

Abstract
In adenovirus E4 mutant infections, viral DNAs form concatemers through a process that requires host Non-homologous End Joining (NHEJ) proteins including DNA Ligase IV (LigIV). Adenovirus proteins E4 34k and E1b 55k form the substrate-selection component of an E3 ubiquitin ligase and prevent concatenation by targeting LigIV for proteasomal degradation. The mechanisms and sites involved in targeting this and other E3 ligase substrates generally are poorly-understood. Through genetic analysis, we identified the α2 helix of one LigIV BRCT domain (BRCT-1) as essential for adenovirus-mediated degradation. Replacement of the BRCT domain of DNA ligase III (LigIII), which is resistant to degradation, with LigIV BRCT-1 does not promote degradation. A humanized mouse LigIV that possesses a BRCT-1 α2 helix identical to the human protein, like its parent, is also resistant to adenovirus-mediated degradation. Thus, both the BRCT-1 α2 helix and an element outside BRCT-1 are required for adenovirus-mediated degradation of LigIV.
Keywords: Ubiquitin ligase, Adenovirus, DNA Ligase IV, Substrate selection, NHEJ
INTRODUCTION
Among the most common post-translational modifications employed by eukaryotic cells to regulate protein levels and activities is ubiquitination, covalent attachment of the 8kDa ubiquitin protein to a lysine residue in the ubiquitinated protein. Ubiquitination of a protein can alter its stability, localization and participation in protein-protein interactions.
Proteins can be conjugated to single ubiquitin moieties (monoubiquitination) or to ubiquitin chains (polyubiquitination). Within polyubiquitin chains, each ubiquitin is connected by its C-terminal glycine to one of the seven lysines (K6, K11, K27, K29, K33, K48, K63) in the preceeding ubiquitin. Proteins modified by polyubiquitin chains of four or more linked via K48 are degraded by the 26S proteasome. Targeted destruction of proteins by the ubiquitin/proteasome pathway is an essential process that is conserved among all eukaryotes (Woelk et al. 2007). Disruption of the ubiquitination/proteasome pathway can be lethal to individual cells or lead to protein buildup and diseases such as Alzheimer’s, Parkinson’s, and Huntington’s diseases (Ciechanover and Iwai 2004).
Modification of a target protein by ubiquitin occurs in three stages. First, ubiquitin is activated in an ATP-dependent manner through covalent attachment to one of two E1 activating enzymes, Ube1 (Uba1) (Haas et al. 1982) and Uba6 (Jin et al. 2007) in mammalian cells, by a thioester bond. Activated ubiquitin can then be transferred to any of at least 40 E2 ubiquitin conjugating enzymes [Pfam Pf00179 ubiquitin conjugation enzyme family (Finn et al. 2010)]. Finally, ubiquitin is transferred to its intended target by one of several hundred E3 ubiquitin ligases (Wei Li et al. 2008), which act as target selectors. There are two classes of E3s. Roughly 95% of human E3’s are within the RING family (Wei Li et al. 2008), which contain a RING (Really Interesting New Gene) finger subunit that binds the E2 (Petroski and Deshaies 2005). The ubiquitin linked to the E2 enzyme is transferred directly onto the substrate without any Ub-E3 intermediate (Deshaies and Joazeiro 2009). The remaining E3 complexes contain a HECT (Homologous to E6-AP C-Terminus) domain, and form a covalent intermediate with ubiquitin before transference to the target (Rotin and Kumar 2009).
The ubiquitination pathway is hijacked by many viruses to make the cellular environment more permissive for viral replication. Adenovirus type 5 (Ad5) (Harada et al. 2002), Herpesviruses (Epstein Barr Virus (Sato et al. 2009), murid Herpesvirus (Rodrigues et al. 2009), and Kaposi’s Sarcoma associated Herpesvirus (Cai et al. 2006)) and Human Immunodeficiency Virus (HIV) (Xianghui Yu et al. 2003), all exploit the ubiquitin pathway by redirecting Cullin 5-containing RING finger E3s to ubiquitinate novel targets that would otherwise interfere with virus propagation. In uninfected cells, Cullin 5 forms a complex with Elongin B, Elongin C, and Rbx1 to create a complete E3 ubiquitin ligase complex that targets Dab1 (Feng et al. 2007), ErbB2 (Ehrlich et al. 2009), Hif1-α (Ehrlich et al. 2009), Rbp1 (Harreman et al. 2009; Yasukawa et al. 2008) and Estrogen Receptor (ERα) (Johnson et al. 2006) for degradation. Upon Ad5 infection, two early proteins, E4 34k (product of E4 ORF 6) and E1b 55k, associate with the Cullin 5/Elongin B/Elongin C/Rbx1 complex to target novel proteins including p53 (E Querido et al. 1997), Mre11 (Stracker et al. 2002), DNA Ligase IV (LigIV) (Baker et al. 2007), BLM (Orazio et al. 2011), and integrin α3 (Dallaire et al. 2009) for ubiquitination and subsequent degradation. E4 34k binds to the E3 complex through its association with Elongin B/C by means of its three BC-box motifs (Blanchette et al. 2004; Ying Cheng et al. 2007). E1b 55k selects and binds the target protein and brings them to the E3 ligase through complex formation with E4 34k. Details of how these host proteins are recognized and targeted for degradation, however, remain unknown.
Three of the five proteins targeted for degradation by the Ad5 ubiquitin ligase are involved in the recognition and subsequent repair of DNA double-strand breaks through Non Homologous End Joining (NHEJ). Mre11 (Rupnik et al. 2009) and p53 (Sengupta and Harris 2005) are involved early in the DNA repair pathway and act as sensors of DNA damage, and LigIV is essential in the last joining step in the NHEJ pathway (Wilson et al. 1997; Critchlow et al. 1997). The presence of viral DNA in infected cells is sufficient to initiate a DNA damage response (Karen et al. 2009) which results in NHEJ mediated adenoviral genome ligation. Because the resultant concatamerized genomes are too large to be packaged into the viral capsid (Bett et al. 1993), the inhibition of NHEJ is essential for viral propagation. Viruses frequently have built-in redundancy for processes that are crucial for viral propagation, and the multiple routes by which adenoviruses inhibits the DNA damage response at both early and late steps clearly shows that inhibition of DNA repair is vital to adenovirus infection.
DNA Ligase IV (LigIV) is a member of the ATP-dependent DNA ligase family. Members of this family all contain a DNA binding domain (DBD), adenylation domain (AdD), and oligonucleotide binding domain (OBD), which together constitute 70% of each protein (Figure 1A) (Pascal et al. 2004; Wei et al. 1995; Cotner-Gohara et al. 2008). Other members of the DNA ligase family include DNA Ligase I, which has the crucial role during DNA replication of ligating together Okazaki fragments (Henderson et al. 1985; Barnes et al. 1992), and DNA Ligase III (LigIII) which has a role in nucleotide excision repair (NER) (Moser et al. 2007). Within the DNA ligase family, only LigIV is targeted for degradation by adenovirus. How LigIV is specifically recognized by adenovirus is unknown.
Figure 1. DNA Ligases.

