chronic degenerative disease characterized by the gradual progression of cell apoptosis and resulting tissue destruction, it is plausible that mitogenic signaling, which can down-regulate apoptosis induced through the caspase pathway (Steelman et al., 2008), may play a role in sustaining the configuration and functions of affected glands. Although it was shown that M3R promotes neuroblastoma cell survival through activation of the ERK signaling pathway (Greenwood and Dragunow, 2010), it remains unclear whether M3R signaling in salivary gland cells is similarly cytoprotective against proinflammatory cytokine-induced apoptosis.
In the present study, we hypothesized that chemical agonist-mediated M3R activation could induce cell survival signaling in salivary gland cells, thereby contributing to the protection of cells against apoptosis caused by inflammatory insult. To test this hypothesis, apoptosis was induced in human salivary gland (HSG) cells through inflammatory stimulation with TNFα/IFNγ. CCh was used as a chemical agonist for M3Rs, on the basis of studies that showed that CCh could efficiently activate M3Rs expressed on cultured HSG cells (Cha et al., 2006; Pauley et al., 2011). We then investigated the cytoprotective effect of CCh-mediated M3R ligation, as well as the molecular mechanism underlying such M3R-mediated cytoprotective effects.
Materials and Methods
Chemicals.
2-[(Aminocarbonyl)oxy]-N,N,N-trimethylethanaminium chloride [carbachol (CCh)] was purchased from Sigma-Aldrich (St. Louis, MO). The following chemicals were used as inhibitors for cell signaling molecules: 1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenylthio)butadiene (U0126; Cell Signaling Technology, Danvers, MA), 2-morpholin-4-yl-8-phenylchromen-4-one (LY294002; Cell Signaling Technology), 4-diphenylacetoxy-N-methylpiperidine methiodide (4-DAMP; Tocris Bioscience, Ellisville, MO), 1,2-bis-(o-aminophenoxy)-ethane-N,N,N′,N′-tetraacetic acid, tetraacetoxymethyl ester (BAPTA-AM; Tocris Bioscience), N-(3-chlorophenyl)-6,7-dimethoxy-4-quinazolinamine (AG1478 Cayman Chemical, Ann Arbor, MI), 4-amino-5-(4-chlorophenyl)-7-(dimethylethyl)pyrazolo[3,4-d]pyrimidine (PP2; Enzo Life Sciences, Inc., Plymouth Meeting, PA), and 2-[1-(3-dimethylaminopropyl)-1H-indol-3-yl]-3-(1H-indol-3-yl)maleimide (GF109203X; Enzo Life Sciences, Inc.).
Cell Culture.
HSG cells established from a human salivary gland (Sato et al., 1985) were used in this study. Cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA), penicillin G solution (100 units/ml), and streptomycin (100 μg/ml) (medium A) and were maintained in a humidified atmosphere of 5% CO2 at 37°C. The HSG cells were seeded at a density of 2.0 × 104 cells per well in 96-well plastic culture plates or 105 cells per well in six-well plastic culture plates. Before the addition of any stimulants, the medium was changed to Dulbecco's modified Eagle's medium supplemented with antibiotics but without fetal bovine serum (medium B), and cells were incubated for 4 h. The cells were then pretreated for 30 min with or without U0126 (2.5 μM), LY294002 (2.5 μM), 4-DAMP (1 μM), BAPTA-AM (10 μM), AG1478 (5 μM), PP2 (5 μM), or a nontoxic variant of diphtheria toxin protein (CRM197, 10 μM; Sigma-Aldrich) in medium B. After pretreatment, the HSG cells were exposed to 100 μM CCh or 100 ng/ml recombinant EGF (PeproTech, Rocky Hill, NJ) for various periods, in the absence or presence of a combination of TNFα (50 ng/ml; PeproTech) and IFNγ (10 ng/ml; PeproTech). For chemical reagents dissolved in dimethylsulfoxide, the appropriate concentration of dimethylsulfoxide was added as a solvent control.
Cell Survival Assay.
The viable cells in each culture were enumerated with the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay by using a CellTiter 96 aqueous nonradioactive proliferation assay kit (Promega, Madison, WI), according to the manufacturer's instructions. TUNEL staining for apoptotic cells was performed by using a DeadEnd fluorometric TUNEL system (Promega). The fluorescence signals were detected with an Olympus FSX100 fluorescence microscopy (Olympus, Tokyo, Japan).
Measurement of Caspase 3/7 Activity.
Caspase 3/7 activity was measured with a Caspase-Glo assay kit (Promega), according to the manufacturer's instructions. Luminescence intensity was monitored with a fluorometric imaging plate reader (Synergy HT; BioTek Instruments, Winooski, VT).
Immunoblotting.
Cells were lysed in buffer containing 25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 0.1% SDS, 1% Nonidet P-40, 10% glycerol, and 1% (v/v) Triton X-100 (Kajiya et al., 2011). The cell lysates were subjected to ultrasonic treatment for 8 s, on ice. The proteins in the cell lysates were separated by using SDS-polyacrylamide gel electrophoresis (12% gel) and were electrophoretically transferred onto a nitrocellulose (NC) membrane (Bio-Rad Laboratories, Hercules, CA). The NC membranes were blocked with 5% nonfat milk for 1 h, followed by overnight reaction at 4°C with rabbit anti-human phosphorylated ERK antibody (1:2000; Cell Signaling Technology), rabbit anti-human total ERK antibody (1:2000; Cell Signaling Technology), rabbit anti-human phosphorylated Akt antibody (1:2000; Cell Signaling Technology), rabbit anti-human total Akt antibody (1:200; Cell Signaling Technology), rabbit anti-human phosphorylated PKCδ antibody (1:500; Cell Signaling Technology), rabbit anti-human total PKCδ antibody (1:1000; Cell Signaling Technology), rabbit anti-human phosphorylated EGFR antibody (1:500; Cell Signaling Technology), rabbit anti-human total EGFR antibody (1:5000; Cell Signaling Technology), rabbit anti-human phosphorylated Src family (Tyr416) antibody (1:500; Cell Signaling Technology), rabbit monoclonal anti-human total c-Src antibody (clone 36D10, 1:4000; Cell Signaling Technology), and horseradish peroxidase-conjugated mouse anti-β-actin antibody (1:5000; AbCam Inc., Cambridge, MA). After extensive washes, the NC membranes were incubated with peroxidase-conjugated donkey anti-rabbit IgG antibody (1:5000; Jackson ImmunoResearch Laboratories Inc., West Grove, PA) for 1 h at room temperature. The localization of specific antibody deposited to the molecule of interest on the NC membranes was detected by developing color with the Immobilon Western chemiluminescent horseradish peroxidase substrate (Millipore Corp., Billerica, MA).
Immunoprecipitation.
Equal amounts of cell lysates (500 μg of total protein in 1 ml of lysis buffer) were incubated overnight at 4°C with rabbit anti-human total EGFR antibody (1:50). GammaBind Plus Sepharose beads (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK) then were applied to the lysate/antibody mixture, which was incubated for 4 h at 4°C. After extensive washing with lysis buffer, proteins captured by the anti-EGFR antibody-coated beads were separated through SDS-polyacrylamide gel electrophoresis and were subjected to Western blot analyses as described above. Total c-Src protein levels were determined with rabbit anti-human total c-Src monoclonal antibody (clone 32G6, 1:500; Cell Signaling Technology).
[Ca2+]i Measurements.
