

Abstract
The use of adeno-associated virus (AAV) as a gene therapy vector is limited by the host neutralizing immune response. The cryo-electron microscopy (EM) structure at 8.5 Å resolution is determined for a complex of AAV-2 with the Fab′ fragment of monoclonal antibody (MAb) A20, the most extensively characterized AAV MAb. The binding footprint is determined through fitting the cryo-EM reconstruction with a homology model following sequencing of the variable domain, and provides a structural basis for integrating diverse prior epitope mappings. The footprint extends from the previously implicated plateau to the side of the spike, and into the conserved canyon, covering a larger area than anticipated. Comparison with structures of binding and non-binding serotypes indicates that recognition depends on a combination of subtle serotype-specific features. Separation of the neutralizing epitope from the heparan sulfate cell attachment site encourages attempts to develop immune-resistant vectors that can still bind to target cells.
Keywords: Adeno-associated virus, Antibody, A20, Epitope, Fab′, Gene therapy, Monoclonal
INTRODUCTION
Gene therapy involves the delivery to cells of DNA designed to alleviate disease of genetic nature or predisposition, DNA that commonly encodes a functional replacement for a mutated gene. AAV has become a leading candidate vector for gene therapy because it is non-pathogenic, induces little inflammatory response, integrates site-specifically into chromosome 19, infects dividing and non-dividing cells, has wide cell tropism, and is producible in large quantity (Carter et al., 2008). AAV’s main disadvantages include a limited gene payload of 5kb (Dong et al., 1996) and seropositivity of 35–80% of the U.S. population to AAV-2 (Calcedo et al., 2009) (vide infra). Current and recent clinical trials using AAV vectors are targeting hemophilia B, prostate and melanoma cancers, Canavan disease, Alzheimer’s, Parkinson’s, muscular dystrophy, rheumatoid arthritis and HIV vaccines (Carter, 2005).
AAV is a non-enveloped virus with a single-stranded DNA genome surrounded by a protein capsid that is composed of 60 protein subunits related by icosahedral symmetry (Xie et al., 2002). Three capsid proteins; VP-1, VP-2, and VP-3 are present in a ratio of 1:1:10, and have molecular weights of 87kD, 73kD, and 62kD, differing (only) in N-terminal extensions of 137 residues (VP-1) and 64 residues (VP-2) relative to VP-3 (Agbandje-McKenna and Kleinschmidt, 2011). It is not clear whether VP-1 & 2 substitute for VP-3 at specific locations in the otherwise 60-fold symmetric capsid.
Immune responses to AAV are mild and non-inflammatory, significantly reducing the possibility of complications during treatment (Bueler, 1999; Chirmule et al., 1999). However, immune response still presents a significant barrier to efficient delivery of the vector during initial treatment, as neutralizing antibodies have been found in up to 60% of the population (Blacklow et al., 1968; Boutin et al., 2010; Calcedo et al., 2009; Chirmule et al., 1999; Moskalenko et al., 2000). Clinical trials involving experimental cystic fibrosis and hemophilia treatments required re-administration of therapies (Flotte and Carter, 1998; Kay et al., 2000). As only ~10% of progenitor cells below the lung epithelium are transduced, transgene expression falls over 6 months as surface cells are turned over (Carter and Flotte, 1996). Exposure to natural AAV or vectors in animals and humans results in development of neutralizing antibodies (Halbert et al., 2000; Moskalenko et al., 2000; Peden et al., 2004; Xiao et al., 1999). This can decrease the efficiency of transgene expression upon subsequent administration(s) of vectors of the same serotype (Davidoff et al., 2005; Manno et al., 2006; Wang et al., 2011; Wang et al., 2010). Cellular immune response is largely directed against the viral capsid, while immune response to the transgene product is rare (Brockstedt et al., 1999; Chirmule et al., 2000; Halbert et al., 1997; Hernandez et al., 1999; Manning et al., 1998). Therefore, it is widely thought that the engineering of antigenic variant vectors will have substantial impact in the development of efficient gene therapy treatments (Flotte, 2005; Peden et al., 2004; Xiao et al., 1997).