A. Domains of the DNA ligase family proteins. DNA Ligases I, III, and IV constitute the mammalian DNA ligase family. All three members have a DNA binding domain (DBD), adenylation domain (AdD), and oligonucleotide binding domain (OBD). DNA Ligase III and IV both contain BRCA C-terminus (BRCT) domains at their C-terminus. Ligase III has a zinc finger (ZnF) domain and Ligase I has a nuclear localization signal (NLS) and proliferating cell nuclear antigen (PCNA) binding domain at the N-terminus. The amino acid position (Cole et al. 2008) of each domain is indicated above each diagram. B. LigIV truncation mutants. All mutants have an N-terminal myc tag and encompass the amino acids indicated in the name. The bars represent the conserved ATP-dependent ligase domains; ovals are BRCT domains. C. Degradation of LigIV truncation mutants by adenovirus. LigIV mutant plasmids were transfected into 293 cells which were subsequently either mock infected (−) or infected with H5dl1013 (+) for 20hrs. Cells were lysed, equal amounts of protein were loaded onto a polyacrylamide gel, and tagged LigIV was visualized by immunoblotting for the myc tag. The loading control is actin, and p53 is a control for adenovirus ubiquitin ligase function. U: Untransfected cells.
Our hypothesis is that a unique structure or amino acid sequence within LigIV, which is absent from the other DNA ligases, is recognized by adenoviral proteins E4 34k and E1b 55k and confers sensitivity to degradation in adenovirus infection. The purpose of this study was to identify these recognition elements. Through truncation analysis, a minimal targeting region sufficient for degradation of LigIV by adenovirus was identified. High resolution mapping through the use of site-directed mutagenesis identified an alpha helix within the first BRCT domain of LigIV (BRCT-1) that is critical for degradation and which is absent in other DNA Ligase family members, providing an explanation for LigIV specificity. This alpha helix, however, is not sufficient for targeted degradation, since neither its insertion into LigIII nor fusion of LigIV’s full-length BRCT-1 domain to LigIII targets the chimeric protein for degradation. Therefore, additional recognition elements exist in LigIV.
RESULTS
Localization of the targeting domain in LigIV
To identify regions in LigIV essential for recognition by the adenovirus ubiquitin ligase, a series of myc-tagged LigIV truncation mutants (Costantini et al. 2007) (Figure 1B) were examined for susceptibility to degradation during Ad5 infection. 293 cells transfected with each truncation mutant were mock infected or infected with H5dl1013 (Bridge and G Ketner 1989), an Ad5 mutant which is deleted for E4 ORFs 1–3 and overproduces E4 34k. Levels of the myc-tagged truncated LigIV proteins were determined by immunoblotting at 20 hours post infection (p.i) (Figure 1C). Actin served as a loading control, and degradation of endogenous p53 as a control for successful adenovirus infection. Truncated proteins containing amino acids 1-790 (myc-LigIV 1-790), 1-748 (myc- LigIV 1-748), and 600-911 (myc-LigIV 600-911) were all degraded as efficiently as full length LigIV. In contrast, the protein truncated at residue 643 (LigIV 1-643) was not degraded. These results indicate that the region 643-748 contains an element necessary for LigIV recognition by the adenovirus ubiquitin ligase complex. They also suggest that the larger region 600-748 contains elements sufficient for LigIV degradation.
Fine-structure mapping of recognition elements
BRCT domains, such as that contained within LigIV 643-748, frequently participate in protein-protein interactions (Rodriguez 2008) and therefore LigIV BRCT-1 is a likely site of interactions that mediate degradation by the adenovirus ubiquitin ligase. Mutation of amino acids that directly participate in interactions with the adenovirus ubiquitin ligase are predicted to interfere with LigIV degradation. Therefore, to define the elements that are essential for adenovirus-mediated degradation of LigIV, we examined degradation of a series of LigIV mutants with amino acid substitutions within the region 600-748.
To guide selection of mutation sites within BRCT-1, we took advantage of the observation that although human and canine LigIV proteins are very similar in amino acid sequence (Figure 2A), canine LigIV is not degraded in Ad5 infected MDCK cells (Figure 2B). Both canine and human Mre11 are efficiently degraded during Ad5 infection (Figure 2B), which indicates that the Ad5 E4 34k and E1b 55k proteins interact successfully with canine Cullin 5 and Elongins B and C to form a functional ubiquitin ligase. We therefore attribute the lack of canine LigIV degradation to differences between canine and human LigIV in essential elements that mediate recognition by the adenovirus ubiquitin ligase or ubiquitination sites. Within the BRCT-1 region, canine and human LigIV differ at four positions, which are indicated in bold in Figure 2A and shown as wire frames on the ribbon diagram of the human LigIV BRCT-1 domain in Figure 3A. Because the tops of BRCT domains tend to participate in protein-protein interactions (Rodriguez 2008) it is of interest that two of the amino acids altered in canine DNA LigIV (673 and 724) are within this region. Human LigIV Q673 also is part of the SQ consensus DNA PK phosphorylation site (Lees-Miller et al. 1990), and human LigIV is phosphorylated at S672 (Yu-Gang Wang et al. 2004). Phosphorylation of this residue could affect ubiquitination (Catic et al. 2004) or interaction with the adenovirus ubiquitin ligase. Canine LigIV contains H in the corresponding position, and is not likely to be phosphorylated there. The remaining two amino acid differences occur at residue 735, located at the opposite end of BRCT-1, and 714, which lies in the structurally conserved α2 helix. Mouse LigIV also contains substitutions at these amino acids (Figure 2A) and similarly is not degraded by adenovirus (data not shown).
Figure 2. LigIV homologues.

A. Alignment of the amino acid sequences of the BRCT-1 domains of human, canine, and murine DNA LigIV. Amino acids that diverge from the human sequence are indicated in bold. The four changes present in canine LigIV, also present in mouse and rat (not shown), are indicated by arrows with position in the human protein indicated beneath. B. Canine DNA LigIV is not degraded in adenovirus infection. Extracts of MDCK canine kidney cells infected with adenovirus were fractionated by SDS PAGE and immunoblotted for LigIV, Mre11, p53, and actin. Mre11 and p53 are efficiently degraded, indicating that the viral ubiquitin ligase functions in canine cells, but LigIV is not.
Figure 3. Site directed mutagenesis of LigIV BRCT-1.
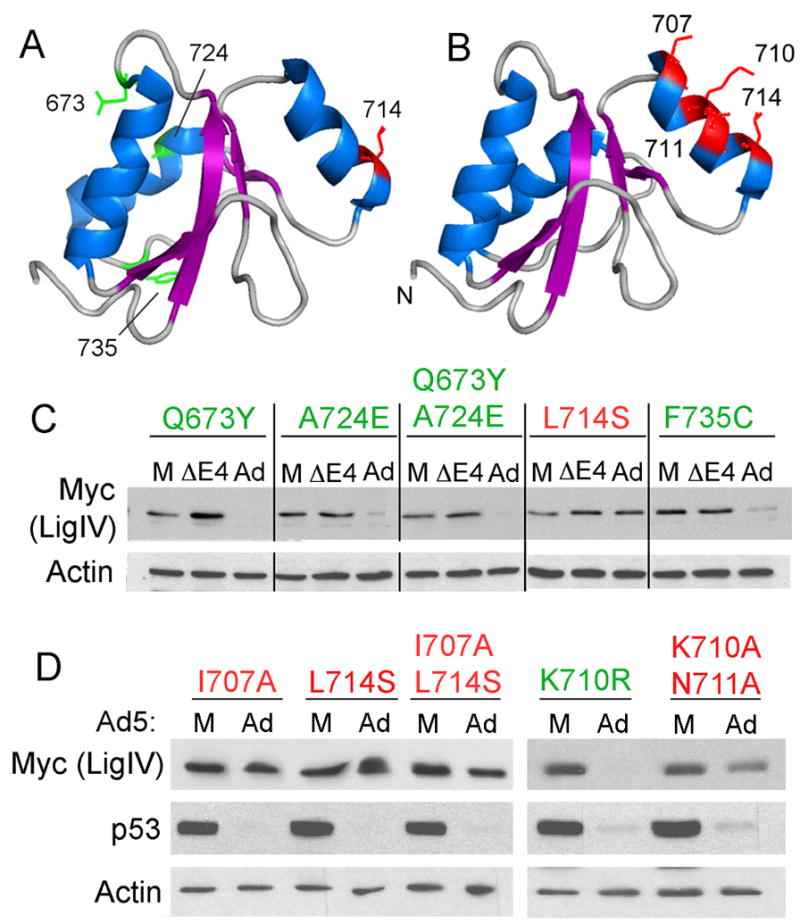
A. Structure of the BRCT-1 domain of human LigIV (PDB ID: 2E2W(Nagashima et al. 2006), rendered by Pymol) with the amino acids that differ from the canine sequence indicated in wire frame format. B. Structure of the BRCT-1 domain of LigIV with solvent-facing amino acids in the α2 helix indicated in wire frame format. C and D. 293 cells were transfected with myc-tagged LigIV 600–911-based expression plasmids containing amino acid substitutions at the indicated positions. Four hours after transfection, cells were either mock infected (M), infected with adenovirus (Ad), or infected with a deletion mutant adenovirus that does not express E4 genes (ΔE4). Cells were harvested 20hrs post infection, lysed, and 20μg soluble protein was run on a polyacrylamide gel that was immunoblotted for myc, actin (C and D), and p53 (D).