[Ca2+]i was measured by using a fluo-8 no-wash calcium assay kit (AAT Bioquest, Sunnyvale, CA), as described previously (Ohta et al., 2011). HSG cells were incubated for 30 min at 37°C in phenol red-free Hanks' buffer with 20 mM HEPES containing fluo-8 no-wash dye-loading solution, in the presence or absence of 4-DAMP (1 μM) or BAPTA-AM (10 μM), and then were incubated for 30 min at room temperature. HSG cells were exposed to 100 μM CCh for 1 min, and the fluorescence intensity (excitation wavelength, 490 nm; emission wavelength, 525 nm) was detected with a fluorometric imaging plate reader. The fluorescence intensities were quantified from three independent cell cultures.
Transfection of siRNA.
Validated PKCδ siRNA, EGFR siRNA, and negative-control siRNA were obtained from Invitrogen (identification nos. PRKCDVHS41574 for PKCδ siRNA, EGFRVHS41680 for EGFR siRNA, and 12935-300 for negative-control siRNA). HSG cells in medium A were seeded at a density of 1.25 × 104 cells per well in 24-well plastic culture plates and were cultured for 24 h at 37°C. Then, 20 nM levels of PKCδ siRNA, EGFR siRNA, or negative-control siRNA were transfected into the cells by using RNAiMAX reagent (Invitrogen), according to the manufacturer's instructions. After 48 h of incubation, the cells treated with or without 100 μM CCh were lysed, and immunoblotting assays were performed as described above.
Statistical Analysis.
Differences between two groups of interest were analyzed with Student's t tests.
Results
CCh Protects HSG Cells from Apoptosis Induced through TNFα/IFNγ Stimulation.
To examine the cytoprotective effects of M3R in HSG cells, the muscarinic receptor agonist CCh was tested with respect to TNFα/IFNγ-induced apoptosis, which was reported previously to activate apoptosis signaling in HSG cells (Kamachi et al., 2002; Kulkarni et al., 2006). Consistent with a previous report, the combined TNFα/IFNγ treatment reduced cell viability among HSG cells (Fig. 1A). However, CCh protected against cell death induced by TNFα/IFNγ, in a dose-dependent manner (Fig. 1A). TUNEL staining showed that TNFα/IFNγ stimulation increased the number of apoptotic cells, whereas CCh treatment significantly abrogated the increase (Fig. 1, B and C). We monitored the effect of CCh on the caspase 3/7 death signal activity induced by the TNFα/IFNγ challenge in HSG cells. As shown in Supplemental Fig. 1A, caspase 3/7 activity reached a significantly higher level than control values at 24 h after stimulation with inflammatory cytokines. Therefore, in the following experiments, measurements of caspase 3/7 activity were performed 24 h after TNFα/IFNγ challenge. It should be noted that the protocol for incubating HSG cells for 24 h with proinflammatory cytokines to induce caspase activity was published previously (Kulkarni et al., 2006). The increased caspase 3/7 activity induced by TNFα/IFNγ (Fig. 1D) was significantly attenuated by the addition of CCh (Fig. 1D). It is noteworthy that ≥4-h pre-exposure to CCh did not demonstrate inhibitory effects of CCh on TNFα/IFNγ-induced caspase 3/7 activity (Supplemental Fig. 1B). However, both simultaneous addition of CCh and 1-h pretreatment with CCh attenuated TNFα/IFNγ-induced caspase 3/7 activity (Supplemental Fig. 1B), which indicates that CCh-mediated antiapoptosis signaling is effective in the 0- to 1-h range. Taken together, these findings suggested that CCh could protect HSG cells from TNFα/IFNγ-induced apoptotic signaling.
Fig. 1.

CCh protects HSG cells from proinflammatory cytokine-induced apoptosis. A, HSG cells were treated with or without various concentrations of CCh (10–1000 μM), in the absence or presence of TNFα (50 ng/ml)/IFNγ (10 ng/ml), and were incubated for 56 h. Cell viability was determined as described under Materials and Methods and is shown as a percentage of the viability of cells grown in medium B. Values represent the mean ± S.D. of four cultures. *, p < 0.05; **, p < 0.01, values differ significantly (t test). B to D, HSG cells were treated with or without 100 μM CCh, in the absence or presence of TNFα (50 ng/ml)/IFNγ (10 ng/ml), and were incubated for 40 h (B and C) or 24 h (D). B and C, TUNEL-positive apoptotic cells (green) are shown under each set of conditions (B), and the graph shows the percentage of TUNEL-positive apoptotic cells (C). Values represent the mean ± S.D. of three cultures. **, p < 0.01, values differ significantly (t test). Similar results were obtained from three experiments. D, caspase 3/7 activity was indicated by luminescence activity, as described under Materials and Methods. Values represent the mean ± S.D. of three cultures. *, p < 0.05; **, p < 0.01, values differ significantly (t test).
CCh Activates ERK and Akt Signaling Cascades, Which Play Crucial Roles in Protecting HSG Cells from Inflammatory Cytokine-Induced Apoptosis.
To understand how CCh-induced signaling protects HSG cells from apoptotic challenge, we explored the influence of CCh on ERK and Akt phosphorylation, which initiates signaling for cell survival in many types of cells (Steelman et al., 2008; Kajiya et al., 2009; Chappell et al., 2011). CCh treatment dramatically increased the phosphorylation of ERK and Akt, with both peaking at 5 min and then gradually decreasing (Fig. 2, A and B). Whereas CCh-induced ERK phosphorylation was inhibited by U0126 (MEK/ERK1/2 inhibitor), LY294002 (PI3K/Akt inhibitor) had no effect (Fig. 2C). In contrast, the up-regulation of Akt phosphorylation caused by CCh was remarkably inhibited by LY294002 but not by U0126 (Fig. 2C). It is noteworthy that neither U0126 nor LY294002 affected TNFα/IFNγ-induced caspase 3/7 activity, whereas those inhibitors clearly attenuated the CCh-mediated protective effect in HSG cells (Fig. 2, D and E). TNFα/IFNγ stimulation transiently increased the phosphorylation of ERK and Akt but phosphorylation decreased below basal levels 20 min after the challenge with inflammatory cytokines (Supplemental Fig. 2), which suggests that the net impact of TNFα/IFNγ on ERK and Akt phosphorylation levels in HSG cells was suppressive. These findings indicate that the up-regulation of both ERK and Akt signaling induced by CCh is responsible for its cytoprotective effect in HSG cells.
Fig. 2.