A20 is a monoclonal antibody which neutralizes AAV-2 and AAV-3B subsequent to primary receptor binding (Wobus et al., 2000). It does not bind to the other predominant serotypes 1, 4, 5, 6, 8 and 9 (Murphy et al., 2008; Wobus et al., 2000; Wu et al., 2006; Xie et al., 2011). It is the most widely studied monoclonal antibody against AAV-2, due in part to its ability to bind only to fully assembled capsid and not to unassembled capsid proteins (Moskalenko et al., 2000; Wistuba et al., 1995; Wobus et al., 2000). It has been suggested that there are at least three neutralizing immunogenic sites on the capsid of AAV-2 (Lochrie et al., 2006), therefore it is unlikely that mutations solely within the A20 footprint will yield a capsid that is completely distinct antigenically. However, two mutations which inhibit A20 neutralization also confer resistance to polyclonal neutralization, suggesting that A20 binding might model neutralization at a dominant epitope (Lochrie et al., 2006). There have been multiple attempts to locate the A20 epitope using various techniques, including PEPSCAN, peptide competition, peptide insertions, and both site-directed and scanning mutagenesis (Girod et al., 1999; Lochrie et al., 2006; Moskalenko et al., 2000; Shi et al., 2001; Wobus et al., 2000; Wu et al., 2000). Lochrie et al. (2006) have drawn attention to inconsistencies between some of the experimental epitope mappings, and between some of the proposed immunogenic sites and the subsequent AAV-2 atomic structure (Xie et al., 2002), noting that inaccuracies result from known limitations of standard mehods (Van Regenmortel, 1992). There has long been an interest in resolving these questions through direct visualization of an AAV-2/A20 complex (Lochrie et al., 2006).
We present here a cryo-EM reconstruction of AAV-2 in complex with A20 monoclonal Fab′ antibody at a resolution of 8.5 Å. The antibody-binding footprint of AAV-2 has been determined from the previously determined crystal structure of AAV-2 by fitting a homology model of A20 Fab′ into the experimental density of the complex. To our knowledge, this is the first structure of an AAV-antibody complex. The binding interface is compared with the corresponding surfaces of closely related viruses for insights into the characteristics of serotypes that might determine whether or not a virus is recognized by A20.
RESULTS AND DISCUSSION
Use of Fab′ fragments
EM imaging takes advantage of the 60-fold icosahedral symmetry, for which it was important to achieve near saturated binding of equivalent binding sites on the virus. At the required stoichiometries, attempts to image the complex of AAV-2 with intact purified MAb A20 were foiled by aggregation, either when virus and antibody were pre-mixed in solution, or when antibody was added to virus pre-loaded onto EM grids. With papain-digested Fab fragments, prepared the conventional way, neither unstained or negatively stained EM revealed fragments bound to the AAV surface. Even large excesses of Fab failed to compete with intact Mab in dot-blot assays using biotinylated Mab A20. These data suggested that the papain-digested fragments were not stable in a native fold. By contrast, pepsin-digested Fab′ fragments competed with MAb in dot-blot assays, and could be seen decorating the surface of AAV-2 in negatively stained EM images.
Electron microscopy reconstruction
The complex of AAV-2 with Fab′ fragments of neutralizing monoclonal antibody A20 (Wistuba et al., 1995) was visualized through cryo-EM and reconstructed to a resolution of 8.5 Å (FSC0.5) (Figure 1). 11,898 particles from 1066 images were combined in a 3-D reconstruction. Class averages of the complex resolved the variable and constant Fab′ domains, with the presumptive variable domain, bound to the capsid surface, better defined than the constant domain (Supplementary Figure 1). The EM reconstruction clearly shows the distinctive features of AAV, previously seen in the uncomplexed virus (O’Donnell et al., 2009; Xie et al., 2002), namely the three-fold peaks, the 5-fold cylinder, 2-fold valley, canyon, and plateau (Figure 2). All of the differences in the capsid density between the complex and native forms could be attributed to differing resolution, i.e. there is no evidence of large scale antibody-induced conformational change in the virus.
Figure 1.

Fourier shell correlation. At a conservative threshold of 0.5, the resolution would be assessed as 8.5 Å. A threshold of 0.143 yields 6.7 Å.
Figure 2.

Reconstruction of AAV-2 complexed with A20 Fab′ (A) compared to a prior cryo-EM reconstruction of native AAV-2 (B) (O’Donnell et al., 2009). The view is along a 2-fold with additional 2-fold axes horizontal and vertical. The asymmetric unit is outlined in panel B with a pentamer at the 5-fold vertex, triangles at the 3-folds and an oval at the 2-fold axis. Reconstructions are colored by radius from the center from blue to red. The canyon surrounding each 5-fold and the depressed area near each 2-fold are blue, the cylinder and plateau are green, and both the 3-fold proximal viral spikes and Fab′ are red. C: Fit of the homology model into the density of the cryo-EM reconstruction. Two symmetry-equivalent Fab′ fragments are shown in blue (pale for the light chain, dark for heavy) with CDRs highlighted according to the color key, H1-3 and L1-3. AAV-2 is shown in green with variable regions VR-I, -III, -VII and -IX, the main CDR contacts, highlighted in color. The view is tangential to the virus surface with yellow arrows indicating symmetry axes, 2-folds on each side, and looking from a 3-fold (front center) towards a 5-fold (out of view). Density is contoured at 2.5 σ. Figure S4 shows a stereographic version of panel C, from which the quality of fit can be more readily appreciated.
