

Abstract
Homohexameric, N-Ethylmaleimide Sensitive Factor (NSF) disassembles Soluble NSF Attachment Protein Receptor (SNARE) complexes after membrane fusion, an essential step in vesicular trafficking. NSF contains three domains (NSF-N, NSF-D1, and NSF-D2), each contributing to activity. We combined electron microscopic (EM) analysis, analytical ultracentrifugation (AU) and functional mutagenesis to visualize NSF’s ATPase cycle. 3D density maps show that NSF-D2 remains stable, whereas NSF-N undergoes large conformational changes. NSF-Ns splay out perpendicular to the ADP-bound hexamer and twist upwards upon ATP binding, producing a more compact structure. These conformations were confirmed by hydrodynamic, AU measurements: NSF-ATP sediments faster with a lower frictional ratio (f/f0). Hydrodynamic analyses of NSF mutants, with specific functional defects, define the structures underlying these conformational changes. Mapping mutations onto our 3D models allows interpretation of the domain movement and suggests a mechanism for NSF binding to and disassembly of SNARE complexes.
Keywords: SNAREs, AAA proteins, Analytical centrifugation, Membrane trafficking, Electron microscopy
1. Introduction
The N-Ethylmaleimide Sensitive Factor (NSF), first purified in 1988 (Block et al., 1988), has been shown to be an essential enzyme required for all membrane trafficking events in a cell. This homohexameric ATPase binds to Soluble NSF Attachment Protein (SNAP)-Receptor (SNARE) complexes and mediates the recycling of spent SNARE complexes for subsequent rounds of membrane fusion (Zhao et al., 2011). As with other ATPases associated with diverse cellular activities (AAA) proteins, NSF couples the hydrolysis of ATP to the conformational modulation of a protein complex, in this case the SNARE complex (Hanson and Whiteheart, 2005). NSF protomers are composed of three domains: NSF-N (1–205); and two AAA domains (NSF-D1, 206–477; NSF-D2, 478–744) (Tagaya et al., 1993). Each uniquely contributes to overall activity. NSF-N is required for SNAP–SNARE binding and positively charged residues at the top of the interface between the NA and NB subdomains are important (Nagiec et al., 1995; Zhao et al., 2009). NSF-D1 and -D2 contain the ~230 amino acid motif that is the hallmark of the AAA family. NSF-D1 catalyzes ATP hydrolysis, which is required for SNARE complex disassembly (Whiteheart et al., 1994; Nagiec et al., 1995). Conserved elements of the AAA motif in NSF-D1 (i.e. Walker A and B box, Arginine Fingers, Sensor 2) have been shown, by mutagenesis analysis, to be critical for NSF function (Zhao et al., 2009). NSF-D2 is not catalytic, but is required for hexamerization.
NSF binds to the SNARE complex via an adaptor protein called α-Soluble NSF Attachment Protein (α-SNAP) (Clary et al., 1990). Three α-SNAPs mediate binding of one NSF hexamer to the SNARE complex (Wimmer et al., 2001). Electron microscopy studies and crystallographic data indicate that SNAP binds along the length of the four-helical, coiled-coils of the SNARE complex in an antiparallel manner, such that its C-terminus is opposite to the SNARE’s transmembrane domains (Hanson et al., 1997; Hohl et al., 1998; Furst et al., 2003). Consistently, mutagenesis suggests that the C-terminus of SNAP is important for NSF binding and the penultimate leucine is required to stimulate NSF’s ATPase activity and thus SNAP–SNARE complex disassembly (Barnard et al., 1996, 1997; Marz et al., 2003). Under non-hydrolysis conditions, the complex of NSF/SNAP/SNARE resembles a “sparkplug” with the NSF hexamer at the wide end and the transmembrane domains at the other (Hohl et al., 1998; Furst et al., 2003). Based on this arrangement, one could envision a mechanism by which NSF untwists the SNARE complex to mediate its disassembly.
Full-length NSF has not been crystallized but structures of NSF-N (May et al., 1999; Yu et al., 1999) and NSF-D2 (Lenzen et al., 1998; Yu et al., 1998) are available, as are several EM analyses of NSF (Hanson et al., 1997; Furst et al., 2003) and the NSF–SNAP–SNARE complex (Hohl et al., 1998; Furst et al., 2003). Despite these structural data, it is not clear how NSF mediates SNAP–SNARE complex disassembly. To advance this endeavor, we have characterized the conformational changes of NSF primed to bind the SNAP–SNARE complex and of NSF at the end of the ATPase cycle using single particle analysis and analytical ultracentrifugation of NSF in the ATP- and ADP-bound states, respectively. We have also characterized the conformations of two NSF mutants that are deficient at specific steps in the SNARE disassembly process. Based on these data, we begin to gain insight into how NSF disassembles SNARE complexes.
2. Materials and methods
2.1. Plasmids, mutagenesis, and protein expression
His6-NSF-myc (1–744) from Chinese Hamster Ovary (CHO) cells was cloned into pQE9 (Qiagen, Valencia, CA). His6-NSF-D1D2-myc fragment (206–744), and His6-NSF-D2 fragment (488–744) were inserted into the pQE9 vector. The NSF-L441A and NSF-R67A mutants were created with the QuickChange, site-directed mutagenesis kit (Stratagene, La Jolla, CA) using wild-type His6-NSF-myc as template. The mutations were confirmed by dideoxy nucleotide sequencing by Davis Sequencing (Davis, CA). NSF wild-type (NSF-WT) or mutants were expressed in the Escherichia coli strain, Rosetta DE3 pLacI, and purified according to the published methods (Zhao et al., 2009). The purified proteins were finally dissolved in Buffer A (50 mM HEPES/KOH, pH 7.4, 300 mM KCl, 1 mM MgCl2, 2mM β-mecaptoethanol, 5% glycerol) containing 0.5 mM ATP and stored at −80 °C. The purity of the proteins was examined on a 12.5% SDS–PAGE gel and stained by Coomassie Blue R-250. NSF or mutants were exchanged to different nucleotide states by Size Exclusion Chromatography (SEC) on a column (0.7 × 30 cm) of Sephadex G-50 (Sigma, St. Louis, MO) at 200 μl/min.