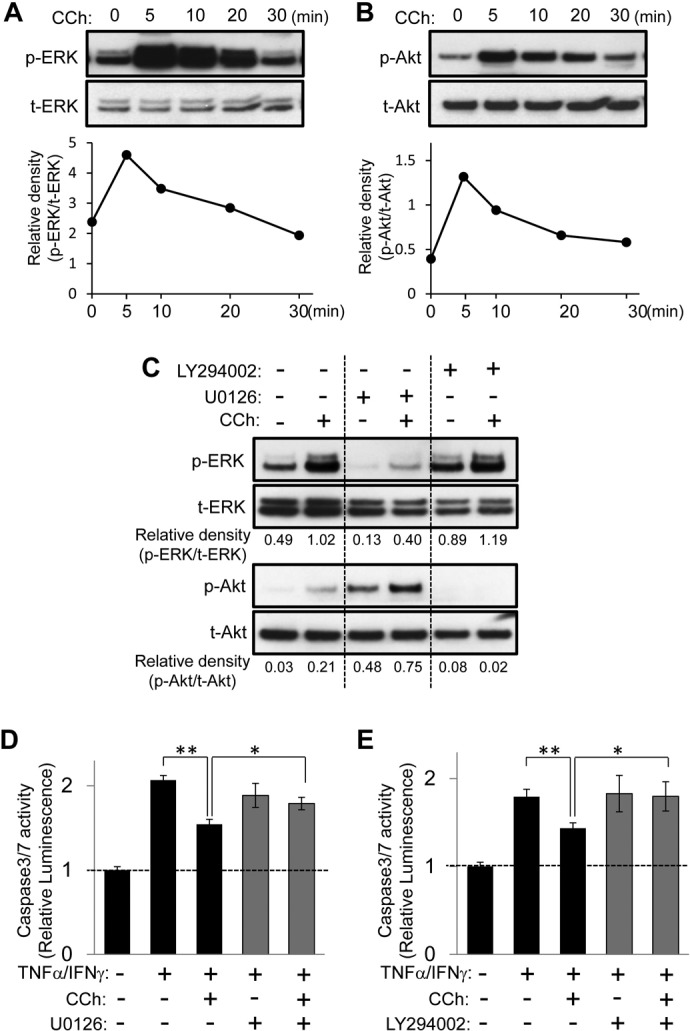
ERK and Akt signaling pathways are essential for CCh-promoted cytoprotection against apoptotic challenges in HSG cells. A and B, HSG cells were exposed to CCh (100 μM) for the indicated times. The phosphorylated (p) and total (t) ERK (A) and Akt (B) levels were analyzed through immunoblotting. C, HSG cells were pretreated with or without U0126 (2.5 μM) or LY294002 (2.5 μM) for 30 min and then were exposed to CCh (100 μM) for 10 min. The phosphorylated and total ERK and Akt levels were analyzed through immunoblotting. Quantification of the band density was performed through densitometric scanning of each band by using National Institutes of Health ImageJ software. D and E, HSG cells were pretreated with or without U0126 (2.5 μM) (D) or LY294002 (2.5 μM) (E) for 30 min. The cells then were treated with or without 100 μM CCh, in the absence or presence of TNFα (50 ng/ml)/IFNγ (10 ng/ml), and were incubated for 24 h. Caspase 3/7 activity was indicated by luminescence activity, as described under Materials and Methods. Values represent the mean ± S.D. of three cultures. *, p < 0.05; **, p < 0.01, values differ significantly (t test).
M3R Activation Is Responsible for CCh-Mediated Cell Survival Signaling in HSG Cells.
To examine the relationship between M3R activation and CCh-induced cell survival signaling, the M3R antagonist 4-DAMP was used. Pretreatment with 4-DAMP abrogated the phosphorylation of both ERK and Akt induced by CCh in HSG cells (Fig. 3A). Furthermore, the CCh-mediated protective effect on TNFα/IFNγ-induced caspase 3/7 activity was significantly inhibited by 4-DAMP. These findings suggest that CCh activates the ERK and Akt mitogenic survival signaling cascade through M3R in HSG cells.
Fig. 3.

M3R is involved in CCh-induced cell survival signaling in HSG cells. A, HSG cells were pretreated with or without 4-DAMP (1 μM) for 30 min and then were exposed to CCh (100 μM) for 10 min. The phosphorylated (p) and total (t) ERK and Akt levels were analyzed through immunoblotting. Quantification of the band density was performed through densitometric scanning of each band by using National Institutes of Health ImageJ software. B, HSG cells were pretreated with or without 4-DAMP (1 μM) for 30 min. The cells then were treated with or without 100 μM CCh, in the absence or presence of TNFα (50 ng/ml)/IFNγ (10 ng/ml), and were incubated for 24 h. Caspase 3/7 activity was indicated by luminescence activity, as described under Materials and Methods. Values represent the mean ± S.D. of three cultures. *, p < 0.05; **, p < 0.01, values differ significantly (t test).
Neither Ca2+ nor PKCδ Signaling Is Associated with CCh-Induced Cytoprotective ERK and Akt Signaling Cascades.
It is well known that Ca2+ signaling, which is caused by M3R activation, plays a pivotal role in fluid secretion in salivary gland cells (Li et al., 2004; Koo et al., 2008); therefore, we investigated whether Ca2+ signaling, in addition to the activation of ERK and Akt signaling described above, might be involved in CCh-induced cell survival signaling in HSG cells. As expected, CCh did increase [Ca2+]i, whereas both a M3R inhibitor (4-DAMP) and a Ca2+-chelator (BAPTA-AM) suppressed CCh-induced increases in [Ca2+]i in HSG cells (Fig. 4A). BAPTA-AM, which completely blocked calcium mobility in HSG cells, failed to inhibit CCh-induced phosphorylation of ERK and Akt (Fig. 4B). Moreover, the CCh-mediated suppressive effect on TNFα/IFNγ-induced caspase 3/7 activity was not affected by BAPTA-AM treatment (Fig. 4C). These findings indicate that CCh causes cell survival mitogenic signaling in a Ca2+-independent manner.
Fig. 4.

Neither Ca2+ nor PKCδ signaling is associated with CCh-induced cytoprotective ERK and Akt signaling cascades. A, HSG cells were pretreated with or without 4-DAMP (1 μM) or BAPTA-AM (10 μM) for 30 min and then were exposed to CCh (100 μM), followed by immediate measurement of 8-fluo fluorescence intensity to monitor intracellular calcium mobilization. Relative fluorescence intensity ratios are presented as the mean ± S.D. of three independent experiments, relative to values for medium alone. Cont, control. **, p < 0.01, values differ significantly (t test). ND, no significant difference. B, HSG cells were pretreated with or without BAPTA-AM (10 μM) for 30 min and then were exposed to CCh (100 μM) for 10 min. The phosphorylated (p) and total (t) ERK and Akt levels were analyzed through immunoblotting. C, HSG cells were pretreated with or without BAPTA-AM (10 μM) for 30 min. The cells then were treated with or without 100 μM CCh, in the absence or presence of TNFα (50 ng/ml)/IFNγ (10 ng/ml), and were incubated for 24 h. Caspase 3/7 activity was indicated by luminescence activity. Values represent the mean ± S.D. of three cultures. *, p < 0.05; **, p < 0.01, values differ significantly (t test). D, HSG cells were exposed to CCh (100 μM) for the indicated times. The phosphorylated and total PKCδ levels were analyzed through immunoblotting. E, HSG cells were pretreated with or without 4-DAMP (1 μM) for 30 min and then were exposed to CCh (100 μM) for 5 min. The phosphorylated and total PKCδ levels were analyzed through immunoblotting. F, HSG cells, having been transfected with negative control (neg) or PKCδ siRNA, were cultured for 48 h in medium A. The levels of PKCδ and β-actin in the cells were analyzed through immunoblotting. G, HSG cells, having been transfected with negative control or PKCδ siRNA, were cultured for 48 h in medium A and then were exposed to CCh (100 μM) for 5 min in medium B. The phosphorylated and total ERK, Akt, and PKCδ levels were analyzed through immunoblotting. Quantification of the band density was performed through densitometric scanning of each band by using National Institutes of Health ImageJ software. H, HSG cells, having been transfected with negative control or PKCδ siRNA, were cultured for 48 h in medium A and were pretreated with or without BAPTA-AM (10 μM) for 30 min. The cells then were treated with or without 100 μM CCh, in the absence or presence of TNFα (50 ng/ml)/IFNγ (10 ng/ml), and were incubated for 24 h. Caspase 3/7 activity was indicated by luminescence activity. Values represent the mean ± S.D. of three cultures. *, p < 0.05; **, p < 0.01, values differ significantly (t test).