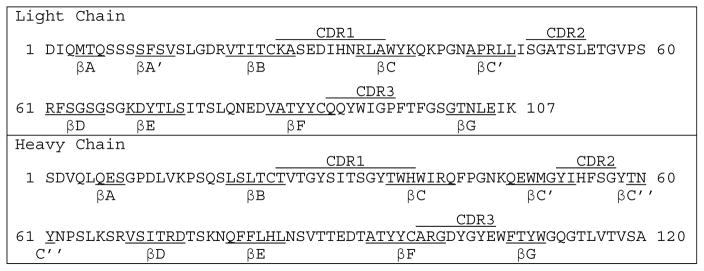
Homology modeling
A20 is a murine Ig3 antibody whose sequence and 3-D structure have not previously been reported. Using sequences determined here for the variable regions of light and heavy chains (Table 1), Modeller (Eswar et al., 2006) was used to create a homology model of the antibody’s variable region. The structure of the complementarity determining regions (CDRs) depends on loop refinement, sometimes a challenging step, so the homology model was checked against the database of CDR conformation based on > 300 antibody structures (North et al., 2011). CDR conformers were used if their database frequency among those with A20-matching sequence motifs exceeded 5% (North et al., 2011). Excluding two conformations of CDR H3 with 5 and 11 Cα outside the A20 density respectively, the frequency-weighted RMS Cα distance between the A20 homology CDRs and matching database conformers was 0.7 Å. Five of the 6 CDRs were in excellent agreement (RMS Cα = 0.4 Å), with greater uncertainty in H3 (RMS Cα = 1.5 Å). In principle, the homology model, built to fit the cryo-EM density (Eswar et al., 2006), should be superior to the un-optimized density-independent cluster structures, and the discrepancy between them provides an upper-limit estimate of the homology model backbone error.
Table 1.
Sequence of the A20 variable regions for both light and heavy chains. The complementarity determining regions (CDR) are annotated for each chain.
|
Analysis of the 3-D reconstruction
Initial rigid group refinement using RSRef (Chapman, 1995) relieved strain in the inter-molecular van der Waals energy (by ~20%) with little change in the quality of the fit (CC = 0.775). (Correlation coefficients are calculated for voxels of the map within 10 Å of any Fab′ or virus atom.) Least-squares fitting of a Butterworth low-pass filter to the model density lowered the residual difference with the experimental density by 1%, and suggested that the effective resolution of modeling was slightly lower (10.6 Å) than would be expected of the FSC0.5 (8.5 Å), noting that the former reflects the limitations of both experiment and modeling. Splitting the Fab′ into variable and constant domains led to only a small improvement in correlation (0.778 to 0.781), but a 40% improvement in intra-protomer van der Waals energy as strain was released. Occupancy of the FAb′ refined robustly to 1.0, indicating saturated binding. Refinement of isotropic group atomic displacement parameters (ADP, “B-factors”) showed that the variable domain was indistinguishable from the capsid protein (<B> = 23.5 Å2), but the more distal constant domain had an optimal B of ~ 300 Å2. These correspond to rms harmonic displacements of U = 0.3 Å and U = 2 Å for the variable and constant domains respectively. Thus, the variable domain can be considered tightly bound with displacement parameters that reflect the experimental resolution. By contrast, constant domain displacements of 2 Å, and perceptibly more diffuse EM density, are consistent with flexibility at the hinge. A final round of positional refinement converged at CC = 0.787 (Figure 2). From start to end of this refinement the rms coordinate change was 0.8 Å.
Binding site
With virus-Fab complexes, it is usual to analyze the interface indirectly, using a model fit to the Fab density, because the constraints of fitting a known fold, like the imposition of stereochemical restraints in crystallographic refinement, can support analysis beyond the nominal experimental resolution (Rossmann, 2000). It does introduce the errors of Fab homology modeling, which are modest for the backbone (see above) and depend mostly on uncertainty in choice of side-chain rotamers. It therefore limits analysis to amino acid level, which is nevertheless a potential improvement upon the 8.5 Å primary data. It was cross-checked in two ways (Table S1). A model-independent footprint was determined by tracing saddle-point vectors between the virus and Fab densities, contoured to correspond to either the solvent-excluded or solvent accessible volumes (Gerstein et al., 2001). Footprints from the solvent excluded density and homology model are similar (Figure 3), sharing 12 of 16 amino acids, and only 2 homology model contacts fall outside the footprint density contoured to approximate the solvent accessible surface. The impact of homology modeling errors was also assessed by comparison with an alternative model (see above), consisting of spliced loops from Dunbrack’s CDR database (North et al., 2011) with matching sequence motifs. 80% of the contact residues were identical. In summary, of 16 footprint residues implicated by the homology model, 12 are robustly implicated by all methods, two are doubtful (Ser658 and Thr660) lying outside the density footprint, and a total of 7 additional residues on the periphery are suggested as possibilities by one or more of the alternative approaches. Discussion will be framed around the core consensus footprint, except as explicitly noted.
Figure 3.