2.2. Electron microscopy and single particle analysis
The samples, in their respective nucleotide-bound states, were incubated with an additional 2 mM AMP-PNP or 2 mM ADP with 2 mM MgCl2 at 0 °C for 30 min, reflecting a 50- to 100-fold excess over the Kd for the two ATP binding nucleotide sites (NSF-D1 and - D2; Matveeva et al., 1997). This ensures that at least 95% of NSF’s binding sites are occupied by the indicated nucleotide. All samples were diluted to ~100 μg/ml in 50 mM HEPES/KOH, 50 mM KCl, pH 7.4, immediately prior to application on freshly glow-discharged carbon coated C-flat grids and preserved with a 1% solution of uranyl formate (Ohi et al., 2004). Two Tecnai F20 Twin transmission electron microscopes were used to acquire all datasets. The microscopes were operating at 120 kV, using a dose of 25 e−/Å2 (ADP, ATP and D1D2), and a nominal underfocus ranging from 0.2 to 2.0 μm. 858 (ATP), 909 (ADP), and 448 (D1D2) images were recorded at a nominal magnification of 62,000× (ATP and ADP) and 50,000× (D1D2) at pixel sizes of 0.137 nm and 0.226 nm, respectively, at the specimen level (Supplementary Fig. 1). Images were recorded with a Tietz F415 4 k × 4 k pixel CCD camera utilizing the Leginon software (Suloway et al., 2005). Experimental data were processed by the Appion software package (Lander et al., 2009). The defoci were estimated using ctffind3 (ATP/ADP) (Mindell and Grigorieff, 2003) and ACE2 (D1D2). The phases were flipped on the whole micrograph. Particles were automatically selected (Roseman, 2004) and extracted at a box size of 160 pixels and binned by a factor of 2 for the final reconstruction. The stacks contained: 113,259 (ATP), 66,846 (ADP) and 57,004 (D1D2) particles. Symmetry-free initial models were created for each dataset independently using Imagic-5 on reference-free aligned 2D class averages (van Heel et al., 1996). To exclude model bias all datasets were also refined against all other starting models, resulting in no change to the final models. A variety of symmetry groups (C1, C3 and C6) were tested during image processing and the resulting maps were in agreement with the final model presented here. Since a C6 point group symmetry was also apparent in reference-free class averages generated by the Xmipp tools: clustering 2D reference-free alignment, and the reference-free maximum likelihood alignment procedure (Scheres et al., 2005a,b; Lander et al., 2009; Sorzano et al., 2010), a C6-pointgroup symmetry was applied to the final refinement cycles. Only particles contributing to the best class averages, as judged by eye, were retained for processing. To further eliminate particles that were not fully assembled, aggregated, or just noise, 3D maximum likelihood, implemented in Xmipp, was applied (Scheres et al., 2008). For each dataset, three models were generated and those that showed clearly distinguishable features, such as the top or bottom view, hexameric shape, or the double stacked rings side views, were used to select the contributing single particles into a new stack for further refinement using standard iterative projection matching in Xmipp and Imagic- 5 (van Heel et al., 1996; Sorzano et al., 2004). For the final refinement round only the best 10–40% from each dataset were used (AMP-PNP: 12,978, ADP: 14,192, D1D2: 20,104). This level of particle inclusion is consistent with the percent hexamer measured by AU for each of the NSF preparations (see Table 1). The angular increment started at 14° and was reduced by 2° every second iteration until the models stabilized at 2°. Resolution was assessed by calculating the Fourier Shell Correlation (FSC) at a cutoff of 0.5, which provided values between 16 and 20 Å resolution (Supplementary Fig. 2).
Table 1.
Measured and Predicted Hydrodynamic Parameters for NSF-WT or Mutants in ADP- and AMP-PNP-Bound States. (Ni2+-NTA)2-Cy3-His6-NSF-Myc wild-type or mutants were in Buffer A containing 0.5 mM ADP or AMP-PNP. Sedimentation velocity was performed at 15,000 rpm and 10° C. Radial intensity scan data were collected, converted to 2 pseudoabsorbance data and fit to the c(s) model by SEDFIT program. The peak s-values have been corrected to reflect water density and viscosity at 20° C (s20,W). The experiments were repeated thrice, all the values in the table were averaged and indicated with the subscript meas. The models in Fig. 2 and the crystal structure of NSF-D2 (PDB entry 1D2 N) were evaluated using the Solution Modeler (SOMO) routine (Fig. 3; Brookes et al., 2010a,b) and hydrodynamic parameters predicted from that analysis are indicated with the subscript pred. Values predicted for the most extended intermediate obtained by morphing between ADP and ATP forms (Movie 3, frame 00:00:04) are indicated with the subscript pred, int. % Hexamer, the percentage of hexamer in solution; s20,W, sedimentation coefficient; ΔAMP-PNP-ADP, s20,W values of ADP state subtracted from that of AMP-PNP bound state; f/f0, frictional ratio; MW, molecular weight.
| % Hexamer
|
s20,W
|
Δ |
f/f0
|
MW (kDa)
|
|||||
|---|---|---|---|---|---|---|---|---|---|
| ADP | AMP-PNP | ADP | AMP-PNP | AMP-PNP-ADP | ADP | AMP-PNP | ADP | AMP-PNP | |
| NSFmeas | 62.01 ± 5.74 | 46.99 ± 3.70 | 11.43 ± 1.00 | 12.11 ± 1.20 | 0.68 | 1.54 ± 0.01 | 1.46 ± 0.02 | 525 ± 92 | 511 ± 95 |
| NSFpred | – | – | 16.37 | 16.91 | 0.54 | 1.33 | 1.28 | – | – |
| NSFpre,int | – | – | – | 14.22 | – | – | 1.37 | – | – |
| D1D2meas | 43.99 ± 2.93 | 14.10 ± 0.31 | 10.13 ± 1.09 | 8.13 ± 0.92 | −2.00 | 1.37 ± 0.001 | 1.66 ± 0.03 | 332 ± 77 | 290 ± 76 |
| D1D2pred | – | – | 13.89 | – | – | 1.27 | – | – | – |
| D2meas | 28.84 ± 1.92 | 10.30 ± 0.63 | 6.66 ± 0.43 | 6.59 ± 0.39 | −0.07 | 1.18 ± 0.003 | 1.19 ± 0.02 | 131 ± 18 | 151 ± 19 |
| D2pred | – | – | – | 7.10 | – | – | 1.31 | – | – |
| L441Ameas | 34.44 ± 1.06 | 34.19 ± 2.78 | 9.94 ± 1.58 | 9.93 ± 1.34 | 30.01 | 1.77 ± 0.07 | 1.78 ± 0.07 | 474 ± 141 | 530 ± 144 |
| R67A meas | 55.59 ± 12.48 | 38.71 ± 4.21 | 11.10 ± 0.96 | 11.85 ± 1.12 | 0.75 | 1.59 ± 0.02 | 1.49 ± 0.004 | 543 ± 86 | 506 ± 87 |
To map amino acids of interest, the D1 domain of NSF was modeled using the I-Tasser webserver (Zhang 2007; Roy and Kucukural, 2010). It generated 5 homology models of the NSF monomer with high confidence values. The best homology model (C-score: −0.06, TM-score 0.71 ± 0.12) was used together with the D2 and N domains of the available X-ray structures of NSF (Lenzen et al., 1998; May et al., 1999). Automatic docking of individual domains was performed using the Chimera software package (Pettersen et al., 2004). In the first step, the available crystal structure of the individual D2 domain was automatically positioned into the density map using the “fit in map” and “search” functions. Five additional copies were then automatically generated according to the 6-fold symmetry. In the second step, the homology model was placed using the same function. The “search” function allows the user to manually scroll through different docking options. Since the highest correlation for this domain was at the same position as the D2 domain, the next best docking solution was chosen. This corresponded to the expected position of the D1 domain. To verify this fitting, two hexamers were docked using the “fit sequence” function in Chimera to minimize overlap between the individual PDBs. These results were very similar to the positions estimated by the docking of individual domains, supporting the docking solution. In this step, the orientation of the D2 hexamer was also tested. The presented orientation of the D2 domain was docked with a correlation coefficient of 8.78 for AMP-PNP- and 8.76 for the ADP-NSF reconstructions; the same hexamer rotated by 180° had values of 8.26 and 8.16, respectively. These data further support the chosen orientation for the D2 domains. The N domains were placed using the same techniques. Finally, to minimize overlaps the “fit sequence” function was again used respecting the 6-fold symmetry. This minimized the overlaps between the individual PDBs. This process was repeated several times with different starting points. After the last optimization step all models were identical.