If any of these four amino acids are essential for LigIV targeted degradation, mutations of human myc-LigIV 600-911 that change the critical residue to that found in canine LigIV should prevent degradation by adenovirus. Expression plasmids bearing individual mutations (Q673H, L714S, A724E, and F735C) were introduced into 293 cells by transfection. Transfected cells subsequently were either mock infected or infected with H5dl1013 and mutant protein levels were assayed in extracts by immunoblotting with anti c-myc (Figure 3C). LigIV Q673H was not detected in transfected cells, and we therefore introduced the mouse 673Y mutation into myc-LigIV 600-911 to assess the role of residue 673 in targeted degradation. Three mutants (Q673Y, A724E, and F735C and the double mutant Q673Y/A724E) were degraded by adenovirus as efficiently as control LigIV 600-911. The fourth, L714S was not.
The α2 helix of BRCT-1 includes residue 714, suggesting that the α2 helix is an essential element in recognition of LigIV by the adenovirus ubiquitin ligase. To test this hypothesis, three additional residues in the a2 helix were mutated to alanine and tested for effects on degradation by adenovirus. As shown in Figure 3B, I707 and L714 mark the ends of a2 helix and both point directly outward into solution. Residues K710 and N711 also point into solution, but slightly to the side. Mutation of I707 individually or K710 and N711 as a pair was able to prevent degradation by adenovirus (Figure 3D). Therefore, the α2 helix is an essential element in targeting LigIV for degradation.
To explore the possibility that the K710A mutation prevented LigIV degradation by destroying a ubiquitination site, K710 was mutated to arginine to prevent ubiquitination but retain charge to minimize disruption of protein-protein interactions. This substitution had no effect on the ability of adenovirus to target the protein for degradation (Figure 3D). Thus, K710 is not an obligatory ubiquitination site for the adenovirus ubiquitin ligase. Mutagenesis has also been performed in the region 600-653, immediately upstream of BRCT-1. Using myc-LigIV 600-911 as a backbone as above, multiple-alanine substitution mutations were made in five regions predicted to be surface exposed by Jpred (Cole et al. 2008): 600–604, 607–609, 620–625, 625–629, and 644–645, constituting 55% of the predicted surface residues. None of these mutations prevented LigIV degradation in adenovirus-infected cells (Supplementary Figure S1), indicating that these 20 amino acids are not essential for recognition and degradation of LigIV.
In all, seven of the 16 lysines within LigIV 600–748 were individually mutated and none prevented degradation. If the ubiquitinations site lies within this region, it could be at any of the nine lysine residues that have yet to be tested. However, the site at which proteins are modified by ubiquitin can be promiscuous and deletion of one ubiquitination site can cause another lysine to become ubiquitinated in its place (Roth and Davis 2000; Petroski and Deshaies 2003).
LigIV BRCT-1 or the BRCT-1 α2 helix is not sufficient for adenovirus mediated degradation
DNA ligase III (LigIII) and LigIV are both members of the ATP-dependent DNA Ligase family of proteins and both contain the N-terminal DBD, AdD, and OBD domains as well as C-terminal BRCT domain(s) (Figure 1A), but only LigIV is targeted for degradation (Figure 1C). Since a fragment containing the first and second BRCT domains of LigIV (600–911) is efficiently degraded by adenovirus, replacement of LigIII BRCT-1 domain with that region from LigIV might promote targeting of LigIII for degradation. We addressed this hypothesis by testing such a DNA LigIII/IV chimeric protein for susceptibility to degradation by adenovirus infection. The site selected for fusion within LigIII, at amino acid 787, is located in a region predicted to be unstructured between the OBD (LigIII 590–771) and BRCT (LigIII 845–922) domains and where manipulations should not disrupt secondary or tertiary structure. Additionally, the region downstream of 787 shows some similarity to the corresponding region in LigIV (600–653; Supplementary Figure S2), further minimizing the chances of structural abnormalities in the fusion protein. To produce the chimera, LigIV DNA encoding amino acids 600–911 was PCR amplified with a primer that added LigIII 773–787 to the N-terminus, and the fusion DNA was inserted into LigIII at a unique endogenous restriction enzyme site within the LigIII segment. The chimera, LigIII/LigIV BRCT 1+2 (LigIII 1-787/LigIV 600–911), was transfected into 293 cells that were subsequently infected with adenovirus. LigIII was not degraded upon infection with adenovirus, but LigIII/LigIV BRCT-1+2 was degraded (Figure 4). These findings indicate that in the context of DNA LigIII, LigIV 600–911 remains sufficient to mediate recognition and degradation by the adenovirus ligase.
Figure 4. Degradation of Ligase III/Ligase IV fusions in adenovirus infection.
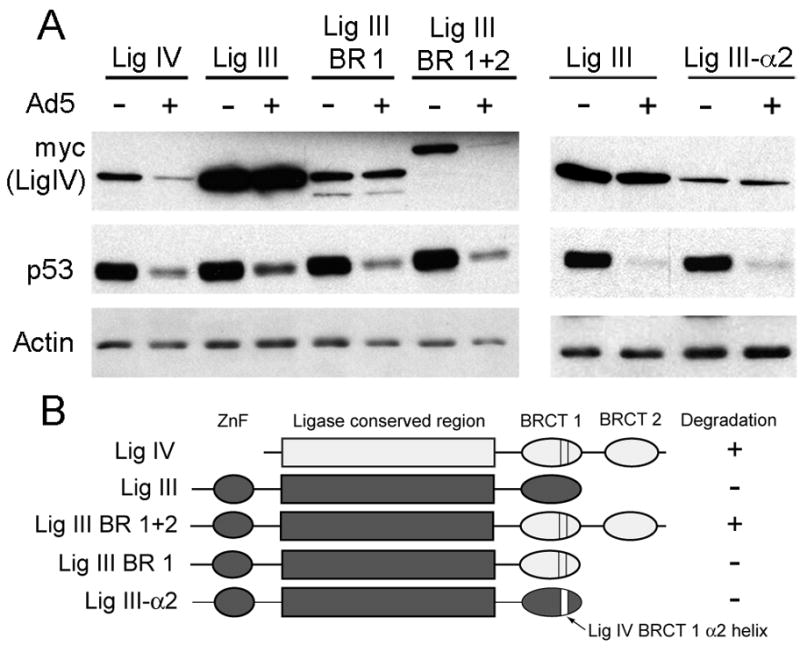
A. 293 cells were transfected with DNAs encoding wild type LigIV and LigIII, or the fusion proteins LigIII 1-787/LigIV 600–748 (BRCT-1), LigIII 1–787/LigIV 600–911 (BRCT-1+2), and LigIII/LigIV 703–720 (α2). Four hours after transfection, cells were either mock infected (−) or infected with adenovirus (+). Cells were harvested 20hrs post infection, lysed, and 20μg soluble protein was loaded onto a polyacrylamide gel that was immunoblotted for myc, actin, and p53. B. Diagrams of the fusion proteins. LigIV sequernces are colored white; LigIII sequences dark gray. Insertion of the LigIV BRCT1 α2 helix in LigIII-α2 is indicated by the white bar. ZnF: LigIII zinc finger region.