Next, we investigated the possible engagement of PKCδ in M3R signaling, because activation of PKC has been linked to muscarinic receptor-induced ERK activation in several cell types (Keely et al., 1998; Jiménez and Montiel, 2005). On the basis of the following lines of evidence, we focused on PKCδ expressed in HSG cells. First, CCh caused cell survival mitogenic signaling in a Ca2+-independent manner (Fig. 4). Second, in contrast to the major isoforms of PKC (α, βI, βII, and γ), which require Ca2+ for full activation, activation of a second class of PKC isoforms, including PKCδ, occurs in a Ca2+-independent manner (Parker and Murray-Rust, 2004). Third, B lymphocytes infiltrating the salivary glands of patients with SjS could cause epithelial cell apoptosis through activation of PKCδ (Varin et al., 2012). We found that CCh increased PKCδ phosphorylation levels in a time-dependent manner (Fig. 4D) and the CCh-induced PKCδ up-regulation was blocked by 4-DAMP (Fig. 4E), which indicates that PKCδ is activated through M3R in HSG cells. Transfection of PKCδ siRNA decreased PKCδ expression (Fig. 4F). Contrary to our expectation, PKCδ knockdown did not affect the CCh-induced phosphorylation of ERK and Akt in HSG cells (Fig. 4G), whereas phosphorylated PKCδ expression was undetectable (data not shown). Furthermore, PKCδ siRNA failed to attenuate the CCh-mediated protective effect against TNFα/IFNγ-induced caspase 3/7 activity in HSG cells (Fig. 4H). These findings demonstrate that CCh-induced cell survival signaling is independent of PKCδ signaling in HSG cells.
EGFR Transactivation Is Involved in CCh-Induced Cell Survival Signaling.
It was reported that some muscarinic receptors could induce ERK activation through transactivation of EGFR in various cell types (Keely et al., 2000; Kanno et al., 2003). Therefore, we tested whether CCh-induced cell survival signaling is mediated by EGFR transactivation. CCh increased the phosphorylation of EGFR in a time-dependent manner (Fig. 5A) but the increase in phosphorylated EGFR levels was blocked by the M3R inhibitor 4-DAMP (Fig. 5B), which indicates that EGFR is transactivated through M3R in HSG cells. To test whether EGFR phosphorylation is involved in CCh-induced cell survival signaling, we next performed an inhibition assay using AG1478, an EGFR kinase-specific inhibitor. Pretreatment of HSG cells with AG1478 abrogated the CCh-induced phosphorylation of EGFR, ERK, and Akt (Fig. 5C). The protective effect of CCh on TNFα/IFNγ-induced caspase 3/7 activity was also significantly decreased by AG1478 (Fig. 5D). To confirm these findings, we performed an RNA interference-based, gene-silencing assay with EGFR siRNA. Transfection of EGFR siRNA decreased EGFR expression levels in HSG cells (Fig. 5E). EGFR siRNA transfection attenuated CCh-induced ERK and Akt phosphorylation (Fig. 5F). These findings suggest that M3R ligation with CCh leads to EGFR transactivation, which in turn initiates cytoprotective survival signaling in HSG cells.
Fig. 5.
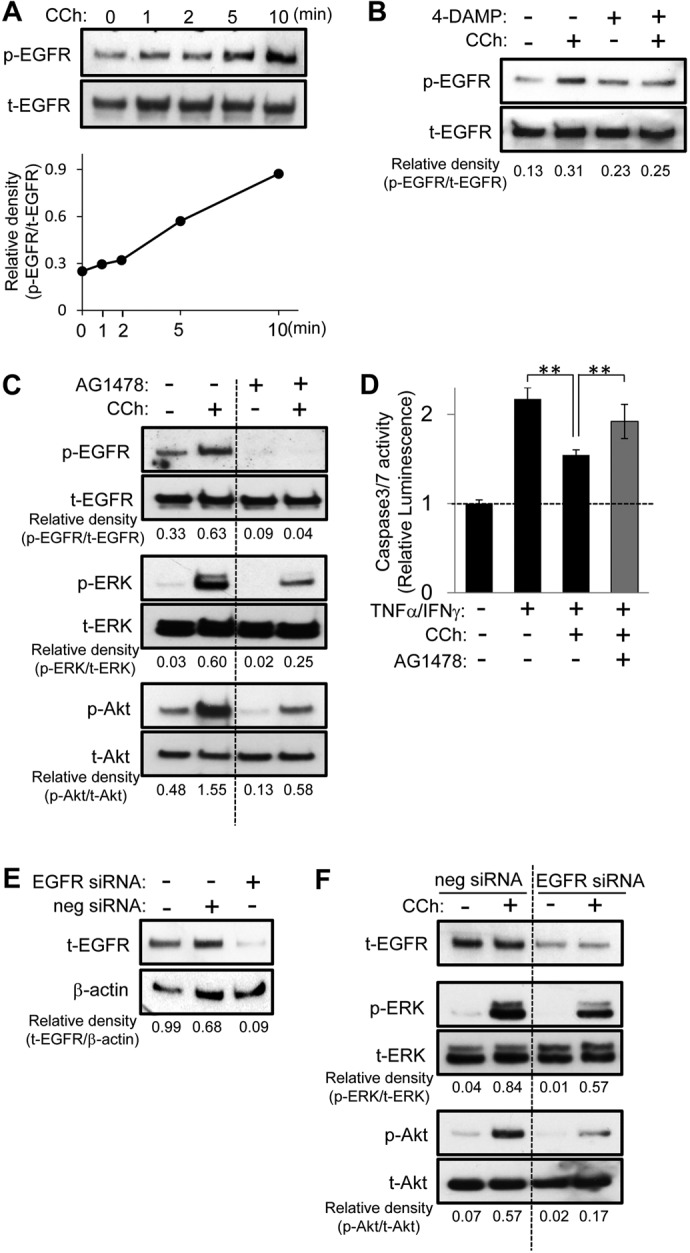
EGFR transactivation is involved in CCh-induced cell survival signaling. A, HSG cells were exposed to CCh (100 μM) for the indicated times. The phosphorylated (p) and total (t) EGFR levels were analyzed through immunoblotting. B and C, HSG cells were pretreated with or without 1 μM 4-DAMP (B) or 5 μM AG1478 (C) for 30 min and then were exposed to CCh (100 μM) for 5 min. The phosphorylated and total EGFR, ERK, and Akt levels were analyzed through immunoblotting. D, HSG cells were pretreated with or without AG1478 (5 μM) for 30 min. The cells then were treated with or without 100 μM CCh, in the absence or presence of TNFα (50 ng/ml)/IFNγ (10 ng/ml), and were incubated for 24 h. Caspase 3/7 activity was indicated by luminescence activity, as described under Materials and Methods. Values represent the mean ± S.D. of three cultures. **, p < 0.01, values differ significantly (t test). E, HSG cells, having been transfected with negative control (neg) or EGFR siRNA, were cultured for 48 h in medium A. The levels of EGFR and β-actin in the cells were analyzed through immunoblotting. F, HSG cells, having been transfected with negative control or EGFR siRNA, were cultured for 48 h in medium A and then were exposed to CCh (100 μM) for 5 min in medium B. The phosphorylated and total ERK, Akt, and EGFR levels were analyzed through immunoblotting. Quantification of the band density was performed through densitometric scanning of each band by using National Institutes of Health ImageJ software.
The Intracellular Signal Adaptor c-Src Intervenes between M3R and EGFR in the CCh-Induced EGFR Transactivation Cascade.