2.3. Ultracentrifugation analyses
Ni2+-loaded, bis-NTA-Cy3 ((Ni2+-NTA)2-Cy3) was synthesized and His6-NSF or mutants (4 μM) were labeled with (Ni2+-NTA)2-Cy3 (1 mole of dye per mole of NSF hexamer) as described (Zhao et al., 2010). The sedimentation velocity measurements of (Ni2+- NTA)2-Cy3-His6-NSF-myc or mutants in Buffer A containing either 0.5 mM ADP or AMP-PNP were performed at 10.0 ± 0.1 ° C in a Beckman XL-A analytical ultracentrifuge (Beckman, Fullerton, CA), using an An-60 Ti rotor at indicated speed (NSF-WT, NSF-L441A and NSF-R67A at 15,000 rpm; NSF-D1D2 at 20,000 rpm; NSF-D2 at 35,000 rpm). The sample volumes were 300 μl with a starting OD550nm of between 0.15 and 0.17. The radial absorbance distributions during the experiment were recorded at 550 nm. The data were fit using a numerical solution of the Lamm equation implemented in the program SEDFIT (Schuck, 2000; Dam and Schuck, 2004).
For differential sedimentation of the AMP-PNP- and ADP-bound forms of (Ni2+-NTA)2-Cy3-His6-NSF-myc wild-type or mutants, solutions placed in the sample and reference sectors were matched for OD550nm and volume. In addition to protein and buffer, the solution in the sample sector contained 0.5 mM AMP-PNP while that in the reference sector contained ADP. The radial intensity distributions during the experiment were recorded at 550 nm. The data were converted to two pseudoabsorbances and fit using a numerical solution of the Lamm equation implemented in the program SEDFIT (Schuck, 2000; Dam and Schuck, 2004).
2.4. Hydrodynamic calculations
Theoretical values of s20,W were computed using the SOMO bead-modeling approach (Rai et al., 2005) available in the US-SOMO implementation (Brookes et al., 2010a,b) (http://www.ultrascan.uthscsa.edu). The program settings were: accessible surface area cut-offs 20 Å and 50% for atomic structures and modeled beads, respectively; synchronous overlap removal in all steps; outward translation of exposed side-chain beads and bead fusion thresholds of 70% between exposed beads. Buried beads were not included in the computations. Stick boundary conditions were assumed throughout.
3. Results
3.1. Visualized by electron microscopy and image analysis
To investigate the structural changes that the NSF hexamer undergoes during nucleotide hydrolysis, we examined its conformation in two different nucleotide-bound states: AMP-PNP- (AMP-PNP is a non-hydrolysable ATP analogue; Fig. 1a) and ADP- (NSF-ADP; Fig. 1b). The AMP-PNP-bound state is considered equivalent to an ATP-bound state. Electron microscopy and single particle analysis were used to visualize the proteins preserved in negative stain (Supplementary Figs. 1 and 3). During the initial stage of the analysis the particles were aligned and averaged in a reference-free manner in two-dimensions (2D; see Methods). The 6-fold symmetry of the complexes is apparent in both top and bottom views. All reconstructions have a diameter of ~115 Å and a height of ~80 Å with a central circular pore (Fig. 1) and six spokes connected to the central pore. Examining the top, bottom, and side views of the maps indicates that NSF is composed of two stacked hexameric rings that are slightly offset with respect to each other. Oblique grooves between the rings characterize the side views (Fig. 1). Comparison of the maps shows that the bottom rings consist of six morphologically similar globular domains; whereas the top rings display marked differences. The upper rings do not contain a visible central pore at mass correlating threshold; however, it is apparent when the threshold is reduced (data not shown).
Fig. 1.
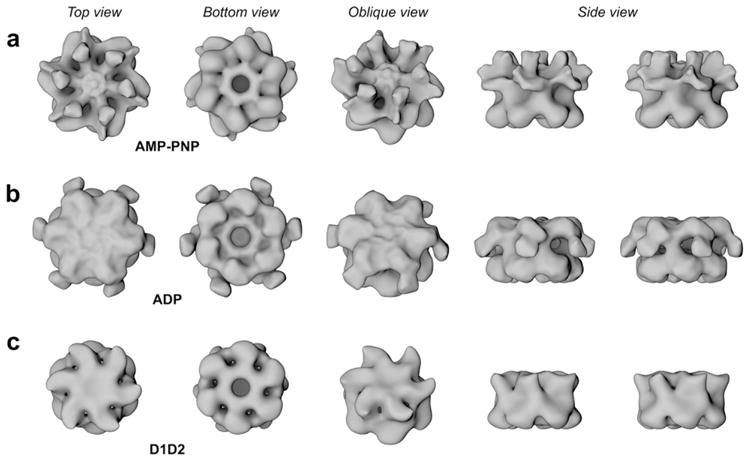
Surface representations of NSF in three different nucleotide-bound states and the N domain deletion mutant D1D2-ADP. Surface representations of the reconstructed density maps at mass correlating thresholds filtered to 18 Å (~510 and ~390 kDa). Rows a–c show NSF-AMP-PNP, -ADP and NSF-D1D2-ADP, respectively. The maps are shown as top, bottom, oblique and side views (from left to right). All maps have a diameter of ~115 Å and a height of ~80 Å for the D1 and D2 domains together. In NSF-AMP-PNP map arm densities protrude upwards, in NSF-ADP they splay outwards, and no arm densities are apparent in the D1D2-ADP map.
The NSF-ADP 3D-density has arm-like structures protruding from the central ring (Fig 1b). These arms were not apparent for the NSF-AMP-PNP (Fig. 1a) state. Densities, protruding from NSF’s main body, were observed in single particle EM and quick-freeze/deep-etch studies (Hanson et al., 1997; Furst et al., 2003). Based on this work, it seems likely that the arms are due to the NSF-N domain. To directly address this point and to verify the topology of NSF, a data set for the ΔN truncation mutant (D1D2) bound to ADP was obtained (Fig. 1c and Supplementary Fig. 4). A comparison of all three 3D-reconstructions shows that bottom views (presumably NSF-D2) are highly similar. The NSF-D1D2-ADP state shows some differences which could be a consequence of the loss of the NSF-N domain. It is also possible that a small degree of misalignment between top and bottom views could explain this slight discrepancy since the two ATP-binding domains (D1 and D2) are so similar. However, as the purpose of generating the D1D2 model was to unambiguously determine the position of the N-domains, the difficulty in distinguishing the top and bottom views does not alter our conclusions. When viewing from the top, the arms of NSF-ADP splay out from the upper ring and point downwards (Figs. 1b and 2c and d). Since these arms are not apparent in the D1D2 map (Fig. 1c and Supplementary Fig. 4), we conclude that these densities correspond to the N domains of the NSF complex. Similar comparisons between the full-length and D1D2 maps show that the densities in the upper ring of the NSF-AMP-PNP differ significantly from NSF-ADP state. In the NSF-AMP-PNP state, which is primed for SNAP–SNARE complex binding, the arm densities protrude upwards and are interspersed with alternating holes extending through the upper ring (Fig. 1).
Fig. 2.