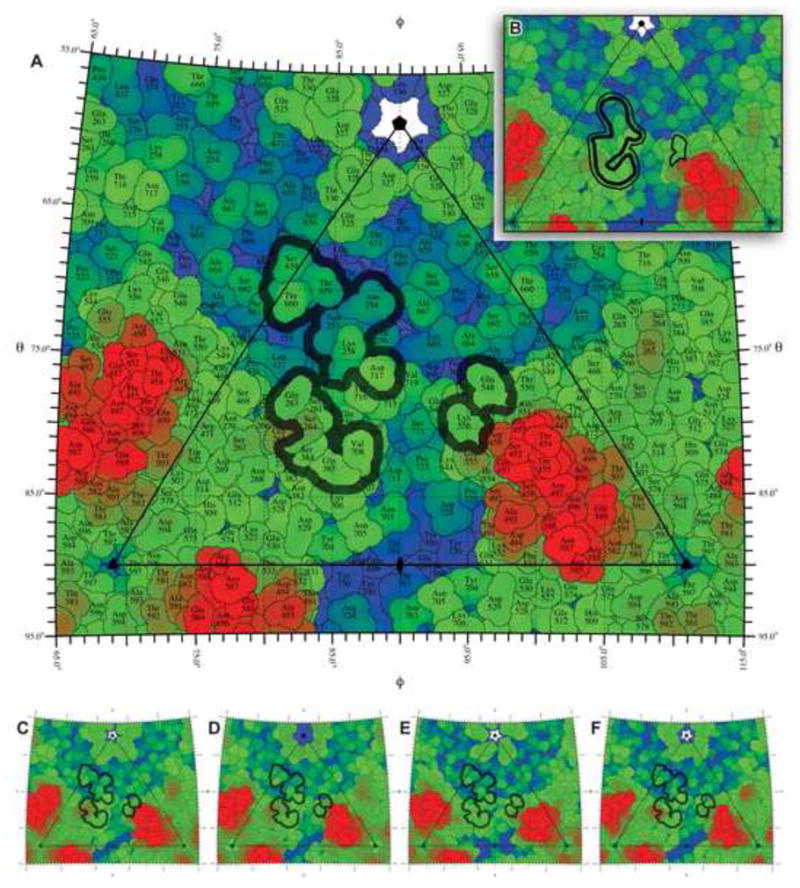
Stereographic polar projections of the Fab′ A20 footprint (outlined in black) onto the AAV asymmetric unit, outlined by the triangle (see Figure 1B). Color indicates distance from the viral center, from blue (105 Å) to red (140 Å). (A) Footprint determined from a homology model fit to the EM density projected onto the surface of AAV-2; (B) Footprint determined directly from the 8.5 Å density, contoured at a level to correspond to the solvent-excluded volume (thick line) or solvent-accessible surface (thin line); (C – F) Projections of the AAV-3B (C), AAV-4 (D), AAV-6 (E), and AAV-8 (F) structures onto which is overlaid the AAV-2 Fab′ footprint from panel A.
Residues implicated in the footprint are listed in Table 2 and Table S1 and illustrated in Figure 3. The footprint is comprised of multiple peptides contributed by three different symmetry-related subunits, explaining why MAb A20 is specific for assembled capsids (Wobus et al., 2000). It encompasses regions of diverse sequence on the plateau and side of the spike, but also nearly as many in the more conserved canyon region. The more exposed parts of the footprint contain residues from 4 of 9 “variable regions” (VR) previously noted to have the highest sequence diversity among AAVs (Agbandje-McKenna and Chapman, 2006; Govindasamy et al., 2006). Earlier difficulties in linear epitope mapping are explained by the finding that 6 distinct segments of polypeptide chain contribute to the footprint; mAb A20 binds to a conformational epitope defined by protein tertiary structure, and not a simple linear sequence of amino acids.
Table 2.
Regions of AAV-2 contacted by MAb A20. Residues were identified that fell within 4 Å of the fitted Fab′ homology model. Most of the footprint is validated by comparison to two independent assessments (Table S1). Parentheses denote residues on the periphery that were not implicated by all three approaches.
| AAV-2 Residues | Subunit | Variable Region | |
|---|---|---|---|
|
|
|||
| Plateau | 261SSQS264 | B | I |
|
| |||
| Ser384, Gln385 | B | III | |
|
| |||
| Val708 | A | IX | |
| Asn717 | A | ||
|
|
|||
| Canyon wall | Lys258 | B | |
|
|
|||
| Canyon | Asn253, Asn254 | B | |
|
| |||
| 658(S)T(T)660 | I | ||
|
|
|||
| Spike | (Glu548, Lys556) | A | VII |
It has been reported that human rhinovirus is neutralized by bivalent antibody attachment in some cases (Hewat and Blaas, 1996; Rossmann et al., 1985). For bivalent attachment adjacent Fab C-termini need to be located within 25–29 Å of each other (Hewat and Blaas, 1996; Hewat et al., 1998; Thouvenin et al., 1997). For the A20 homology model, the closest-symmetry related C-terminal ends of A20 Fab′ are 47 Å apart. The resolution and distinctive features of the reconstruction leave little ambiguity in the orientation of the Fab′, with the C-termini of the heavy chain Fab′ fragments pointing outwards away from their nearest neighbor. Bivalent attachment of an intact antibody would require implausible levels of distortion at the elbow (relative to the observed configuration), and displacements that would be 10-fold greater than any disorder implied in the constant domain B-factors. Thus, although symmetry-related antibody footprints come within 12 Å of each other, the structure is compatible with only monovalent attachment.