It is known that EGFR transactivation induced by GPCRs (such as muscarinic receptors) is dependent on activation of the cell signal adaptor c-Src, a nonreceptor tyrosine kinase, in various cell types (Rosenblum et al., 2000; Yeh et al., 2005). Therefore, we investigated whether c-Src might be associated with CCh-induced transactivation of EGFR signaling in HSG cells. CCh caused rapid transient phosphorylation of Src at Tyr416, indicating the active state of the kinase, which began to be expressed as early as 1 min after stimulation with CCh (Fig. 6A). The immunoprecipitation assay also showed that CCh stimulation enhanced the association between c-Src and EGFR in a time-dependent manner, which was detectable after 1 min (Fig. 6B). Moreover, 4-DAMP treatment blocked both CCh-induced phosphorylation of c-Src at Tyr416 (Fig. 6C) and the association of c-Src with EGFR (Fig. 6D). It is noteworthy that the Src tyrosine kinase inhibitor PP2 reduced CCh-induced phosphorylation of EGFR, ERK, and Akt in HSG cells (Fig. 6E). Finally, the protective effect of CCh against TNFα/IFNγ-induced caspase 3/7 activity was significantly attenuated by PP2 (Fig. 6F). Taken together, these findings demonstrate that CCh-induced Src activation through M3R causes EGFR transactivation, leading to cell survival signaling mediated by ERK and Akt phosphorylation in HSG cells.
Fig. 6.

Src activation is essential for the CCh-induced EGFR transactivation cascade, resulting in cytoprotection against apoptotic challenges. A and B, HSG cells were exposed to CCh (100 μM) for the indicated times. C and D, HSG cells were pretreated with or without 4-DAMP (1 μM) for 30 min and then were exposed to CCh (100 μM) for 2 min. A and C, the phosphorylated (p) and total (t) Src levels were analyzed through immunoblotting. B and D, cell lysates were prepared and used for immunoprecipitation (IP) with antibody to total EGFR. The levels of c-Src protein coimmunoprecipitated with EGFR and the total amounts of EGFR were visualized through immunoblotting (IB). E, HSG cells were pretreated with or without PP2 (5 μM) for 30 min and then were exposed to CCh (100 μM) for 2 min. The phosphorylated and total EGFR, ERK, and Akt levels were analyzed through immunoblotting. Quantification of the band density was performed through densitometric scanning of each band by using National Institutes of Health ImageJ software. F, HSG cells were pretreated with or without PP2 (5 μM) for 30 min. The cells then were treated with or without 100 μM CCh, in the absence or presence of TNFα (50 ng/ml)/IFNγ (10 ng/ml), and were incubated for 24 h. Caspase 3/7 activity was indicated by luminescence activity, as described under Materials and Methods. Values represent the mean ± S.D. of three cultures. **, p < 0.01, values differ significantly (t test).
Exogenously Applied EGF Emulates CCh-Induced Cytoprotective Cell Signaling.
If it is true that transactivation of EGFR through M3R can induce survival mitogenic signaling, then direct (i.e., exogenous) activation of EGFR should induce mitogenic survival signaling in HSG cells. To test this supposition, we examined the effect of exogenously applied EGF on EGFR, ERK, and Akt phosphorylation and TNFα/IFNγ-induced caspase 3/7 activity in HSG cells. Exogenously applied EGFR increased the levels of phosphorylated EGFR (Fig. 7A), ERK (Fig. 7B), and Akt (Fig. 7C) in a time-dependent manner. Most importantly, EGF treatment attenuated TNFα/IFNγ-induced caspase 3/7 activity (Fig. 7D). It is noteworthy that both the MEK-ERK inhibitor U0126 and the PI3K/Akt inhibitor LY294002 significantly decreased the cytoprotective effects of EGF (Fig. 7D). These findings clearly suggest that EGFR activation plays a critical role in the protection of salivary gland cells from apoptotic challenge.
Fig. 7.

Exogenously applied EGF activates both ERK and Akt signaling cascades, which results in inhibition of caspase 3/7 activity induced by an inflammatory apoptotic challenge in HSG cells. A—C, HSG cells were exposed to exogenously applied EGF (100 ng/ml) for the indicated times. The phosphorylated (p) and total (t) EGFR (A), ERK (B), and Akt (C) levels were analyzed through immunoblotting. Quantification of the band density was performed through densitometric scanning of each band by using National Institutes of Health ImageJ software. D, HSG cells were pretreated with or without U0126 (2.5 μM) or LY294002 (2.5 μM) for 30 min. The cells then were treated with or without 100 ng/ml EGF, in the absence or presence of TNFα (50 ng/ml)/IFNγ (10 ng/ml), and were incubated for 24 h. Caspase 3/7 activity was indicated by luminescence activity, as described under Materials and Methods. Values represent the mean ± S.D. of three cultures. *, p < 0.05; **, p < 0.01, values differ significantly (t test).
Discussion
The present study revealed that ligation of M3R, through transactivation of EGFR, could up-regulate not only ERK but also Akt, which suppresses the caspase 3/7 death signal, as well as apoptosis of HSG cells induced by an inflammatory insult (Fig. 8). Although Akt and ERK signaling are major intracellular signaling pathways for cell survival (Steelman et al., 2008; Kajiya et al., 2009; Chappell et al., 2011), many previous studies on the pathophysiological features of HSG cells focused on understanding M3R-induced Ca2+ signaling in the fluid secretion system (Li et al., 2004; Koo et al., 2008; Tobin et al., 2009) and not the underlying cytoprotective mechanism. Soltoff and Hedden (2010) demonstrated that activation of M3R promoted ERK phosphorylation in salivary gland cells, which supports our finding of CCh-mediated activation of ERK and Akt phosphorylation. However, the finding in the present study (i.e., that activation of M3R elicited cytoprotective signals through transactivation of EGFR) provides a novel scientific foundation for understanding the pathophysiological characteristics of SjS and offers new molecular targets for SjS drug discovery.
Fig. 8.

The CCh-induced M3R signaling cascade in HSG cells is summarized. P, phosphate.
The fact that some GPCR agonists can transactivate EGFR was originally reported in 1996 (Daub et al., 1996), and this theory is now applied to a wide variety of GPCR ligands (Gschwind et al., 2001). This paradigm requires the intervention of the intracellular signal adaptor molecule c-Src, which activates the intracellular tyrosine kinase domain of EGFR (Amorino et al., 2007; Rozengurt, 2007), as shown in this study (Fig. 6). However, studies demonstrated that transactivation of EGFR by GPCR ligands could occur through both intracellular c-Src intervention and extracellular EGFR-ligand transfer (Ohtsu et al., 2006; Bhola and Grandis, 2008). More specifically, activation of GPCR induces the expression of extracellular enzymes that cleave the ectodomain of heparin-binding (HB) EGF (cell membrane-bound form of EGFR ligands). Subsequently, the released (HB) EGF binds and activates EGFR (Ohtsu et al., 2006; Bhola and Grandis, 2008). To test whether such an alternative mechanism of extracellular M3R-EGFR transactivation is involved in CCh-stimulated HSG cells, we explored the effect of the HB EGF inhibitor CRM197 on CCh-induced EGFR, ERK, and Akt phosphorylation (Supplemental Fig. 3). However, because the HB EGF inhibitor blocked neither CCh-induced EGFR transactivation nor ERK/Akt phosphorylation (Supplemental Fig. 3), M3R-induced EGFR transactivation in HSG cells seemed to be solely regulated by intracellular c-Src intervention between M3R and EGFR.
In addition to CCh-induced EGFR transactivation involving Src activation, the present study demonstrated that EGFR activation induced by exogenously applied EGF is involved in cytoprotective signaling in HSG cells. Although exogenously applied EGF elicited full inhibition of caspase 3/7 activity induced by TNFα/IFNγ (Fig. 7D), it is noteworthy that the CCh-mediated suppression of caspase 3/7 activity was rather modest (∼50–60% inhibition) (Fig. 2, D and E). CCh also induced differences in the time course and signal intensity of EGFR, ERK, and Akt phosphorylation (Figs. 2, A and B, and 5A), compared with that induced by EGF (Fig. 7, A–C). More specifically, EGF induced EGFR phosphorylation and mitogenic signaling activity more rapidly and dramatically than did CCh. These findings indicated that the intensity of CCh-evoked EGFR transactivation was attenuated through the intervention of the M3R-Src signaling process, compared with direct activation of EGFR with EGF.