Docking of the individual domains into the surface representations. Results of the automated docking procedure are depicted for the ATP- (a and b) and ADP-states (c and d) of full-length NSF as side views with (a and c) and without the 3D-densities (b and d). Only one out of the six monomers is shown for clarity. Alternative views of the docking of the full hexamer are presented in lower panel (e and f) and in Movies 1 and 2. The automated positioning of the individual PDBs is in good agreement with the individual density maps. No significant overlap between individual domains is observed. N domains (PDB entry 1QDN) are colored in blue, D1 in orange and D2 (PDB entry 1D2N) in. Note the stability of D2, relative to D1, which tilts and rotates: the N arms pivoting by almost 180° around the α-helical part of D1 (Movie 3). Residues of special interest are labeled: yellow (R10, K104, K105, D142, K143) and red (R67) residues: important for SNAP/SNARE binding; purple: Sensor 2 (E440, L441, E442); blue: Arg-fingers (R385, R388); green: pore residues (Y296, G298).
3.2. Molecular docking assigns the domain architecture of NSF
Molecular docking was carried out using the available (NSF-N and -D2) and modeled (NSF-D1) PDB structures (see Section 2). Automated docking of individual D2 domains showed that they fit into the lower rings of all density maps, revealing an arrangement that was almost identical to the expected hexamer (Lenzen et al., 1998; Yu et al., 1998). The β-sheet subdomain faces the central pore with the α-helical domain at the edges. In all models, the orientation of the NSF-D2 hexamer is identical with that of VCP/p97. Docking of NSF-D1 and NSF-N domains was performed using the NSF-AMP-PNP (Fig. 2a and b and Movie 2) and NSF-ADP (Fig. 2c and d and Movie 1) maps. The modeled NSF-D1 hexamer is stacked with its C-termini facing the N-termini of the NSF-D2 hexamer with a ~17° offset along the central vertical axis (Fig. 2 and Supplementary Figs. 4 and 5). This orientation contradicts the previous finding that described a C-termini to C-termini arrangement of the NSF-D1 and -D2 domains (Furst et al., 2003). Comparison of the maps for NSF-ADP and NSF-AMP-PNP show that the NSF-D1 domains are slightly rearranged. Using the nucleotide-binding site as a central anchoring point, a twist of ~10° is measured in the horizontal plane, as well as a ~20° tilt along the vertical axis. Additionally, the domains are translated by ~5 Å in the ADP conformation. At the present resolution of our maps, we did not take the liberty of sub-domain or flexible fitting to optimize the model. The putative N-domains in the AMP-PNP map had less mass than expected from the corresponding crystal structure. Since this is not observed in the ADP state we can presume that this region in the AMP-PNP state is more flexible (Fig. 2). The NSF-N domains undergo a significant conformational shift; pivoting almost 180° upwards from the ADP to AMP-PNP state (Figs. 1 and 2 and Movie 3). When our 3D reconstruction of NSF-AMP-PNP was superimposed with the 20S particle EM map (Furst et al., 2003), we observed significant overlap of the NSF-D1- and NSF-N-domains (Supplementary Fig. 5). In addition, residues that contribute to the SNAP–SNARE binding site: yellow (R10, K104, K105, D142, K143) and red (R67) on the N-domain and green residues (Y296 and G298), in the pore (Zhao et al., 2009), are easily accessible and face the solvent in our molecular model of NSF-AMP-PNP (Fig. 2 and Movies 1 and 2). On the N-domain, the residues point sideways towards the neighboring domain rather than directly towards the central pore. These data unequivocally establish that there are significant, nucleotide-dependent, conformational changes in NSF. These changes are almost exclusively in the ATP hydrolyzing, NSF-D1 domain and in the SNAP–SNARE-binding, N-domain. NSF-D2 remains stable.
3.3. The conformational changes of NSF detected by analytical ultracentrifugation
To further define the conformational changes that NSF undergoes in solution, NSF in different nucleotide states was examined by sedimentation velocity analytical ultracentrifugation (AU). Since the absorbance of a buffer containing 0.5 mM nucleotide is ~7.7 A260 cm−1, NSF was labeled with (Ni2+-NTA)2-Cy3 through binding to its N-terminal His6-tag. The movement of NSF in solution was then detected by measuring the absorbance of Cy3 at 550 nm (Zhao et al., 2010). We examined the sedimentation coefficients (s20,W) and frictional ratios (f/f0) of NSF, D1D2, and D2 in their AMP-PNP- or ADP-bound state. NSF-AMP-PNP, compared to NSF-ADP, has larger s20,W (12.11 vs. 11.43) and a smaller f/f0 ratio (1.46 vs. 1.54) which indicates that the AMP-PNP bound form is more compact in solution (Table 1). The D2 domain alone displays almost the same s20,W (6.59 vs. 6.66) and f/f0 (1.19 vs. 1.18) in either nucleotide state, suggesting that NSF-D2 exhibits little, nucleotide-dependent conformational change in solution. These data are consistent with those observed in the single particle analysis: the NSF-D1 and NSF-N showed rearrangements based on nucleotide binding, but NSF-D2 did not. The NSF-D1D2 (Δ N) mutant has different hydrodynamic parameters. NSF-D1D2 sediments more slowly in its AMP-PNP-bound state, with a smaller s20,W value (8.13 vs. 10.13). This differential is opposite to that seen for full-length NSF in the two nucleotide states, indicating that the AMP-PNP- bound state of the truncation mutant is less compact. Given the lack of changes in NSF-D2, it appears that NSF-D1 is responsible for the differences between AMP-PNP- and ADP-D1D2. These data imply that NSF-D1 is flexible and that the N domain directly affects its conformation (as noted above). Without the constraints offered by NSF-N, D1 may not pack into a hexamer and thus may extend from the stable D2 base.
Using the structural models generated from the EM data (Fig. 2), we constructed hydrodynamic bead models to assess the congruence between the EM and AU analyses (Fig. 3). The models were evaluated using the Solution Modeler (SOMO) routine (Brookes et al., 2010a,b). Excellent agreement was found between the measured and predicted values of s20,W for the compact D2 hexamer (s20,W,pred = 7.10 vs. s20,W,meas = 6.66 ± 0.43, Table 1). In contrast, for NSF and the NSF-D1D2 mutant, The predicted values of s20,W were uniformly larger and f/f0 values were uniformly smaller than those measured by AU (Table 1). This generalized difference between predicted and measured values suggests that, in solution, the ensembles of NSF structures are on average less compact than the structural models predict. Our models were built from compact crystalline domains arranged to fit the negative stain EM envelopes. Thus, they may not capture features of the most extended conformations available to NSF. Indeed, our hypothetical conformational trajectory between ATP and ADP states is populated with extended conformers giving smaller s-values than the end states (Movie 3 and Table 1). This suggests that differences between observed and predicted hydrodynamics may reflect sampling of states along the normal conformational pathway. Finally, the hydrodynamic models do predict that NSF-ATP should sediment more rapidly than NSF-ADP, which is observed experimentally (Table 1). The theoretical s-value difference between the two states (~0.6 s) is in very good agreement with experimental observation (0.68 s) measured for full-length NSF.
Fig. 3.