MAb A20 neutralizes at a step subsequent to cellular attachment (Wobus et al., 2000). AAV enters cells endosomally, after initial attachment to a heparan sulfate proteoglycan (HSPG) (Summerford and Samulski, 1998), and subsequent binding to one of several possible protein co-receptors (Agbandje-McKenna and Kleinschmidt, 2011). The HSPG binding footprint has recently been mapped by electron microscopy (O’Donnell et al., 2009). Co-receptor footprints are currently unknown. (A site suggested by mutations affecting transduction but not heparin-binding (Lochrie et al., 2006) actually fell within the HSPG footprint (O’Donnell et al., 2009), so the sensitivity of heparin binding assays may not support such specific designations.) Here, we see that the MAb A20 and heparin binding footprints do not overlap. This is consistent with the earlier finding that A20 does not inhibit cellular attachment (Wobus et al., 2000). It remains to be seen whether A20’s neutralization results from interference with co-receptor mediated entry, or conceivably some later step. A20 occludes considerably more of the surface than the footprint residues making direct contact and thus could affect other interactions essential to AAV’s life cycle. A20’s variable domain extends deep into the canyon and is nestled between the 5-fold cylinder and the 3-fold spikes. Although none of the residues of the cylinder and only two of the spike make direct contact, the gap between virus and antibody is narrow enough here to shield additional residues from interacting with macromolecules that might be essential to AAV’s life cycle.
Comparison with autonomous parvoviruses
There are not yet other Dependovirus-antibody complexes with which to compare, but there are several cryo-EM structures from the autonomous parvoviruses of the sister genus Parvovirus. The greatest similarities are with antibodies that bind to epitope B of the closely related pair, canine parvovirus (CPV) and feline panleukopenia virus (FPV) (Hafenstein et al., 2009; Wikoff et al., 1994) (Figure S1). The AAV-2 A20 binding site therefore contrasts with the binding site of MAb B7 of minute virus of mouse (MVM) which is directly above the 3-fold axis of symmetry (Kaufmann et al., 2007). The surface topology of the autonomous viruses at the B7 site differs from AAV with loops from adjacent subunits coming together to form more of a single elevated “massif”. AAV has 3 distinct protrusions, between which are valleys that meet at the 3-fold, so the B7 site is less accessible. However, some common themes are emerging. None of the complexes, including AAV-2/A20, are compatible with bivalent attachment, and several of the binding sites: MVM B7, CPV/FPV B-site and AAV2 A20, are at interfaces where loops from neighboring subunits come together to define the viral structure.
Cryo-EM-defined footprints for six CPV/FPV MAbs overlap at site “B”, comprised mostly of loop 3, a region implicated by mutagenesis to be antigenic (Hafenstein et al., 2009; Wikoff et al., 1994). CPV residues 298 to 302 superimpose over the AAV-2 A20 footprint if aligned by icosahedral symmetry. The B site and VR1 of the AAV-2 A20 footprint are located on the plateaus of their respective viral surfaces. In CPV/FPV, bound antibodies overlap with the binding site of the transferrin receptor, blocking both viral attachment and subsequent entry steps (Hafenstein et al., 2007), but the steps of AAV entry and mechanisms of A20 neutralization are not as well characterized. The considerable similarities appear to result from convergent evolution, because AAV’s VR1 does not correspond in primary structure with loop 3 of CPV/FPV. VR1 is in the loop between β-strands B and C, which in CPV/FPV is pushed up from the plateau as part of the 3-fold massif, and comprises epitope A, to which a distinct set of 3 MAbs bind (Hafenstein et al., 2009). Paradoxically, epitope B (on the plateau of CPV and FPV) is formed by residues from the loop between strands G and H whose equivalents are at the exposed tip of the 3-fold protrusions in AAV. In the autonomous viruses, the plateau extends further out from the 3-fold axis, and the footprints of B-site antibodies all fall within the bounds of the plateau. With AAV-2, the topography is more varied, and for the first time we see an Fab binding site extend over the edge of the plateau into a more prominent and less exposed canyon.