We next wanted to determine whether either the LigIV α2 helix or LigIV BRCT-1 alone is sufficient to target LigIII for degradation. To test that hypothesis we inserted the LigIV α2 helix into LigIII in place of the short loop that occupies the corresponding position in LigIII. After comparison of the crystal structures of LigIII BRCT and LigIV BRCT-1 and identification of equivalent sites and unique restriction endonuclease sites surrounding the loop and α2 helix, LigIV 703–720 was inserted between PmlI and BspEI sites within LigIII to replace amino acids 893 to 904. To address the possibility that the BRCT-1 domain is sufficient, a LigIII 1–787/LigIV 600–748 chimera was prepared essentially as described above for LigIII 1–787/LigIV 600–911.
293 cells were transfected with LigIII/LigIV α2 (LigIII1–892/LigIV 703–720/LigIII 904–922) or LigIII/LigIV BRCT-1 (LigIII 1–787/LigIV 600–748) and subsequently infected with adenovirus. In neither case was the chimeric protein a target for adenovirus-mediated degradation (Figure 4B). This suggests that neither the α2 helix nor BRCT-1 alone is sufficient to mediate a productive interaction with the adenovirus ubiquitin ligase. However, it is unknown if in these chimeric proteins whether the α2 helix in LigIV BRCT-1 is exposed. When BRCT domains are present in pairs, as in native LigIV or LigIII 1–787/LigIV 600–911, they usually form a dimer. In the LigIII fusion proteins, LigIV BRCT-1 is present alone and cannot dimerize and thus could be oriented differently than in the presence of LigIV BRCT2. Moreover, the possibility that steric hindrance from some region of LigIII prevents exposure of α2 helix cannot be ruled out.
Canine and mouse LigIV are not targeted for degradation by adenovirus (Figures 2B and 5). Within the α2 helix, canine and mouse LigIV present a serine at position 714 in place of the human LigIV leucine (Figure 2A). Mutation of human LigIV L714 to the canine/mouse serine to create L714S prevented degradation by adenovirus (Figure 3D). These observations raise the possibility that the human α2 helix in the context of a mouse or canine LigIV protein might confer sensitivity to the adenovirus E3 ligase. To test this hypothesis, the BRCT-1 α2 helix of mouse LigIV was humanized by mutating S714 to leucine through site directed mutagenesis. 293 cells were transfected with mouse wild type or mutant LigIV (mLigIVS714L) and subsequently infected with adenovirus. Neither wild-type nor mutant mouse LigIV was targeted for degradation (Figure 5). Presence of a human α2 helix therefore was not sufficient to target mouse LigIV for degradation.
Figure 5. The LigIVα2 helix is not sufficient for targeted degradation by adenovirus.
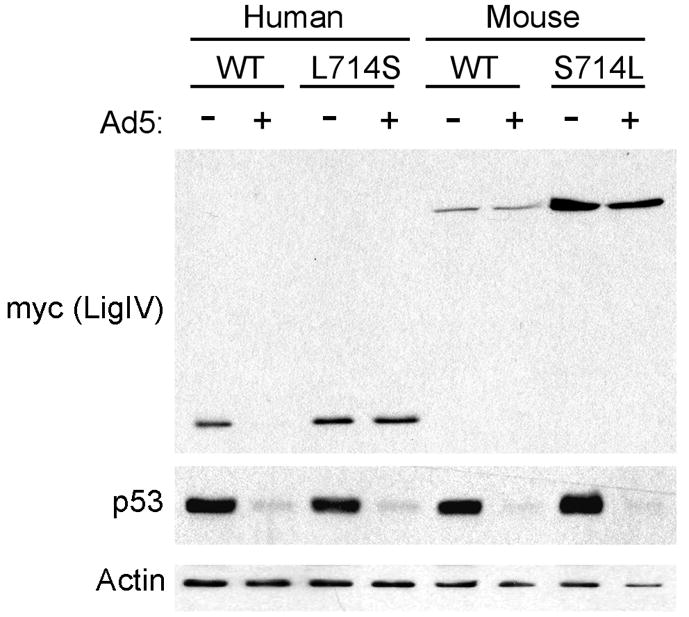
Human LigIV 600–911 was mutated at residue 714 to the mouse amino acid (L714S), and full-length mouse LigIV was mutated at residue 714 to the human amino acid (S714L). 293 cells were transfected with wild type (WT) human (600–911) or mouse (1–911) LigIV and the 714 plasmids. Four hours after transfection, cells were either mock infected (−) or infected with adenovirus (+) for 2hrs. Cells were harvested 20hrs post infection, lysed, and 20μg soluble protein was run on a polyacrylamide gel that was immunoblotted with anti-myc, anti-p53, and anti-actin.
Taken together, these observations indicate that the DNA LigIV α2 helix contains elements essential for adenovirus-mediated degradation, but that sequences outside that region are also required.
DISCUSSION
Adenovirus type 5 inhibits the Non-Homologous End Joining (NHEJ) DNA repair pathway through targeted degradation of DNA Ligase IV (LigIV) (Baker et al. 2007). Viral proteins E4 34k and E1b 55k capture the host Cullin 5 E3 ubiquitin ligase complex to target LigIV as well as p53 (E Querido et al. 1997), Mre11 (Stracker et al. 2002), BLM (Orazio et al. 2011), and integrin α3 (Dallaire et al. 2009) for ubiquitination and subsequent proteasome-mediated degradation. The structural and sequence elements within these proteins required for recognition and subsequent degradation by adenovirus is unknown, aside from p53 (Roth et al 1998). In this study, human LigIV was examined to identify the determinants for targeted degradation and a region that is essential but not sufficient for adenovirus-targeted degradation was identified.
LigIV is a member of a family of ATP-dependent DNA ligases, but only LigIV is targeted for degradation by adenovirus. All three DNA ligase family members contain recognizable DNA binding domain (DBD), adenylation domain (AdD), and oligonucleotide binding domain (OBD) motifs (Figure 1). However, amino acid sequence conservation among family members is poor: alignment of the primary amino acid sequences of DNA ligases I, III and IV revealed that these proteins share only 23% overall sequence identity. Thus, protein sequence does not provide clues regarding the elements that determine the unique susceptibility of DNA ligase IV to the adenovirus ubiquitin ligase.