Hydrodynamic bead models. Hydrodynamic bead models of full-length NSF in AMP-PNP and ADP-bound states and of D1D2-ADP and D2-ANP-PNP were constructed based on the models in Fig. 2 and the crystal structure of NSF-D2 (PDB entry 1D2N). The models were built using the Solution Modeler (SOMO) routine (Brookes et al., 2010a,b) and presented as top and side views. Bead colors reflect degree of surface exposure as assigned by SOMO.
3.4. Analysis of the R67 and Sensor 2 (L441A) mutants
Since NSF-N and NSF-D1 undergo conformational changes during ATP hydrolysis, we focused on residues in these two domains. One, R67 in NSF-N, is located on the positively charged surface near the apex of the interface between NSF-NA and NSF-NB (red residues in Fig. 2). R67A/E mutations disrupt NSF binding to SNAP–SNARE complexes (Zhao et al., 2009). The positional conservation of R67 in many Class 2 AAA proteins with N domains indicates a key functional role (Zhao et al., 2011). Studies of VCP/p97 suggest that the residue might be part of a “latch” controlling the position of the VCP-N domain by linking it to VCP-D1 (Brunger and DeLaBarre, 2003; DeLaBarre and Brunger, 2003). Since NSF-N is important for SNAP–SNARE binding, any disruption in a “latch” could affect binding. If R67 is part of a “latch”, we would expect R67 mutants to disrupt nucleotide-induced conformational changes. Alternatively, if R67 is only part of a binding site with its adjacent residues, mutating it might not affect the nucleotide-dependent rearrangements. In Fig. 4a and b, the R67A mutant showed similar, nucleotide- dependent conformational changes as did wild-type NSF. The downward deflection of the differential scan indicates that the R67A-AMP-PNP form sediments faster than the ADP-bound form (Fig. 4b). Consistently, the relative difference in s20,W and f/f0 values between R67A-ADP or R67-AMP-PNP were similar to those for NSF-WT in the two nucleotide-bound states (Table 1). These conformational changes in the R67A mutant are not consistent with R67 serving as an anchoring latch that restricts N domain movement; but, do support the contention that R67, together with its adjacent, positively charged residues, may serve as a SNAP–SNARE binding site.
Fig. 4.
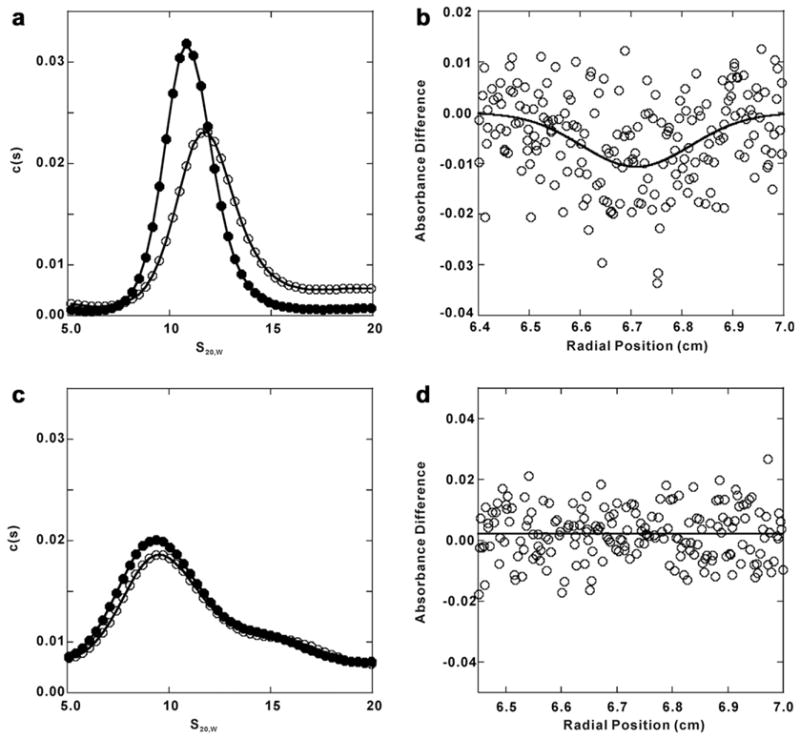
Sedimentation velocity characterization of NSF mutant conformation in ADP or AMP-PNP state. Panel a: sedimentation velocity analysis of NSF-R67A in ADP- and AMP-PNP- bound states. His6-NSF-R67A in ADP (closed circles) or AMP-PNP (open circles) was labeled with (Ni2+-NTA)2-Cy3 and sedimentation velocity was performed as in the Methods. Radial intensity scan data were collected, converted to 2-pseudoabsorbance data and fitted to the c(s) model by SEDFIT program, returning the spectrum of s-values shown. Panel b: differential scan of sedimentation velocity of the AMP-PNP and ADP forms of (Ni2+-NTA)2-Cy3-His6-NSF-R67 (see Section 2). Solutions placed in the sample and reference sectors of the rotor were matched for OD550nm and volume. The downward deflection in the scan, taken after 80 min of sedimentation, indicates that NSF-R67A in its AMP-PNP-bound form sediments more rapidly. Panel c: the c(s) spectrum of (Ni2+-NTA)2-Cy3-His6-NSF-L441A-ADP (closed circles) or L441A-AMP-PNP (open circles) derived from the sedimentation velocity data as in panel A. Panel d: differential sedimentation of the AMP-PNP and ADP forms of (Ni2+-NTA)2-Cy3-His6-NSF-L441A. The lack of deflection in the scan indicates that NSF-L441A in its AMP-PNP-bound form sediments in the same speed as that in the ADP-bound form.
The Sensor 2 region is adjacent to the nucleotide-binding site and thus may control the position of the α-helical subdomain relative to the nucleotide and the α/β subdomain in NSF-D1. In this position, Sensor 2 could detect the nucleotide bound and thus discriminate between ADP and ATP in the binding pocket. Mutations in Sensor 2 partially affect SNAP–SNARE binding; moreover, they lack SNAP–SNARE-stimulated ATPase and the ability to disassemble SNAP–SNARE complexes (Zhao et al., 2009). To test the hypothesis that Sensor 2 affects nucleotide-dependent conformational changes, we analyzed the L441A mutant by sedimentation velocity AU. Interestingly, the L441A mutant did not show significant alterations upon the binding of ATP or ADP, as indicated by the overlapping sedimentation profiles and the flatness of the differential scan (Fig. 4c and d, respectively). In fact, both L441A-ADP and L441A-AMP-PNP had lower s20,W and higher f/f0 values indicating that the hexamer is less compact than either nucleotide-bound states of NSF-WT (Table 1). These data imply that Sensor 2 controls the nucleotide-dependent conformational changes of NSF. Without that action, the N domains may be even more splayed than in the wild type NSF-ADP conformation.