Ligation of M3R with its cognate agonist induces classic Gq protein-mediated phospholipase C β activation leading to the production of the second messengers inositol trisphosphate and diacylglycerol, which causes increases in [Ca2+]i and activation of PKCs (Singer et al., 1997). In accordance with this classic theory, CCh stimulated intracellular Ca2+ mobilization and PKCδ phosphorylation through M3R in HSG cells (Fig. 4). Although these cell signaling cascades were demonstrated to regulate saliva formation and secretion, CCh mediation of these classic pathways was not associated with cell survival signaling in HSG cells (Fig. 4) (Ambudkar et al., 1993; Soltoff and Toker, 1995; Soltoff et al., 1998).
Because the PKC family is composed of more than 15 isozymes, we tested, in addition to PKCδ siRNA, the effects of the pan-PKC inhibitor GF109203X on CCh-induced cytoprotective signaling in HSG cells. Consistent with the results from the PKCδ siRNA experiments (Fig. 4), the PKC inhibitor failed to attenuate CCh-induced ERK and Akt phosphorylation or its cytoprotective effect against TNFα/IFNγ-induced caspase 3/7 activation (Supplemental Fig. 4). It is still possible that other PKCs are associated with the CCh-induced mitogenic signaling, because GF109203X inhibits PKCα, PKCβI, PKCδ, and PKCε but has little or no effect on other PKC isoforms, such as PKCβII and PKCγ. A total absence of PKCβII in the acinar epithelial cells of patients with SjS has been reported (Törnwall et al., 1997), which suggests that PKCβII activation may not be associated with inflammation-induced caspase 3/7 activation in the context of SjS lesions. However, it is possible that other GF109203X-resistant PKC isoforms, such as PKCγ, may participate in CCh-mediated cytoprotective signaling. For example, when the effects of the EGFR inhibitor AG1478 (Fig. 5C) on the CCh-elicited phosphorylation of EGFR, ERK, and Akt were compared with those of the Src inhibitor PP2 (Fig. 6E), discrepancies in the levels of suppression of phosphorylation by these two drugs were evident, which suggests the possible intervention of other signaling pathways between Src and EGFR. Additional comprehensive studies will be required to elucidate the signaling networks that are involved in CCh-induced cytoprotective events.
Lin et al. (2008) reported that CCh stimulates ERK phosphorylation through PKC activity without EGFR transactivation, whereas another muscarinic agonist, pilocarpine, could up-regulate ERK phosphorylation through c-Src-dependent EGFR transactivation in the human salivary gland cell line HSY. Unlike the present study, which addressed the effects of CCh on cytoprotection, the study by Lin et al. (2008) focused only on CCh-induced ERK signaling, without considering the downstream outcomes of ERK activation. The HSG cells used in the present study predominantly express M3R among all muscarinic receptor isoforms (Nagy et al., 2007), whereas HSY cells express M1 and M3 receptors to equal degrees (Lin et al., 2008). To explain the discrepancy in the findings on the actions of CCh between the study by Lin et al. (2008) and the present study, it is thought that CCh binding to M1 receptors in HSY cells might have induced ERK phosphorylation without EGFR transactivation, whereas CCh-mediated M3R activation resulted in EGFR transactivation-dependent ERK activation in the present study. The study by Lin et al. (2008) supports our key finding (i.e., that activation of M3R can lead to cytoprotective ERK activation signaling through EGFR transactivation in HSG cells).
In native salivary gland tissue, CCh-induced elevations of [Ca2+]i stimulate fluid secretion in salivary acinar cells by activating apically located Cl− channels and basolaterally located K+ channels (Romanenko et al., 2006). In this context, CCh seems to increase the turnover of Na+/K+-ATPase and reduce intracellular ATP levels, which promotes the activation of AMP-activated protein kinase (AMPK) (Soltoff, 2004). It was reported that adiponectin could prevent IFNγ-induced apoptosis of salivary gland cells through AMPK activation (Katsiougiannis et al., 2010), which suggests that signaling that can activate AMPK, including CCh-evoked M3R stimulation, may also protect salivary gland cells from apoptosis. In contrast, immunohistochemical analysis of diseased gland tissue from a patient with SjS demonstrated that the surviving salivary gland cells strongly exhibited phosphorylated EGFR expression, although the number of apoptotic cells was increased (Nakamura et al., 2007). Dang et al. (2008) reported that salivary gland injury induced in rats up-regulated EGFR phosphorylation in salivary acinar cells, which protected cells against apoptosis. The latter two reports indicate that EGFRs of salivary gland cells are activated in the physiological context of SjS lesions. Finally, the finding that anti-EGFR monoclonal antibodies, such as cetuximab, can suppress the recurrence and/or metastasis of salivary gland carcinoma (Locati et al., 2009) strongly supports the finding that EGFR-evoked signaling is engaged in salivary gland cell survival in native tissue. Therefore, the application of EGF or the ligation of receptors (including M3R) that can trigger the transactivation of EGFR may constitute a novel therapeutic regimen to ameliorate the gland destruction observed among patients with SjS.
Although M3R chemical agonists, such as pilocarpine and cevimeline, are often used clinically to stimulate salivary secretion in patients with SjS (Mavragani and Moutsopoulos, 2007), no studies have addressed whether such treatment can also protect against the progression of gland destruction. On the basis of our results, it is plausible that clinical treatment using M3R chemical agonists could yield cytoprotective effects against proinflammatory cytokine-induced apoptosis in salivary gland cells, particularly in the context of the tissue destruction that occurs in patients with SjS.
In summary, CCh-stimulated M3R transactivates EGFR through a signal intervention mediated by c-Src, which results in phosphorylation of both ERK and Akt. The culmination of these signaling events attenuates TNFα/IFNγ-induced caspase 3/7 activity and protects the salivary gland cells from apoptosis (Fig. 8). This study sheds light on the EGFR transactivation system and introduces novel molecular targets to the search for a therapeutic chemical compound that can protect HSG cells from inflammation-induced apoptosis, potentially leading to the development of novel therapeutic interventions against SjS.
Supplementary Material
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
This study was supported by the National Institutes of Health National Institute of Dental and Craniofacial Research [Grant DE019644].
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
- SjS
- Sjögren's syndrome
- HSG
- human salivary gland
- M3R
- muscarinic type 3 receptor
- CCh
- carbachol
- ERK
- extracellular signal-regulated kinase
- EGFR
- epidermal growth factor receptor
- EGF
- epidermal growth factor
- siRNA
- small interfering RNA
- MEK
- mitogen-activated protein kinase kinase
- NC
- nitrocellulose
- TNFα
- tumor necrosis factor α
- IFNγ
- interferon γ
- AMPK
- AMP-activated protein kinase
- HB
- heparin-binding
- TUNEL
- terminal deoxynucleotidyl transferase dUTP nick end labeling
- GPCR
- G protein-coupled receptor
- PKC
- protein kinase C
- PI3K
- phosphatidylinositol 3-kinase
- 4-DAMP
- 4-diphenylacetoxy-N-methylpiperidine methiodide
- BAPTA-AM
- 1,2-bis-(o-aminophenoxy)-ethane-N,N,N′,N′-tetraacetic acid, tetraacetoxymethyl ester
- LY294002
- 2-morpholin-4-yl-8-phenylchromen-4-one
- AG1478
- N-(3-chlorophenyl)-6,7-dimethoxy-4-quinazolinamine
- PP2
- 4-amino-5-(4-chlorophenyl)-7-(dimethylethyl)pyrazolo[3,4-d]pyrimidine
- GF109203X
- 2-[1-(3-dimethylaminopropyl)-1H-indol-3-yl]-3-(1H-indol-3-yl)maleimide
- MTS
- 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- U0126
- 1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenylthio)butadiene.