To localize the essential targeting elements within the 911aa LigIV protein we utilized a series of previously described truncation mutants (Costantini et al. 2007). The shortest truncated protien (600–911aa) and the N terminal fragment 1–748 were both targeted for degradation, suggesting that elements sufficient for LigIV degradation lie in the region between residues 600–748 (Figure 1C). The first BRCT domain of LigIV lies between amino acids 654 and 748, and our data therefore suggested the hypothesis that BRCT-1 contains elements necessary for adenovirus-targeted degradation. BRCT repeats are composed of 4 central beta-sheets surrounded by 3 alpha-helices. Mutation of solvent exposed amino acids within the second alpha helix (α2) of BRCT-1 prevented degradation by adenovirus (Figure 3C and D) and confirmed the importance of the α2 helix in targeted degradation of LigIV by adenovirus. The BRCT-1 α2 helix of LigIV is a unique feature within the ATP-dependent DNA ligase family. DNA ligase I lacks BRCT domains entirely and the single BRCT domain of ligase III (LigIII) contains only the first and third α-helices, with a short loop where LigIV has the α2 helix (Krishnan et al. 2001). The fact that LigIV alone possesses an α2 helix explains the mechanism by which LigIV is the only DNA ligase in human cells to be targeted for adenovirus-mediated degradation.
This α2 helix however is not sufficient to target LigIV for degradation, since insertion of α2 into LigIII’s BRCT domain and fusion of LigIV BRCT-1 to LigIII were both unable to promote degradation of LigIII (Figure 4). These data suggest that LigIV sequences that lie outside of the BRCT1 α2 helix are required for targeted degradation. Since targeted degradation occurs when the LigIV BRCT-1 domain is accompanied by either LigIV C-terminal sequences 749–911 or LigIV N-terminal sequences 1–600, each of those segments individually must contain features that satisfy the second requirement. Alternatively, the LigIV BRCT-1 α2 helix may be sufficient to direct adenovirus targeted degradation, but in LigIII/LigIV fusion proteins with short LigIV segments the critical α2 helix might be inaccessible due to misfolding of the chimeric protein and thus unavailable for directing targeted degradation.
These competing interpretations of our data represent two mutually exclusive hypotheses: 1) the LigIV BRCT-1 α2 helix is one of at least two determinants for targeted degradation, and 2) the LigIV BRCT-1 α2 helix is sufficient to target a polypeptide for degradation, provided it is accessible. To distinguish between these hypotheses, we used murine LigIV to test the functionality of the LigIV BRCT-1 α2 helix in the context of a closely-related but degradation-resistant LigIV protein.
Human and murine LigIV share 92% similarity, and within the α2 helix, they differ only at the amino acid corresponding to human LigIV L714, where the murine sequence contains serine, a substitution shown above to prevent adenovirus-induced degradation. We created a murine LigIV S714L substitution mutant (mLigIVS714L), which has a BRCT-1 α2 helix that is identical to the human α2 helix and, significantly, is in the context of a native LigIV polypeptide. Folding of mLigIVS714L is expected to preserve the overall LigIV structure and, presumably, accessibility of the BRCT-1 α2 helix. mLigIVS714L is resistant to degradation during adenovirus infection (Figure 5). These data clearly demonstrate that while the α2 helix of LigIV BRCT-1 is one contributing determinant of adenovirus-targeted LigIV degradation, the α2 helix alone is not sufficient to mediate targeted degradation. Examination of the ~60 remaining residues that are dissimilar between human and murine LigIV may identify additional determinants for adenovirus-targeted LigIV degradation.
How might the BRCT1 domain of human LigIV act as a determinant for E4 34k/E1b55k dependent polyubiquitination? BRCT repeats have been characterized as phosphoserine/threonine (pS/T) binding domains (Xiaochun Yu et al. 2003; Williams et al. 2005) where the TxK motif in the α2 helix and an SG motif directly after the first beta strand (Clapperton et al. 2004; Rodriguez 2008) mediate binding to a pS/TxxΩ (Ω: hydrophobic) motif in the binding partner. While human LigIV BRCT-1 contains only the SG motif, it has been shown to bind to a generic phosphoserine peptide (Xiaochun Yu et al. 2003) and thus LigIV BRCT-1 may participate in phosphorylation-dependent interactions. Phosphorylation of both E4 34K and E1b 55K has been observed in vivo (Boivin et al. 1999), (Takayesu et al. 1994) and both viral proteins independently bind LigIV (Baker et al. 2007; Jayaram et al. 2008). Only E4 34K, though, contains consensus BRCT binding sites (68–71 SVGF, 205–8 SFGY), and the actual sites of E4 34k phosphorylation have yet to be determined. Nevertheless, these data suggest the testable hypothesis that adenovirus-targeted degradation of LigIV is mediated by interaction of phosphorylated adenoviral proteins with the BRCT repeats of human LigIV.
LigIV is part of a multiprotein complex and LigIV-binding proteins may play a role in LigIV targeted degradation. The best-known LigIV binding partner is XRCC4, which is required for the stability and catalytic activity of LigIV in vitro and in vivo (Critchlow et al. 1997; Grawunder et al. 1997). Because LigIV 1–748 lacks the XRCC4-interaction domain of LigIV (767–783 (Grawunder et al. 1998)) and is effectively degraded during adenovirus infection (Figure 1C), we conclude that the LigIV/XRCC4 interaction is not required for targeted degradation of human LigIV during adenovirus infection. LigIV’s BRCT-2 α2 helix does not contact XRCC4 (Wu et al. 2009), but instead faces into solution, and is probably accessible for protein-protein interactions. Because LigIV can either bind XRCC4 or E4-34k, but not both (Jayaram et al. 2008) it is likely that the XRCC4/LigIV complex is disassembled before LigIV polyubiquitination. Whether disruption of the LigIV/XRCC4 complex is a requirement for or a consequence of binding of viral proteins to LigIV is currently unknown.
It is likely that yet unknown host proteins are targeted by the adenovirus ubiquitin ligase. The identification of the α2 helix as required for the recognition of LigIV by the ubiquitin ligase suggested that presence of this element might be useful in identifying other adenovirus ubiquitin ligase targets. Therefore, we examined the list of proteins believed to contain one or more BRCT repeats to determine if other LigIV BRCT-1 α2-like helices might be found. We screened BRCT containing proteins found in the Pfam database (Finn et al. 2010) (PF00533) for an 8 amino acid long region roughly where the α2 helix should be that contains terminal hydrophobic residues, analogous to LigIV amino acids I707 and L714, and a centrally located charged residue analogous to K710. This search returned 30 candidate degradation targets. Where published structural information was available we manually examined the crystal structures for candidate targets to confirm the 8aa region was actually within an α2 helix. For evaluation of candidate targets lacking published structural information we used Jpred to predict the secondary structure (Cole et al. 2008) of candidate BRCT repeats and manually examined the output for a predicted alpha helix overlapping the entire candidate 8aa region. This search yielded a single high probability candidate for adenovirus-targeted degradation: topoisomerase-II binding protein 1 (TopBP1).
TopBP1 contains nine BRCT repeats, which are distributed throughout the length of the protein (Yamane et al. 1997; Rappas et al. 2011) and it is the second of these repeats that contains a LigIV α2like helix that matches our criteria for adenovirus targeted degradation. Recent work by Blackford showed that TopBP1 is not degraded during Ad5 infection, but is a target for degradation by E4 34k and Cullin 2 in an E1b 55k-independent mechanism employed by adenovirus type 12 (Ad12) (Blackford et al. 2010). Ad12 E4 34k bound TopBP1, which is similar to the observed binding of LigIV by Ad5 E4 34k (Jayaram et al. 2008) and while these observations suggest similar mechanisms for E4 34k-target recognition more needs to be done before a bona fide E4 34k-binding motif can be considered.