4. Discussion
We have examined the conformational changes that NSF undergoes in two different stages of its hydrolytic cycle by comparing the structures of NSF in the ATP- and ADP-bound states (AMP-PNP- bound states is assumed to reflect the ATP-bound state). We observe a strong reorientation of the N and D1 domains between states, while the D2 remains remarkably stable. The most significant change observed between states was the movement of NSF-N, from positions near the top of the hexamer axis in the AMP-PNP- bound state, to ones down and away from the NSF-D1 in the ADP-bound state (Figs. 1 and 2 and Movies 1–3). These conformation changes were consistent with hydrodynamic analyses, which showed that, in solution, the ADP-bound form is less compact with a lower s20,W. The data indicate that in response to ATP hydrolysis, the NSF-N arms pivot around the α-helical sub domain of D1 by almost 180° (Fig. 2 and Movie 3). ATP-bound NSF is competent to bind SNAP–SNARE complexes but the ADP-bound form is not; thus, this observed reorientation of NSF-N is likely to be important for SNAP–SNARE complex binding. In the ATP-bound state, the positively- charged residues (Fig. 2, Movie 2; yellow and red residues) in NSF N, which are required for SNAP–SNARE binding (Zhao et al., 2009), are positioned so that they could interact with the negatively charged C-terminal region of the three α-SNAPs from the SNAP–SNARE complex (Barnard et al., 1996, 1997; Wimmer et al., 2001). Such an arrangement of NSF, SNAP, and SNAREs is consistent with the available structures of the NSF–SNAP–SNARE complex (Hohl et al., 1998; Furst et al., 2003; Supplementary Fig. 5). Given the difference in position of the NSF-N domains between the ATP and ADP-bound conformations (Supplementary Fig. 4), it appears that ATP hydrolysis leads to an outward pivoting motion of the N domains suggesting a mechanism by which SNAP–SNARE complexes could be disassembled (see below).
Structures of other AAA ATPases, obtained by various methods, show significant differences between the nucleotide states (Tucker and Sallai, 2007). Several studies have described conformational changes in the closely related VCP/p97. EM analysis of VCP/p97 shows highly flexible N domains (Rouiller et al., 2002; Beuron et al., 2003, 2006). Beuron and coworkers proposed a model in which the N domain of VCP/p97 rotates by ~100° upon binding to the p47 adaptor complex (Beuron et al., 2006). Different nucleotide states of VCP/p97 have also been described by Small Angle X-ray Scattering (SAXS) experiments (Davies et al., 2005, 2008). In these studies the D1 and D2 domains form a stable central core, while the N domains undergo significant conformational changes upon ATP binding, albeit less than observed in our study of NSF. It is likely that the relative motion of NSF will be stronger than that of VCP/p97 due to differences in the mechanisms of these two ATPases. This assumption is based on the fact that the catalytically active domains are switched. In VCP/p97 the peripheral N domains are separated from the D2 hexamer by the catalytically inactive D1 hexamer. In NSF the N domains are contiguous with the active D1 hexamer. Thus it is likely that the combined motion of N and D1 in NSF will result in more significant changes (Brunger and DeLaBarre, 2003). Structural studies of VCP/p97 using X-ray crystallography also reveal conformational changes for the different stages in the hydrolysis cycle. However, these changes are not as strong as was reported using SAXS or EM analysis. It is possible that the structural constraints imposed by the crystal lattice restrict the conformations (Davies et al., 2008). Lee and coworkers observe significant conformational changes, as the AAA ATPase ClpB goes through its hydrolysis cycle (Lee et al., 2007). Superposition of their EM maps onto the ones presented here reveal similarities within the basic architecture of both proteins (data not shown). Interestingly, the ~17° offset between the D1 and D2 domains seems to be conserved. In the cryo-EM structure of Vps4p, the N domains are reported to form a central protrusion above the upper ring on the symmetry axis (Yu et al., 2008). Taken together with the data reported here, these studies indicate a great variability in N domain positioning in response to different nucleotide states.
4.1. How NSF might use ATP hydrolysis to disassemble the SNAP–SNARE complex
From biochemical analysis of NSF, it is clear that the nucleotide state of NSF-D1 determines function. ATP-binding is essential for SNAP–SNARE binding and ATP hydrolysis is essential for complex disassembly. This argues that the two states (AMP-PNP- and ADP-bound) examined here, represent the binding competent start (Fig. 5a, b) and the post-disassembly finish of NSF’s catalytic cycle (Fig. 5e and f). In the ATP-bound state, NSF-N domains are positioned near the apex of the hexamer, thus presenting six potential binding surfaces to the three SNAPs coating the SNARE coiled coils (Fig. 5b). Based on mutagenesis data, R67 and the surrounding residues (Fig. 5, yellow bar) constitute a significant part of the interacting surface. Consistently, this surface is accessible and correctly oriented in the ATP-bound state. Upon ATP hydrolysis, (inferred from the ADP-bound state) this binding surface is moved down and away to the periphery of the hexamer. Such a movement might represent the “power stroke” required for pulling or twisting the SNAPs and thus facilitating SNARE complex disassembly (Fig. 5c and d). This “power stroke” is, in part, controlled by the Sensor 2 region of NSF-D1. This region monitors the nucleotide bound in the active site and propagates the nucleotide-dependent conformational changes, perhaps through repositioning of the α-helical domain of the AAA motif. Further analysis will be required to determine how this repositioning mediates the changes in NSF-N domains observed here.
Fig. 5.
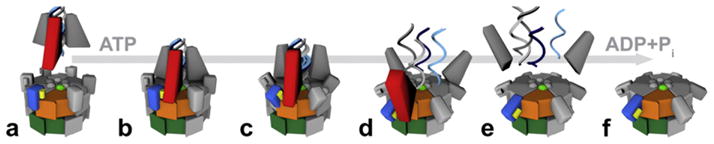
Potential mechanism of NSF. NSF-ATP binds to the C-terminus of α-SNAP (red wedge), which is bound to the coiled coil of the SNARE complex (a and b). Upon ATP hydrolysis the N-domains (blue block) pivot around the D1-domain, away from the center pulling, the α-SNAP and SNARE complex (c and d). This radial force serves to separate the SNAREs and thus disassemble the complex (e and f). The positively charged surface of NSF-N, which is important for SNAP–SNARE binding, is depicted as a yellow bar. The pore residues in NSF-D1 are depicted as green spheres. NSF-D1 and NSF-D2 are depicted as orange and green blocks, respectively.
Of note are the pore residues (Fig. 5, green spheres). This region, like NSF-N, has been shown to contribute to SNAP–SNARE complex binding (Zhao et al., 2009). The relative positions of the pore residues and the N domains suggest that the pore could serve as a fulcrum against which the N domains pull during ATP hydrolysis and complex disassembly. In this scenario, the SNAREs could be held in place by the pore, while the N-domains pull on the SNARE-bound SNAPs. The resulting radial force could be sufficient to unwind the coiled-coil domains of the SNARE complex. While this is speculative, such a model does explain how a soluble NSF homohexamer could mediate the disassembly of a membrane-anchored complex. This model will be useful in formulating future cross-linking and single molecule experiments, which will be required to determine if the pore and the N domain work together as fulcrum and lever, respectively.
5. Conclusions
In this study, we have shown that NSF undergoes significant conformational changes in its N and D1 domains during the ATPase cycle. In the ATP-bound state, which is competent to bind SNAP–SNARE complexes, the N domains are positioned at the top of the hexamer ring. In this configuration the N domains are positioned to bind the membrane-distal end of the cylindrical SNAP–SNARE complex. In the ADP-bound state, which represents the post-complex- disassembly state, the N domains have rotated almost 180° and are positioned along the side of the ring. The magnitude of these conformational changes is supported by analytical ultracentrifugation analysis and by hydrodynamic modeling. AU analyses of a NSF mutant (L441A) in the Sensor 2 suggest that this portion of the AAA motif of D1 mediates these nucleotide-dependent alterations. Further mapping of functionally-defined, NSF mutations onto the structures presented here, suggest a molecular mechanism by which this AAA protein uses ATP hydrolysis to disassemble SNAP–SNARE complexes.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The electron microscopy and image analysis presented here was conducted at the National Resource for Automated Molecular Microscopy which is supported by the National Institutes of Health though the National Center for Research Resources’ P41 program (RR017573). The work was also supported by Grants HL56652 and HL091893 from the National Institutes of Health to S.W.W. A.M. received support from the NIH Roadmap Award P50 GM073197 and E.W. and B.C. received support from GM095573. We thank Dr. Lance Hellman for his help with the analytical ultracentrifugation experiments and Dr. Elena A. Matveeva for her insightful comments on the manuscript.