Epitope
As the first AAV neutralizing monoclonal antibody (Wobus et al., 2000), MAb A20 has been the subject of repeated epitope mappings using multiple approaches (vide infra). The AAV-2 crystal structure (Xie et al., 2002) showed that not all regions implicated by peptide scanning (Moskalenko et al., 2000; Wobus et al., 2000) could fall within a typical antibody footprint, or, indeed were near the outer surface of the virus. Thus, structure has already helped integrate the molecular data, complementing scanning approaches and their known limitations (Van Regenmortel, 1992). Visualization of the A20-AAV2 binding interface provides an additional perspective. Exact consistency with molecular approaches should not be expected, due to ambiguities in each approach, including the potential for wider conformational change in escape mutants, and limited precision in structural studies at 8.5 Å resolution (see above). Nevertheless, integration of a physical epitope from EM visualization with the functional molecular approaches can provide a more robust understanding of immunogenicity.
There has been wide variation in surface sites implicated in A20 binding, but the plateau has been implicated more often than other regions. Single residue substitutions at 64 surface-exposed sites revealed a cluster that affected A20 binding or neutralization: Q263, S264, S384, Q385 and V708 (Lochrie et al., 2006). These are all within the EM footprint, and are joined in close proximity by the sites of peptide insertions after S261, A266, N244 and S247, the first three of which inhibited A20 binding (Girod et al., 1999; Wu et al., 2000). Thus, from this molecular data and our structure, a consistent picture emerges that an important part of the A20 epitope is the plateau where the VR-I and VR-III loops from one subunit and VR-IX from a neighbor pack together.
The EM reconstruction shows the A20-AAV2 binding surface extending into the canyon, a region that was not tested by Lochrie et al. (2006) or other studies. To the EM footprint are added S261, S262 and N717 on the edge of the plateau, K258 on the wall, then N253, N254, T659 and possibly S658 and T660 in the canyon. As in the complex of human rhinovirus (HRV) 14 with Fab17-1A (Smith et al., 1996), the CDRs of MAb A20 penetrate a less exposed, more conserved region of the surface, although in AAV, the region has not yet been associated with any viral function. Extending the binding site into the canyon approximately doubles the size of the footprint implicated by Lochrie’s cluster of plateau mutants.
The core residues of the footprint are implicated both in model-based calculation of contacts, and in projections of experimental Fab′ density onto AAV-2 that are not dependent on modeling the Fab′. A few residues on the periphery are less certain, because their designation is sensitive to the choice of solvent probe or density contour level. E548 and K556 come within 4 Å of modeled A20 atoms, but are connected by density only at lower contours corresponding to the solvent accessible surface. Mutation of E548 has been seen to yield a small 3-fold resistance to A20 neutralization (Lochrie et al., 2006) or, like K556 (ibid), to have no impact upon binding (Wu et al., 2000). It seems likely that the observed proximity of the Fab′ to two residues on the side of an adjacent AAV-2 spike is incidental and not critical to the binding interactions. Indeed, the spike residues are apparently not part of the epitope, because the contact would be with conserved framework residues towards the side of the immunoglobulin domain, not CDR loop residues usually associated with antigen recognition.
Distinctiveness of AAV-2 and AAV-3B
AAV-2 and AAV-3B are the only serotypes which are bound strongly by A20. A search was made for distinguishing characteristics of the footprint that are conserved between AAV-2 & AAV-3B, but distinct for non-bound serotypes. As at 8.5 Å resolution, side chain conformation is not defined for the Fab′, comparison of atomic interactions was not possible, but high resolution structures for AAV serotypes supported comparison of viral surface properties within the footprint region.
In sequence, AAV-3B differs from AAV-2 only at three binding site residues, all on the edge of the A20 footprint: S658 is replaced by P659 in the canyon, while E548 and K556 of AAV-2 are replaced by T549 and N557 in AAV-3B on the side of the spike (Figure 3). Lochrie et al. (2006) found only modest impact on A20-binding of even non-conservative mutations at E548 and K556. The contact at E548/K556 in the AAV-2 complex is superficial, relative to other interactions. Density bridges only when contoured to enclose the solvent accessible, but not the van der Waals surface (Table S1). Furthermore the contact is made by a conserved (non CDR) part of A20, all suggesting that the contact might be incidental. Thus, the core of the footprint is conserved between AAV-2 and -3B, and differences are restricted to peripheral residues that have at most modest impact on binding (Lochrie et al., 2006).
Most of the differences in the surface topology of other serotypes lie outside the binding site. Within the canyon, AAV-2, -3B, -4, -6, and -8 are very similar. There are subtle differences on the plateau, but these are not obviously correlated to A20 binding. For example, AAV-6 (non-binding) is more similar to AAV-3B (binding) than is AAV-2 (binding). In the footprint region, there are not systematic differences in amino acid polarity or electrostatic charge that would clearly disrupt binding. Local changes are exemplified by a threonine insertion after S264 and/or a Q263A substitution in AAV-6, -1 and an AAV-2/-1 chimeric construct (Hauck and Xiao, 2003). These would not create large steric conflicts with A20 (Figure 3A & C). Analysis of subtle changes in interactions, that are presumably important, requires greater precision in A20 side chain structure than available in an 8.5 Å homology-based model.