Is there a protein sequence or structural element common to all adenovirus degradation targets that distinguishes them from other host proteins? Among identified adenovirus ubiquitin ligase targets, only LigIV contains a BRCT repeat or a BRCT-1 α2 helix-like sequence, which indicates that other determinants of degradation must exist. We have discussed the data indicating that the N-terminal and C-terminal domains of LigIV contain one or more elements necessary for adenovirus-targeted degradation.
To date, work similar to that conducted here to determine the specific requirements for degradation in adenovirus targets has only been conducted on one other protein: p53. The region from amino acids 24–28 in p53 was found to be sufficient to promote degradation of the p53 homologue p73 (Roth et al 1998). This region shares nothing obvious with the LigIV α2 helix. Each of the adenovirus target proteins need share only one requirement for degradation: binding to the target selector protein E1b 55k. This consensus motif would therefore likely be an E1b 55k interaction domain. There is evidence, however, that Mre11, LigIV, and p53 may interact with E1b 55k in differing manners (Schwartz et al. 2008). Identification of the remaining determinants for LigIV degradation, as well as determinants for Mre11 and BLM may help us to develop a consensus sequence motif or set of structural requirements that identify potential targets for adenoviral degradation.
Adenovirus specifically promotes degradation LigIV, the only member of the DNA Ligase family that interferes with viral propagation, and does so without affecting other DNA ligases. This specificity is conferred, in part, by the LigIV BRCT-1 α2 helix, which is unique to LigIV. The lack of a LigIV BRCT-1 α2-like helix in other adenovirus degradation targets and the observation that LigIV contains additional elements required for degradation indicates that multiple mechanisms exist for viral protein selection of degradation targets during adenovirus infection.
MATERIALS AND METHODS
Cell culture
293 (FL Graham et al. 1977) and Madin-Darby canine kidney (MDCK) cells were propagated in in Eagle’s minimal essential media (Lonza) supplemented with 10% fetal bovine serum (Gibco), penicillin (50 units/ml), and streptomycin (50mg/ml).
Transfection, infection, and immunoblots
In a six-well plate, 293 cells were plated at 6x105 cells per well, allowed to grow overnight, then transfected with 2mg of each DNA Ligase IV DNA, E1b 55k DNA (where appropriate) and Lipofectamine 2000 for four hours. For cells that were subsequently infected with adenovirus, media was replaced with 1ml fresh serum-free Eagle’s Minimum Essential Medium (EMEM), and the cells were infected with adenovirus at an MOI of 20 plaque-forming units (PFU) for 2hrs. Media was replaced with complete EMEM + 10% FBS, and cells were harvested 24hrs after the beginning of transfection.
For immunoprecipitations, cells were harvested by scraping into phosphate buffered saline (PBS) twenty four hours after beginning transfection, and lysed in 200μL NET buffer (50mM Tris 8.0, 150mM NaCl, 10% Glycerol, 1mM DTT, 2mM EDTA, 0.1% Nonidet P40 (Sigma N3516)). One hundred μL of lysate was incubated with 200mL 2A6 (E1b 55k) hybridoma supernatant overnight at 4°C. The next day, 50μL of a 50% slurry of protein A Sepharose was added, and rocked for 2hrs at 4°C. The beads were washed three times for 15 minutes in 1mL NET buffer at room temperature. Protein sample buffer was added to the beads to liberate the immunoprecipitated proteins, then the supernatant was loaded onto an 8% SDS polyacrylamide gel. 7% of the soluble lysate used for each immunoprecipitation was loaded onto a polyacrylamide gel as the load fraction.
Mouse monoclonal antibodies used in immunoblots were the following: Actin (1:400, Calbiochem), p53 (1:1000, Ab-6, Abcam), 9E10 Myc hybridoma (Evan et al. 1985) supernatant (undiluted), 2A6 E1b 55k hybridoma (Sarnow et al. 1982) supernatant (undiluted).
Site directed mutagenesis and chimeric genes
Mutagenesis of pCR2.1-myc-LigIV-600–911 was carried out either by the Sigma QuikChange Site-Directed Mutagenesis method as recommended in the manual, or by the general megaprimer method. Oligonucleotides are listed in Supplementary Table 1. Mutant clones were sequenced and inserted into the pCI vector between NheI and NotI sites for expression.
LigIII/LigIV chimeric genes were produced by domain swaps as described in the Supplementary Methods.
Supplementary Material
Acknowledgments
The authors thank Cynthia Wolberger and Kasey Karen for thoughtful comments on the work
FUNDING:
This work was supported by the National Institutes of Health [Grant numbers 1R01AI079132, T32CA009110]
Footnotes
Supplementary Data are available at NAR online: Supplementary methods, Supplementary Table 1, Supplementary figures 1 and 2.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baker A, Rohleder KJ, Hanakahi LA, Gary Ketner. Adenovirus E4 34k and E1b 55k Oncoproteins Target Host DNA Ligase IV for Proteasomal Degradation. J Virol. 2007;81:7034–7040. doi: 10.1128/JVI.00029-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes DE, Tomkinson AE, Lehmann Alan R, Webster ADB, Lindahl T. Mutations in the DNA ligase I gene of an individual with immunodeficiencies and cellular hypersensitivity to DNA-damaging agents. Cell. 1992;69:495–503. doi: 10.1016/0092-8674(92)90450-q. [DOI] [PubMed] [Google Scholar]
- Bett AJ, Prevec L, Graham FL. Packaging capacity and stability of human adenovirus type 5 vectors. J Virol. 1993;67:5911–5921. doi: 10.1128/jvi.67.10.5911-5921.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackford AN, Patel RN, Forrester NA, Theil K, Groitl P, Stewart GS, Taylor AMR, Morgan IM, Dobner T, Grand RJA, et al. Adenovirus 12 E4orf6 inhibits ATR activation by promoting TOPBP1 degradation. Proc Natl Acad Sci USA. 2010;107:12251–12256. doi: 10.1073/pnas.0914605107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette P, Cheng CY, Yan Q, Gary Ketner, Ornelles DA, Dobner T, Conaway RC, Weliky Conaway Joan, Branton Philip E. Both BC-Box Motifs of Adenovirus Protein E4orf6 Are Required To Efficiently Assemble an E3 Ligase Complex That Degrades p53. Mol Cell Biol. 2004;24:9619–9629. doi: 10.1128/MCB.24.21.9619-9629.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boivin D, Morrison MR, Marcellus Richard C, Emmanuelle Querido, Branton Philip E. Analysis of Synthesis, Stability, Phosphorylation, and Interacting Polypeptides of the 34-Kilodalton Product of Open Reading Frame 6 of the Early Region 4 Protein of Human Adenovirus Type 5. J Virol. 1999;73:1245–1253. doi: 10.1128/jvi.73.2.1245-1253.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridge E, Ketner G. Redundant control of adenovirus late gene expression by early region 4. J Virol. 1989;63:631–638. doi: 10.1128/jvi.63.2.631-638.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q-L, Knight JS, Verma SC, Zald P, Robertson ES. EC5S Ubiquitin Complex Is Recruited by KSHV Latent Antigen LANA for Degradation of the VHL and p53 Tumor Suppressors. PLoS Pathog. 2006;2:e116. doi: 10.1371/journal.ppat.0020116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catic A, Collins C, Church GM, Ploegh HL. Preferred in vivo ubiquitination sites. Bioinformatics. 2004;20:3302–3307. doi: 10.1093/bioinformatics/bth407. [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Iwai K. The Ubiquitin System: From Basic Mechanisms to the Patient Bed. IUBMB Life. 2004;56:193–201. doi: 10.1080/1521654042000223616. [DOI] [PubMed] [Google Scholar]