Abbreviations
- AAA
ATPase associated with diverse cellular activities
- AU
analytical ultracentrifugation
- EM
electron microscopy
- NSF
N-Ethylmaleimide Sensitive Factor
- PDB
Protein Data Base
- SNAP
Soluble NSF Attachment Protein
- SNARE
SNAP Receptor
- VCP
Valosin Containing Protein
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.jsb.2011.12.018.
Footnotes
Competing financial interests
The authors declare no competing financial interests.
References
- Barnard RJ, Morgan A, Burgoyne RD. Domains of alpha-SNAP required for the stimulation of exocytosis and for N-ethylmalemide-sensitive fusion protein (NSF) binding and activation. Molecular Biology of the Cell. 1996;7 (5):693–701. doi: 10.1091/mbc.7.5.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard RJ, Morgan A, Burgoyne RD. Stimulation of NSF ATPase activity by alpha-SNAP is required for SNARE complex disassembly and exocytosis. Journal Cell Biology. 1997;139 (4):875–883. doi: 10.1083/jcb.139.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuron F, Dreveny I, Yuan X, Pye VE, Mckeown C, et al. Conformational changes in the AAA ATPase p97–p47 adaptor complex. The EMBO Journal. 2006;25 (9):1967–1976. doi: 10.1038/sj.emboj.7601055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuron F, Flynn TC, Ma J, Kondo H, Zhang X, et al. Motions and negative cooperativity between p97 domains revealed by cryo-electron microscopy and quantised elastic deformational model. Journal of Molecular Biology. 2003;327 (3):619–629. doi: 10.1016/s0022-2836(03)00178-5. [DOI] [PubMed] [Google Scholar]
- Block MR, Glick BS, Wilcox CA, Wieland FT, Rothman JE. Purification of an N-ethylmaleimide-sensitive protein catalyzing vesicular transport. Proceedings of the National Academy of Sciences of the United States of America. 1988;85 (21):7852–7856. doi: 10.1073/pnas.85.21.7852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes E, Cao W, Demeler B. A two-dimensional spectrum analysis for sedimentation velocity experiments of mixtures with heterogeneity in molecular weight and shape. European Biophysics Journal. 2010a;39 (3):405–414. doi: 10.1007/s00249-009-0413-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes E, Demeler B, Rocco M. Developments in the US-SOMO bead modeling suite: new features in the direct residue-to-bead method, improved grid routines, and influence of accessible surface area screening. Macromolecular Bioscience. 2010b;10 (7):746–753. doi: 10.1002/mabi.200900474. [DOI] [PubMed] [Google Scholar]
- Brookes E, Demeler B, Rosano C, Rocco M. The implementation of SOMO (SOlution MOdeller) in the UltraScan analytical ultracentrifugation data analysis suite: enhanced capabilities allow the reliable hydrodynamic modeling of virtually any kind of biomacromolecule. European Biophysics Journal. 2010c;39 (3):423–435. doi: 10.1007/s00249-009-0418-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, DeLaBarre B. NSF and p97/VCP: similar at first, different at last. FEBS Letters. 2003;555 (1):126–133. doi: 10.1016/s0014-5793(03)01107-4. [DOI] [PubMed] [Google Scholar]
- Clary DO, Griff IC, Rothman JE. SNAPs, a family of NSF attachment proteins involved in intracellular membrane fusion in animals and yeast. Cell. 1990;61 (4):709–721. doi: 10.1016/0092-8674(90)90482-t. [DOI] [PubMed] [Google Scholar]
- Dam J, Schuck P. Calculating sedimentation coefficient distributions by direct modeling of sedimentation velocity concentration profiles. Methods in Enzymology. 2004;384:185–212. doi: 10.1016/S0076-6879(04)84012-6. [DOI] [PubMed] [Google Scholar]
- Davies JM, Brunger AT, Weis WI. Improved structures of full-length p97, an AAA ATPase: implications for mechanisms of nucleotide-dependent conformational change. Structure/Folding and Design. 2008;16 (5):715–726. doi: 10.1016/j.str.2008.02.010. [DOI] [PubMed] [Google Scholar]
- Davies JM, Tsuruta H, May AP, Weis WI. Conformational changes of p97 during nucleotide hydrolysis determined by small-angle X-Ray scattering. Structure/Folding and Design. 2005;13 (2):183–195. doi: 10.1016/j.str.2004.11.014. [DOI] [PubMed] [Google Scholar]
- DeLaBarre B, Brunger AT. Complete structure of p97/valosin-containing protein reveals communication between nucleotide domains. Nature Structure and Molecular Biology. 2003;10 (10):856–863. doi: 10.1038/nsb972. [DOI] [PubMed] [Google Scholar]
- Furst J, Sutton RB, Chen J, Brunger AT, Grigorieff N. Electron cryomicroscopy structure of N-ethylmaleimide sensitive factor at 11 A resolution. The EMBO Journal. 2003;22 (17):4365–4374. doi: 10.1093/emboj/cdg420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson PI, Roth R, Morisaki H, Jahn R, Heuser JE. Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick-freeze/deep-etch electron microscopy. Cell. 1997;90 (3):523–535. doi: 10.1016/s0092-8674(00)80512-7. [DOI] [PubMed] [Google Scholar]
- Hanson PI, Whiteheart SW. AAA+ proteins: have engine, will work. Nature Review Molecular and Cellular Biology. 2005;6 (7):519–529. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- Hohl TM, Parlati F, Wimmer C, Rothman JE, Sollner TH, et al. Arrangement of subunits in 20 S particles consisting of NSF, SNAPs, and SNARE complexes. Molecular Cell. 1998;2 (5):539–548. doi: 10.1016/s1097-2765(00)80153-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander GC, Stagg SM, Voss NR, Cheng A, Fellmann D, et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. Journal of Structural Biology. 2009;166 (1):95–102. doi: 10.1016/j.jsb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Choi J, Tsai FT. Visualizing the ATPase cycle in a protein disaggregating machine: structural basis for substrate binding by ClpB. Molecular Cell. 2007;25 (2):261–271. doi: 10.1016/j.molcel.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenzen C, Steinmann D, Whiteheart S, Weis WI. Crystal structure of the hexamerization domain of N-ethylmaleimide-sensitive fusion protein. Cell. 1998;94 (4):525–536. doi: 10.1016/s0092-8674(00)81593-7. [DOI] [PubMed] [Google Scholar]
- Marz KE, Lauer JM, Hanson PI. Defining the SNARE complex binding surface of alpha-SNAP: implications for SNARE complex disassembly. The Journal of Biological Chemistry. 2003;278 (29):27000–27008. doi: 10.1074/jbc.M302003200. [DOI] [PubMed] [Google Scholar]
- Matveeva EA, He P, Whiteheart SW. N-Ethylmaleimide-sensitive fusion protein contains high and low affinity ATP-biding sites that are functionally distinct. The Journal of Biological Chemistry. 1997;272 (42):26413–26418. doi: 10.1074/jbc.272.42.26413. [DOI] [PubMed] [Google Scholar]