Authorship Contributions
Participated in research design: Kajiya, Jin, Yu, Cha, and Kawai.
Conducted experiments: Kajiya, Ichimonji, Min, Zhu, and Almazrooa.
Contributed new reagents or analytic tools: Kajiya and Cha.
Performed data analysis: Kajiya, Ichimonji, Min, Zhu, and Almazrooa.
Wrote or contributed to the writing of the manuscript: Kajiya, Jin, Yu, Cha, and Kawai.
References
- Ambudkar IS. (2000) Regulation of calcium in salivary gland secretion. Crit Rev Oral Biol Med 11:4–25 [DOI] [PubMed] [Google Scholar]
- Ambudkar IS, Hiramatsu Y, Lockwich T, Baum BJ. (1993) Activation and regulation of calcium entry in rat parotid gland acinar cells. Crit Rev Oral Biol Med 4:421–425 [DOI] [PubMed] [Google Scholar]
- Amorino GP, Deeble PD, Parsons SJ. (2007) Neurotensin stimulates mitogenesis of prostate cancer cells through a novel c-Src/Stat5b pathway. Oncogene 26:745–756 [DOI] [PubMed] [Google Scholar]
- Baum BJ. (1993) Principles of saliva secretion. Ann NY Acad Sci 694:17–23 [DOI] [PubMed] [Google Scholar]
- Bhola NE, Grandis JR. (2008) Crosstalk between G-protein-coupled receptors and epidermal growth factor receptor in cancer. Front Biosci 13:1857–1865 [DOI] [PubMed] [Google Scholar]
- Cha S, Singson E, Cornelius J, Yagna JP, Knot HJ, Peck AB. (2006) Muscarinic acetylcholine type-3 receptor desensitization due to chronic exposure to Sjögren's syndrome-associated autoantibodies. J Rheumatol 33:296–306 [PubMed] [Google Scholar]
- Chappell WH, Steelman LS, Long JM, Kempf RC, Abrams SL, Franklin RA, Bäsecke J, Stivala F, Donia M, Fagone P, et al. (2011) Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget 2:135–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang H, Elliott JJ, Lin AL, Zhu B, Katz MS, Yeh CK. (2008) Mitogen-activated protein kinase up-regulation and activation during rat parotid gland atrophy and regeneration: role of epidermal growth factor and β2-adrenergic receptors. Differentiation 76:546–557 [DOI] [PubMed] [Google Scholar]
- Daub H, Weiss FU, Wallasch C, Ullrich A. (1996) Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature 379:557–560 [DOI] [PubMed] [Google Scholar]
- Fox RI, Kang HI. (1992) Pathogenesis of Sjögren's syndrome. Rheum Dis Clin North Am 18:517–538 [PubMed] [Google Scholar]
- Fox RI, Kang HI, Ando D, Abrams J, Pisa E. (1994) Cytokine mRNA expression in salivary gland biopsies of Sjögren's syndrome. J Immunol 152:5532–5539 [PubMed] [Google Scholar]
- Fox RI, Stern M. (2002) Sjögren's syndrome: mechanisms of pathogenesis involve interaction of immune and neurosecretory systems. Scand J Rheumatol Suppl 116:3–13 [PubMed] [Google Scholar]
- Greenwood JM, Dragunow M. (2010) M3 muscarinic receptors promote cell survival through activation of the extracellular regulated kinase (ERK1/2) pathway. Eur J Pharmacol 640:38–45 [DOI] [PubMed] [Google Scholar]
- Gschwind A, Zwick E, Prenzel N, Leserer M, Ullrich A. (2001) Cell communication networks: epidermal growth factor receptor transactivation as the paradigm for interreceptor signal transmission. Oncogene 20:1594–1600 [DOI] [PubMed] [Google Scholar]
- Jiménez E, Montiel M. (2005) Activation of MAP kinase by muscarinic cholinergic receptors induces cell proliferation and protein synthesis in human breast cancer cells. J Cell Physiol 204:678–686 [DOI] [PubMed] [Google Scholar]
- Kajiya M, Komatsuzawa H, Papantonakis A, Seki M, Makihira S, Ouhara K, Kusumoto Y, Murakami S, Taubman MA, Kawai T. (2011) Aggregatibacter actinomycetemcomitans Omp29 is associated with bacterial entry to gingival epithelial cells by F-actin rearrangement. PLoS One 6:e18287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajiya M, Shiba H, Fujita T, Takeda K, Uchida Y, Kawaguchi H, Kitagawa M, Takata T, Kurihara H. (2009) Brain-derived neurotrophic factor protects cementoblasts from serum starvation-induced cell death. J Cell Physiol 221:696–706 [DOI] [PubMed] [Google Scholar]
- Kamachi M, Kawakami A, Yamasaki S, Hida A, Nakashima T, Nakamura H, Ida H, Furuyama M, Nakashima K, Shibatomi K, et al. (2002) Regulation of apoptotic cell death by cytokines in a human salivary gland cell line: distinct and synergistic mechanisms in apoptosis induced by tumor necrosis factor alpha and interferon gamma. J Lab Clin Med 139:13–19 [DOI] [PubMed] [Google Scholar]
- Kanno H, Horikawa Y, Hodges RR, Zoukhri D, Shatos MA, Rios JD, Dartt DA. (2003) Cholinergic agonists transactivate EGFR and stimulate MAPK to induce goblet cell secretion. Am J Physiol Cell Physiol 284:C988–C998 [DOI] [PubMed] [Google Scholar]
- Katsiougiannis S, Tenta R, Skopouli FN. (2010) Activation of AMP-activated protein kinase by adiponectin rescues salivary gland epithelial cells from spontaneous and interferon-gamma-induced apoptosis. Arthritis Rheum 62:414–419 [DOI] [PubMed] [Google Scholar]
- Keely SJ, Calandrella SO, Barrett KE. (2000) Carbachol-stimulated transactivation of epidermal growth factor receptor and mitogen-activated protein kinase in T84 cells is mediated by intracellular Ca2+, PYK-2, and p60src. J Biol Chem 275:12619–12625 [DOI] [PubMed] [Google Scholar]
- Keely SJ, Uribe JM, Barrett KE. (1998) Carbachol stimulates transactivation of epidermal growth factor receptor and mitogen-activated protein kinase in T84 cells: implications for carbachol-stimulated chloride secretion. J Biol Chem 273:27111–27117 [DOI] [PubMed] [Google Scholar]
- Kolkowski EC, Reth P, Pelusa F, Bosch J, Pujol-Borrell R, Coll J, Jaraquemada D. (1999) Th1 predominance and perforin expression in minor salivary glands from patients with primary Sjögren's syndrome. J Autoimmun 13:155–162 [DOI] [PubMed] [Google Scholar]
- Koo NY, Li J, Hwang SM, Choi SY, Lee SJ, Oh SB, Kim JS, Lee EB, Song YW, Park K. (2008) Functional epitope of muscarinic type 3 receptor which interacts with autoantibodies from Sjogren's syndrome patients. Rheumatology (Oxford) 47:828–833 [DOI] [PubMed] [Google Scholar]
- Kroneld U, Halse AK, Jonsson R, Bremell T, Tarkowski A, Carlsten H. (1997) Differential immunological aberrations in patients with primary and secondary Sjögren's syndrome. Scand J Immunol 45:698–705 [DOI] [PubMed] [Google Scholar]