- Clapperton JA, Manke IA, Lowery DM, Ho T, Haire LF, Yaffe MB, Smerdon SJ. Structure and mechanism of BRCA1 BRCT domain recognition of phosphorylated BACH1 with implications for cancer. Nat Struct Mol Biol. 2004;11:512–518. doi: 10.1038/nsmb775. [DOI] [PubMed] [Google Scholar]
- Cole C, Barber JD, Barton GJ. The Jpred 3 secondary structure prediction server. Nucl Acids Res. 2008;36:W197–201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini S, Woodbine L, Andreoli L, Jeggo PA, Vindigni A. Interaction of the Ku heterodimer with the DNA ligase IV/Xrcc4 complex and its regulation by DNA-PK. DNA Repair. 2007;6:712–722. doi: 10.1016/j.dnarep.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Cotner-Gohara E, Kim I-K, Tomkinson AE, Ellenberger T. Two DNA-binding and Nick Recognition Modules in Human DNA Ligase III. J of Biol Chem. 2008;283:10764–10772. doi: 10.1074/jbc.M708175200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Critchlow SE, Bowater RP, Jackson SP. Mammalian DNA double-strand break repair protein XRCC4 interacts with DNA ligase IV. Cur Biol. 1997;7:588–598. doi: 10.1016/s0960-9822(06)00258-2. [DOI] [PubMed] [Google Scholar]
- Dallaire F, Blanchette P, Groitl P, Dobner T, Branton Philip E. Identification of Integrin {alpha}3 as a New Substrate of the Adenovirus E4orf6/E1B 55-Kilodalton E3 Ubiquitin Ligase Complex. J Virol. 2009;83:5329–5338. doi: 10.1128/JVI.00089-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies RJ, Joazeiro CAP. RING Domain E3 Ubiquitin Ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- Ehrlich ES, Wang T, Luo K, Xiao Z, Niewiadomska AM, Martinez T, Xu W, Neckers L, Yu X-F. Regulation of Hsp90 client proteins by a Cullin5-RING E3 ubiquitin ligase. Proc Natl Acad Sci U S A. 2009;106:20330–20335. doi: 10.1073/pnas.0810571106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evan GI, Lewis GK, Ramsay G, Bishop JM. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol Cell Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Allen NS, Simo S, Cooper JA. Cullin 5 regulates Dab1 protein levels and neuron positioning during cortical development. Genes & Dev. 2007;21:2717–2730. doi: 10.1101/gad.1604207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, et al. The Pfam protein families database. Nucl Acids Res. 2010;38:D211–222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham F, Smiley J, Russell W, Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol. 1977;36:59–74. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- Grawunder U, Wilm M, Wu X, Kulesza P, Wilson TE, Mann M, Lieber MR. Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature. 1997;388:492–495. doi: 10.1038/41358. [DOI] [PubMed] [Google Scholar]
- Grawunder U, Zimmer D, Lieber MR. DNA ligase IV binds to XRCC4 via a motif located between rather than within its BRCT domains. Cur Biol. 1998;8:873–879. doi: 10.1016/s0960-9822(07)00349-1. [DOI] [PubMed] [Google Scholar]
- Haas AL, Warms JV, Hershko A, Rose IA. Ubiquitin-activating enzyme. Mechanism and role in protein-ubiquitin conjugation. J Biol Chem. 1982;257:2543–2548. [PubMed] [Google Scholar]
- Harada JN, Anna Shevchenko, Andrej Shevchenko, Pallas DC, Berk AJ. Analysis of the Adenovirus E1B-55K-Anchored Proteome Reveals Its Link to Ubiquitination Machinery. J Virol. 2002;76:9194–9206. doi: 10.1128/JVI.76.18.9194-9206.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harreman M, Taschner M, Sigurdsson S, Anindya R, Reid J, Somesh B, Kong SE, Banks CAS, Conaway RC, Conaway Joan W, et al. Distinct ubiquitin ligases act sequentially for RNA polymerase II polyubiquitylation. Proc Natl Acad Sci USA. 2009;106:20705–20710. doi: 10.1073/pnas.0907052106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson LM, Arlett CF, Harcourt SA, Lehmann AR, Broughton BC. Cells from an immunodeficient patient (46BR) with a defect in DNA ligation are hypomutable but hypersensitive to the induction of sister chromatid exchanges. Proc Natl Acad Sci USA. 1985;82:2044–2048. doi: 10.1073/pnas.82.7.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaram S, Gilson T, Ehrlich ES, Yu X-F, Gary Ketner, Hanakahi L. E1B 55k-independent dissociation of the DNA ligase IV/XRCC4 complex by E4 34k during adenovirus infection. Virology. 2008;382:163–170. doi: 10.1016/j.virol.2008.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Li X, Gygi SP, Harper JW. Dual E1 activation systems for ubiquitin differentially regulate E2 enzyme charging. Nature. 2007;447:1135–1138. doi: 10.1038/nature05902. [DOI] [PubMed] [Google Scholar]
- Johnson AE, Le IP, Buchwalter A, Burnatowska-Hledin MA. Estrogen-dependent growth and estrogen receptor (ER)-α concentration in T47D breast cancer cells are inhibited by VACM-1, a cul 5 gene. Mol Cell Biochem. 2006;301:13–20. doi: 10.1007/s11010-006-9392-3. [DOI] [PubMed] [Google Scholar]
- Karen KA, Hoey PJ, Young CSH, Hearing P. Temporal Regulation of the Mre11-Rad50-Nbs1 Complex during Adenovirus Infection. J Virol. 2009;83:4565–4573. doi: 10.1128/JVI.00042-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan VV, Thornton KH, Thelen MP, Cosman M. Solution Structure and Backbone Dynamics of the Human DNA Ligase IIIα BRCT Domain. Biochemistry. 2001;40:13158–13166. doi: 10.1021/bi010979g. [DOI] [PubMed] [Google Scholar]
- Lees-Miller SP, Chen YR, Anderson CW. Human cells contain a DNA-activated protein kinase that phosphorylates simian virus 40 T antigen, mouse p53, and the human Ku autoantigen. Mol Cell Biol. 1990;10:6472–6481. doi: 10.1128/mcb.10.12.6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Bengtson MH, Ulbrich A, Matsuda A, Reddy VA, Orth A, Chanda SK, Batalov S, Joazeiro CAP. Genome-Wide and Functional Annotation of Human E3 Ubiquitin Ligases Identifies MULAN, a Mitochondrial E3 that Regulates the Organelle’s Dynamics and Signaling. PLoS ONE. 2008;3:e1487. doi: 10.1371/journal.pone.0001487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser J, Kool H, Giakzidis I, Keith Caldecott, Mullenders LHF, Fousteri MI. Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase III alpha in a cell-cycle-specific manner. Mol Cell. 2007;27:311–323. doi: 10.1016/j.molcel.2007.06.014. [DOI] [PubMed] [Google Scholar]