- May AP, Misura KM, Whiteheart SW, Weis WI. Crystal structure of the amino-terminal domain of N-ethylmaleimide-sensitive fusion protein. Nature Cell Biology. 1999;1 (3):175–182. doi: 10.1038/11097. [DOI] [PubMed] [Google Scholar]
- Mindell JA, Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. Journal of Structural Biology. 2003;142 (3):334–347. doi: 10.1016/s1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- Nagiec EE, Bernstein A, Whiteheart SW. Each domain of the N-ethylmaleimide-sensitive fusion protein contributes to its transport activity. Journal of Biological Chemistry. 1995;270 (49):29182–29188. doi: 10.1074/jbc.270.49.29182. [DOI] [PubMed] [Google Scholar]
- Ohi M, Li Y, Cheng Y, Walz T. Negative Staining and Image Classification - Powerful Tools in Modern Electron Microscopy. Biological Procedures Online. 2004;6:23–34. doi: 10.1251/bpo70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, et al. UCSF Chimera–a visualization system for exploratory research and analysis. Journal of Computational Chemistry. 2004;25 (13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Rai N, Nollmann M, Spotorno B, Tassara G, Byron O, et al. SOMO (SOlution MOdeler) differences between X-Ray- and NMR-derived bead models suggest a role for side chain flexibility in protein hydrodynamics. Structure. 2005;13 (5):723–734. doi: 10.1016/j.str.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Roseman AM. FindEM – a fast, efficient program for automatic selection of particles from electron micrographs. Journal of Structural Biology. 2004;145 (1–2):91–99. doi: 10.1016/j.jsb.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Rouiller I, DeLaBarre B, May AP, Weis WI, Brunger AT, et al. Conformational changes of the multifunction p97 AAA ATPase during its ATPase cycle. Nature Structure and Molecular Biololgy. 2002;9 (12):950–957. doi: 10.1038/nsb872. [DOI] [PubMed] [Google Scholar]
- Roy A, Kucukural A. I-TASSER: a unified platform for automated protein structure and function prediction. Nature Protocols. 2010;5 (4):725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH, Nunez-Ramirez R, Sorzano CO, Carazo JM, Marabini R. Image processing for electron microscopy single-particle analysis using XMIPP. Nature Protocols. 2008;3 (6):977–990. doi: 10.1038/nprot.2008.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SHW, Valle M, Carazo J-M. Fast maximum-likelihood refinement of electron microscopy images. Bioinformatics. 2005a;21(Suppl 2):ii243–ii244. doi: 10.1093/bioinformatics/bti1140. [DOI] [PubMed] [Google Scholar]
- Scheres SHW, Valle M, Nuñez R, Sorzano COS, Marabini R, et al. Maximum-likelihood multi-reference refinement for electron microscopy images. Journal of Molecular Biology. 2005b;348 (1):139–149. doi: 10.1016/j.jmb.2005.02.031. [DOI] [PubMed] [Google Scholar]
- schuck p. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophysical Journal. 2000;78 (3):1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorzano CO, Marabini R, Velazquez-Muriel J, Bilbao-Castro JR, Scheres SH, et al. Xmipp: a new generation of an open-source image processing package for electron microscopy. Journal of Structural Biology. 2004;148 (2):194–204. doi: 10.1016/j.jsb.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Sorzano COS, Bilbao-Castro JR, Shkolnisky Y, Alcorlo M, Melero R, et al. A clustering approach to multireference alignment of single-particle projections in electron microscopy. Journal of Structural Biology. 2010;171 (2):197–206. doi: 10.1016/j.jsb.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, et al. Automated molecular microscopy: the new Leginon system. Journal of Structural Biology. 2005;151 (1):41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Tagaya M, Wilson DW, Brunner M, Arango N, Rothman JE. Domain structure of an N-ethylmaleimide-sensitive fusion protein involved in vesicular transport. Journal of Biological Chemistry. 1993;268 (4):2662–2666. [PubMed] [Google Scholar]
- Tucker PA, Sallai L. The AAA+ superfamily – a myriad of motions. Current Opinion in Structural Biology. 2007;17 (6):641–652. doi: 10.1016/j.sbi.2007.09.012. [DOI] [PubMed] [Google Scholar]
- van Heel M, Harauz G, Orlova EV, Schmidt R, Schatz M. A new generation of the IMAGIC image processing system. Journal of Structural Biology. 1996;116 (1):17–24. doi: 10.1006/jsbi.1996.0004. [DOI] [PubMed] [Google Scholar]
- Whiteheart SW, Rossnagel K, Buhrow SA, Brunner M, Jaenicke R, et al. N-Ethylmaleimide-sensitive fusion protein: a trimeric ATPase whose hydrolysis of ATP is required for membrane fusion. J Cell Biology. 1994;126 (4):945–954. doi: 10.1083/jcb.126.4.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer C, Hohl TM, Hughes CA, Muller SA, Sollner TH, et al. Molecular mass, stoichiometry, and assembly of 20 S particles. Journal of Biological Chemistry. 2001;276 (31):29091–29097. doi: 10.1074/jbc.M011292200. [DOI] [PubMed] [Google Scholar]
- Yu RC, Hanson PI, Jahn R, Brunger AT. Structure of the ATP-dependent oligomerization domain of N-ethylmaleimide sensitive factor complexed with ATP. Nature Structure Biology. 1998;5 (9):803–811. doi: 10.1038/1843. [DOI] [PubMed] [Google Scholar]
- Yu RC, Jahn R, Brunger AT. NSF N-terminal domain crystal structure: models of NSF function. Molecular Cell. 1999;4 (1):97–107. doi: 10.1016/s1097-2765(00)80191-4. [DOI] [PubMed] [Google Scholar]
- Yu Z, Gonciarz M, Sundquist W, Hill C, Jensen GJ. Cryo-EM structure of dodecameric Vps4p and its 2: 1 complex with Vta1p. Journal of Molecular Biology. 2008;377 (2):364–377. doi: 10.1016/j.jmb.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 2007;69 (Suppl 8):108–117. doi: 10.1002/prot.21702. [DOI] [PubMed] [Google Scholar]
- Zhao C, Hellman LM, Zhan X, Bowman WS, Whiteheart SW, et al. Hexahistidine-tag-specific optical probes for analyses of proteins and their interactions. Analytical Biochemistry. 2010;399 (2):237–245. doi: 10.1016/j.ab.2009.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Matveeva EA, Ren Q, Whiteheart SW. Dissecting the N-ethylmaleimide- sensitive Factor: required elements of the N and D1 domains. Journal of Biological Chemistry. 2009;285 (1):761–772. doi: 10.1074/jbc.M109.056739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Smith EC, Whiteheart SW. Requirements for the catalytic cycle of the N-ethylmaleimide-Sensitive Factor (NSF) Biochimica et Biophysica Acta. 2011;1823 (1):159–171. doi: 10.1016/j.bbamcr.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.