For future efforts to engineer gene therapy vectors with reduced susceptibility to immune neutralization, the reported structure is encouraging. The footprint is larger than previously anticipated, providing a wider choice of residues to mutate for immune neutralization escape. Relatively modest changes can affect antibody binding, and there is no overlap for binding sites of receptor HSPG and this model antibody, increasing the prospects for engineering the vector without disrupting cell attachment.
METHODS
Preparation of AAV2 and A20 Fab′
AAV2 was produced as described previously (Xie et al., 2004). Stocks of hybridoma cells for A20 were generously provided by Jürgen Kleinschmidt. They were grown by seeding 25×106 cells in 15mL of 90% RPMI media (Sigma-Aldrich) and 10% FetalClone 1 serum (Hyclone) in a CELLine CL 1000 bioreactor (Integra). The bioreactor was maintained using 98% RPMI and 10% FetalClone 1 serum. Antibodies were harvested after 1 week. Cells were pelleted out by centrifugation at 70× g for 5 min and the supernatant was passed through a 0.45 μm filter. Secreted antibodies were purified with a HiTrap protein G affinity column (GE Healthcare).
The antibody solution was dialyzed into 20 mM sodium acetate (pH 4.5) and digested with immobilized pepsin (Thermo Scientific) as per the manufacturer’s protocol to yield F(ab2)′. The F(ab2)′ solution was dialyzed overnight into 20 mM sodium phosphate (pH 7.2). Undigested IgG and Fc fragments were removed using a HiTrap protein A affinity column (GE Healthcare). Flow-through fractions containing F(ab2)′ were dialyzed into 150mM PBS and 5 mM EDTA (pH 7.2) using a Float-A-Lyzer G2 (Spectra/Por) with a molecular weight 50 kD weight cutoff.
The F(ab2)′ solution was reduced with Mercaptoethylamine-HCl (2-MEA; Thermo Scientific) according to the manufacturer’s protocol. The resultant crude Fab′ solution was immediately purified by size exclusion chromatography with Superdex 200 (GE Healthcare) using a running buffer of 150 mM sodium chloride, 50 mM sodium phosphate, and 5 mM EDTA (pH 7.2). Gel electrophoresis with silver staining was used to assess purity.
Preparation and cryo-EM of AAV2-A20 Fab′ complex
AAV-2 in 100 mM HEPES, 50 mM magnesium chloride, and 5% glycerol (pH 7.2) was incubated with a 4-fold excess of purified Fab′ (240 Fab′ fragments per 60-fold symmetric virus) for 30 min at 25 °C. Small aliquots of this mixture were applied to holey carbon grids (C-flat). The sample was then flash-frozen by plunging using a Vitrobot (FEI) at 100% humidity and 4°C and using a 2 sec blot time. 1503 images of the specimen were collected at 37,000× magnification and 120 keV on a FEI Titan Krios equipped with a Gatan Ultrascan 4k × 4k CCD camera using the Leginon system (Suloway et al., 2005). The final pixel size was 2.225 Å.
Reconstruction of AAV2-A20 Fab′ complex
Appion (Lander et al., 2009) was used for particle picking, CTF estimation, and stack making. Initial particle selection was performed using the difference of Gaussians method (Voss et al., 2009) and particles over carbon were manually deselected. CTF estimation was performed with the ACE (Automated CTF Estimation) software package (Mallick et al., 2005). Images with an ACE CTF estimation confidence value of less than 0.7 were removed from the data set after which a 35,543 particle stack was made with the phases flipped for individual particles according to their ACE-estimated defocus.
EMAN (Tang et al., 2007) was used for subsequent refinement and reconstruction. Refinement first yielded a reconstruction with a resolution of 9.8 Å by the FSC0.5 criterion. Inspection of the class averages from the refinement suggested conformational heterogeneity in the Fabs. Subclassification was employed to test for and, as necessary, remove heterogeneity. Multivariate statistical analysis was performed on each class of particle projection following each iteration of refinement. Correspondence analysis was performed with hierarchical ascendant classification on aligned particles in a given orientation. This generated 2–6 subclasses for each projection. Only the subclass correlating best with the projection of the current model was included in the reconstruction for that iteration. After refinement, this procedure resulted in an 8.5 Å resolution (FSC0.5) final reconstruction based on 11,898 particles. The whole refinement was repeated using two different starting models: a prior reconstruction of the native virus (O’Donnell et al., 2009); and a model created de novo using the EMAN STARTICOS program. These independent refinements converged on the same solution.