- Nagashima T, Hayashi F, Yokoyama S. Solution structure of the first BRCT domain of human DNA ligase IV. RIKEN Structural Genomics Initiative (RSGI) 2006 [Google Scholar]
- Orazio NI, Naeger CM, Karlseder J, Weitzman MD. The Adenovirus E1b55K/E4orf6 Complex Induces Degradation of the Bloom Helicase during Infection. J Virol. 2011;85:1887–1892. doi: 10.1128/JVI.02134-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascal JM, O’Brien PJ, Tomkinson AE, Ellenberger T. Human DNA ligase I completely encircles and partially unwinds nicked DNA. Nature. 2004;432:473–478. doi: 10.1038/nature03082. [DOI] [PubMed] [Google Scholar]
- Petroski MD, Deshaies RJ. Context of Multiubiquitin Chain Attachment Influences the Rate of Sic1 Degradation. Mol Cell. 2003;11:1435–1444. doi: 10.1016/s1097-2765(03)00221-1. [DOI] [PubMed] [Google Scholar]
- Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- Querido E, Marcellus RC, Lai A, Charbonneau R, Teodoro JG, Ketner G, Branton PE. Regulation of p53 levels by the E1B 55-kilodalton protein and E4orf6 in adenovirus-infected cells. J Virol. 1997;71:3788–3798. doi: 10.1128/jvi.71.5.3788-3798.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappas M, Oliver AW, Pearl LH. Structure and function of the Rad9-binding region of the DNA-damage checkpoint adaptor TopBP1. Nucl Acids Res. 2011;39:313–324. doi: 10.1093/nar/gkq743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues L, Filipe J, Seldon MP, Fonseca L, Anrather J, Soares MP, Simas JP. Termination of NF-[kappa]B activity through a gammaherpesvirus protein that assembles an EC5S ubiquitin-ligase. EMBO J. 2009;28:1283–1295. doi: 10.1038/emboj.2009.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez MC. BRCT Domains, phosphopeptide binding and signaling modules. Front Biosci. 2008;13:5905. doi: 10.2741/3125. [DOI] [PubMed] [Google Scholar]
- Roth J, Konig C, Wienzek S, Weigel S, Ristea S, Dobbelstein M. Inactivation of p53 but not p73 by Adenovirus type 5 E1B 55-Kilodalton and E4 34-Kilodalton Oncoproteins. J Virol. 1998;71:8510–8516. doi: 10.1128/jvi.72.11.8510-8516.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth AF, Davis NG. Ubiquitination of the PEST-like Endocytosis Signal of the Yeast a-Factor Receptor. J Biol Chem. 2000;275:8143–8153. doi: 10.1074/jbc.275.11.8143. [DOI] [PubMed] [Google Scholar]
- Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol. 2009;10:398–409. doi: 10.1038/nrm2690. [DOI] [PubMed] [Google Scholar]
- Rupnik A, Lowndes NF, Grenon M. MRN and the race to the break. Chromosoma. 2009;119:115–135. doi: 10.1007/s00412-009-0242-4. [DOI] [PubMed] [Google Scholar]
- Sarnow P, Sullivan CA, Levine AJ. A monoclonal antibody detecting the adenovirus type 5 E 1 b-58Kd tumor antigen: Characterization of the E 1 b-58Kd tumor antigen in adenovirus-infected and -transformed cells. Virology. 1982;120:510–517. doi: 10.1016/0042-6822(82)90054-x. [DOI] [PubMed] [Google Scholar]
- Sato Y, Kamura T, Shirata N, Murata T, Kudoh A, Iwahori S, Nakayama S, Isomura H, Nishiyama Y, Tsurumi T. Degradation of Phosphorylated p53 by Viral Protein-ECS E3 Ligase Complex. PLoS Pathog. 2009;5:e1000530. doi: 10.1371/journal.ppat.1000530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz R, Lakdawala S, Eshleman H, Russell M, Carson C, Weitzman M. Distinct Requirements of Adenovirus E1b55k Protein for Degradation of Cellular Substrates. J Virol. 2008;82:9043. doi: 10.1128/JVI.00925-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S, Harris CC. p53: traffic cop at the crossroads of DNA repair and recombination. Nat Rev Mol Cell Biol. 2005;6:44–55. doi: 10.1038/nrm1546. [DOI] [PubMed] [Google Scholar]
- Stracker TH, Carson CT, Weitzman MD. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature. 2002;418:348–352. doi: 10.1038/nature00863. [DOI] [PubMed] [Google Scholar]
- Takayesu D, Teodoro JG, Whalen SG, Branton PE. Characterization of the 55K adenovirus type 5 E1B product and related proteins. J Gen Virol. 1994;75 ( Pt 4):789–798. doi: 10.1099/0022-1317-75-4-789. [DOI] [PubMed] [Google Scholar]
- Wang Y-G, Nnakwe C, Lane WS, Modesti M, Frank KM. Phosphorylation and Regulation of DNA Ligase IV Stability by DNA-dependent Protein Kinase. J Biol Chem. 2004;279:37282–37290. doi: 10.1074/jbc.M401217200. [DOI] [PubMed] [Google Scholar]
- Wei YF, Robins P, Carter K, Caldecott K, Pappin DJ, Yu GL, Wang RP, Shell BK, Nash RA, Schär P. Molecular cloning and expression of human cDNAs encoding a novel DNA ligase IV and DNA ligase III, an enzyme active in DNA repair and recombination. Mol Cell Biol. 1995;15:3206–3216. doi: 10.1128/mcb.15.6.3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RS, Bernstein N, Lee MS, Rakovszky ML, Cui D, Green R, Weinfeld M, Glover JNM. Structural basis for phosphorylation-dependent signaling in the DNA-damage response. Biochem Cell Biol. 2005;83:721–727. doi: 10.1139/o05-153. [DOI] [PubMed] [Google Scholar]
- Wilson TE, Grawunder U, Lieber MR. Yeast DNA ligase IV mediates non-homologous DNA end joining. Nature. 1997;388:495–498. doi: 10.1038/41365. [DOI] [PubMed] [Google Scholar]
- Woelk T, Sigismund S, Penengo L, Polo S. The ubiquitination code: a signalling problem. Cell Div. 2007;2:11. doi: 10.1186/1747-1028-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P-Y, Frit P, Meesala S, Dauvillier S, Modesti M, Andres SN, Huang Y, Sekiguchi J, Calsou P, Salles B, et al. Structural and Functional Interaction between the Human DNA Repair Proteins DNA Ligase IV and XRCC4. Mol Cell Biol. 2009;29:3163–3172. doi: 10.1128/MCB.01895-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane K, Kawabata M, Tsuruo T. A DNA-Topoisomerase-11-Binding Protein with Eight Repeating Regions Similar to DNA-repair Enzymes and to a Cell-Cycle Regulator. Eur J Biochem. 1997;250:794–799. doi: 10.1111/j.1432-1033.1997.00794.x. [DOI] [PubMed] [Google Scholar]
- Yasukawa T, Kamura T, Kitajima S, Conaway RC, Conaway Joan W, Aso T. Mammalian Elongin A complex mediates DNA-damage-induced ubiquitylation and degradation of Rpb1. EMBO J. 2008;27:3256–3266. doi: 10.1038/emboj.2008.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Cheng C, Blanchette P, Branton Philip E. The adenovirus E4orf6 E3 ubiquitin ligase complex assembles in a novel fashion. Virology. 2007;364:36–44. doi: 10.1016/j.virol.2007.02.012. [DOI] [PubMed] [Google Scholar]
- Xianghui Yu, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu X-F. Induction of APOBEC3G Ubiquitination and Degradation by an HIV-1 Vif-Cul5-SCF Complex. Science. 2003;302:1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- Xiaochun Yu, Chini CCS, He M, Mer G, Chen J. The BRCT Domain Is a Phospho-Protein Binding Domain. Science. 2003;302:639–642. doi: 10.1126/science.1088753. [DOI] [PubMed] [Google Scholar]
WEB REFERENCES
- [Last accessed June 2010.];Jpred website. http://www.compbio.dundee.ac.uk/www-jpred/index.html.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.