- Kulkarni K, Selesniemi K, Brown TL. (2006) Interferon-gamma sensitizes the human salivary gland cell line, HSG, to tumor necrosis factor-alpha induced activation of dual apoptotic pathways. Apoptosis 11:2205–2215 [DOI] [PubMed] [Google Scholar]
- Lee BH, Tudares MA, Nguyen CQ. (2009) Sjögren's syndrome: an old tale with a new twist. Arch Immunol Ther Exp (Warsz) 57:57–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Ha YM, Kü NY, Choi SY, Lee SJ, Oh SB, Kim JS, Lee JH, Lee EB, Song YW, et al. (2004) Inhibitory effects of autoantibodies on the muscarinic receptors in Sjögren's syndrome. Lab Invest 84:1430–1438 [DOI] [PubMed] [Google Scholar]
- Lin AL, Zhu B, Zhang W, Dang H, Zhang BX, Katz MS, Yeh CK. (2008) Distinct pathways of ERK activation by the muscarinic agonists pilocarpine and carbachol in a human salivary cell line. Am J Physiol Cell Physiol 294:C1454–C1464 [DOI] [PubMed] [Google Scholar]
- Locati LD, Bossi P, Perrone F, Potepan P, Crippa F, Mariani L, Casieri P, Orsenigo M, Losa M, Bergamini C, et al. (2009) Cetuximab in recurrent and/or metastatic salivary gland carcinomas: a phase II study. Oral Oncol 45:574–578 [DOI] [PubMed] [Google Scholar]
- Mavragani CP, Moutsopoulos HM. (2007) Conventional therapy of Sjogren's syndrome. Clin Rev Allergy Immunol 32:284–291 [DOI] [PubMed] [Google Scholar]
- Nagy K, Szlávik V, Rácz G, Ovári G, Vág J, Varga G. (2007) Human submandibular gland (HSG) cell line as a model for studying salivary gland Ca2+ signalling mechanisms. Acta Physiol Hung 94:301–313 [DOI] [PubMed] [Google Scholar]
- Nakamura H, Kawakami A, Ida H, Koji T, Eguchi K. (2007) EGF activates PI3K-Akt and NF-κB via distinct pathways in salivary epithelial cells in Sjögren's syndrome. Rheumatol Int 28:127–136 [DOI] [PubMed] [Google Scholar]
- Ohta K, Laborde NJ, Kajiya M, Shin J, Zhu T, Thondukolam AK, Min C, Kamata N, Karimbux NY, Stashenko P, et al. (2011) Expression and possible immune-regulatory function of ghrelin in oral epithelium. J Dent Res 90:1286–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsu H, Dempsey PJ, Eguchi S. (2006) ADAMs as mediators of EGF receptor transactivation by G protein-coupled receptors. Am J Physiol Cell Physiol 291:C1–C10 [DOI] [PubMed] [Google Scholar]
- Park K, Case RM, Brown PD. (2001) Identification and regulation of K+ and Cl− channels in human parotid acinar cells. Arch Oral Biol 46:801–810 [DOI] [PubMed] [Google Scholar]
- Parker PJ, Murray-Rust J. (2004) PKC at a glance. J Cell Sci 117:131–132 [DOI] [PubMed] [Google Scholar]
- Pauley KM, Gauna AE, Grichtchenko II, Chan EK, Cha S. (2011) A secretagogue-small interfering RNA conjugate confers resistance to cytotoxicity in a cell model of Sjögren's syndrome. Arthritis Rheum 63:3116–3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce KL, Premont RT, Lefkowitz RJ. (2002) Seven-transmembrane receptors. Nat Rev Mol Cell Biol 3:639–650 [DOI] [PubMed] [Google Scholar]
- Romanenko V, Nakamoto T, Srivastava A, Melvin JE, Begenisich T. (2006) Molecular identification and physiological roles of parotid acinar cell maxi-K channels. J Biol Chem 281:27964–27972 [DOI] [PubMed] [Google Scholar]
- Rosenblum K, Futter M, Jones M, Hulme EC, Bliss TV. (2000) ERKI/II regulation by the muscarinic acetylcholine receptors in neurons. J Neurosci 20:977–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozengurt E. (1998) Signal transduction pathways in the mitogenic response to G protein-coupled neuropeptide receptor agonists. J Cell Physiol 177:507–517 [DOI] [PubMed] [Google Scholar]
- Rozengurt E. (2007) Mitogenic signaling pathways induced by G protein-coupled receptors. J Cell Physiol 213:589–602 [DOI] [PubMed] [Google Scholar]
- Sato M, Hayashi Y, Yanagawa T, Yoshida H, Yura Y, Azuma M, Ueno A. (1985) Intermediate-sized filaments and specific markers in a human salivary gland adenocarcinoma cell line and its nude mouse tumors. Cancer Res 45:3878–3890 [PubMed] [Google Scholar]
- Singer WD, Brown HA, Sternweis PC. (1997) Regulation of eukaryotic phosphatidylinositol-specific phospholipase C and phospholipase D. Annu Rev Biochem 66:475–509 [DOI] [PubMed] [Google Scholar]
- Soltoff SP. (2004) Evidence that tyrphostins AG10 and AG18 are mitochondrial uncouplers that alter phosphorylation-dependent cell signaling. J Biol Chem 279:10910–10918 [DOI] [PubMed] [Google Scholar]
- Soltoff SP, Avraham H, Avraham S, Cantley LC. (1998) Activation of P2Y2 receptors by UTP and ATP stimulates mitogen-activated kinase activity through a pathway that involves related adhesion focal tyrosine kinase and protein kinase C. J Biol Chem 273:2653–2660 [DOI] [PubMed] [Google Scholar]
- Soltoff SP, Hedden L. (2010) Isoproterenol and cAMP block ERK phosphorylation and enhance [Ca2+]i increases and oxygen consumption by muscarinic receptor stimulation in rat parotid and submandibular acinar cells. J Biol Chem 285:13337–13348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltoff SP, Toker A. (1995) Carbachol, substance P, and phorbol ester promote the tyrosine phosphorylation of protein kinase C delta in salivary gland epithelial cells. J Biol Chem 270:13490–13495 [DOI] [PubMed] [Google Scholar]
- Steelman LS, Abrams SL, Whelan J, Bertrand FE, Ludwig DE, Bäsecke J, Libra M, Stivala F, Milella M, Tafuri A, et al. (2008) Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia 22:686–707 [DOI] [PubMed] [Google Scholar]
- Tobin G, Giglio D, Lundgren O. (2009) Muscarinic receptor subtypes in the alimentary tract. J Physiol Pharmacol 60:3–21 [PubMed] [Google Scholar]
- Törnwall J, Konttinen YT, Tuominen RK, Törnwall M. (1997) Protein kinase C expression in salivary gland acinar epithelial cells in Sjögren's syndrome. Lancet 349:1814–1815 [DOI] [PubMed] [Google Scholar]
- Varin MM, Guerrier T, Devauchelle-Pensec V, Jamin C, Youinou P, Pers JO. (2012) In Sjögren's syndrome, B lymphocytes induce epithelial cells of salivary glands into apoptosis through protein kinase C delta activation. Autoimmun Rev 11:252–258 [DOI] [PubMed] [Google Scholar]
- Yeh CK, Ghosh PM, Dang H, Liu Q, Lin AL, Zhang BX, Katz MS. (2005) β-Adrenergic-responsive activation of extracellular signal-regulated protein kinases in salivary cells: role of epidermal growth factor receptor and cAMP. Am J Physiol Cell Physiol 288:C1357–C1366 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.