Scaling
Correction of the relative magnification proved to be critical. After data collection, it was discovered that the relevant magnification calibration parameter had not been set appropriately in this first structure determined using a newly commissioned microscope. Thus, such a large correction to the relative magnification (1.075) had not been anticipated. Three approaches yielded the same value for the relative magnification. Firstly, a search was made for the best agreement between the density of the complex and an earlier reconstruction for the native virus (O’Donnell et al., 2009), masking out Fab′ density through use of radial cut-offs of 72 & 120 Å. Secondly, a search was made for the highest correlation between the experimental map of the complex, and the local density calculated from the AAV-2 (uncomplexed) crystallographic structure (Chapman, 1995; Xie et al., 2002). Finally, the magnification was least-squares refined by optimizing the agreement of the full atomic model of the complex (see below) and the experimental EM density. Correction of the magnification had a dramatic impact on the flatness of difference maps, and the model-map correlation, improving from 0.2 to 0.7.
Creating a homology model
Initially, the Fab′ was modeled using an arbitrarily selected antibody from the protein data bank (PDBid 1A6T), without reference to the actual sequence of A20. This was sufficient to uniquely define the approximate orientation of the Fab′ (Figure S3), but a more accurate binding footprint would require a homology model with CDRs of length and conformation appropriate to the sequence of MAb A20. RNA was extracted and cloned from snap-frozen hybridoma cells for PCR amplification using degenerate VH and VL primers and bidirectional cDNA sequencing of the A20 variable domains (Molecular Cloning Laboratories). Modeller (Eswar et al., 2006) was used to create a homology model of the antibody’s variable region. 19 structures from the protein databank with a sequence similarity of 58% or greater were used as templates. 1000 models were produced by aligning the templates to the A20 sequence. Of these, the top 100 models had DOPE scores ranging from −12337 to −12149 and were fitted into the density by Modeller. Of these, the top 10 models had a correlation coefficient of 0.85–0.86. The model with the highest combined DOPE score and correlation coefficient was used as the variable domain in an initial homology model for A20. To model the constant region, of known IgG Fab crystal structures, mouse monoclonal antibody 184.1 (PDBid 1osp (Li et al., 1997)) was chosen on the basis of the highest sequence identity.
CDR database models
Alternative models for the variable domain were generated from the Dunbrack database of CDR loop conformations compiled from > 300 non-redundant high resolution crystal structures (North et al., 2011). For CDRs of a given length, conformations fall into a handful of clusters with sequence fingerprints that have been characterized. For A20 CDRs L1 – L3 and H1 -H2, there was a unique sequence match to a database cluster, and for H3, four were possible, two of which could be eliminated later as segments of five or eleven amino acids extended outside the A20 electron density. Median structures coming from the best database cluster for each CDR were spliced together into a single A20 model after superimposing their constant regions. The process was repeated for the alternate H3 conformation. The sequences were then changed to that of A20, and, where necessary, new side chain rotamers were selected to resolve clashes or bring side chains into density. No additional optimization of these database-derived models was performed. They were used to assess uncertainties in the homology model and in the model-derived footprint.
Docking & Refinement of the homology model
The FAb A20 homology model was fit approximately into a difference map calculated by subtracting a native cryo-EM reconstruction from that of the complex (Figure S3). Initial rigid-group conjugate gradient refinement was performed with the Flex-EM option of Modeller-9 (Topf et al., 2008). This revealed two locations where the automatic homology modeling could be improved, residues 209-210 that overlapped with residues 263-264 of AAV-2, and Lys69 which extended beyond the difference map and clashed with AAV2 325. Residues 205-216 were remodeled using another high-scoring homolog (model 554) that did not conflict, and an alternate favored rotamer (Dunbrack, 2002) was selected for Lys69.
The structure was further optimized using a new implementation of the real-space refinement RSRef (Chapman, 1995), embedded in CNS (Brünger et al., 1998). From this point, instead of using a difference map, the Fab′ model was refined in the presence of AAV-2 into the reconstruction of the complex. Additional refined parameters included the relative EM magnification, and the “soft” resolution limit of a 5th order Butterworth low-pass filter (Frank et al., 1996) applied when calculating the density of the atomic model. The experimental map had been corrected (sharpened) by application of the inverse envelope function with EMB-factor (Fernandez et al., 2008). The low-pass filter allowed the model density to replicate the resulting smooth, but non-Gaussian attenuation near the resolution limit. In refinement, the squared difference between observed and calculated electron density levels was minimized using all map grid points within 10 Å of any model atom, and considering density contributions from atoms up to 25 Å away. Icosahedral symmetry was imposed as a constraint on both Fab′ A20 and the AAV-2 capsid protein. The virus structure was fixed, aligned to the icosahedral symmetry of the map. The Fab′ was refined first as a single rigid-group, optimizing a weighted (~20:1) sum of the density residual and CNS van der Waals repulsion terms. The Fab′ was then split into variable and constant domains for rigid group positional and group B-factor refinement.
Modeling of the A20 structure led to a 0.8 Å clash at AAV-2 Lys258. This was resolved by choosing a different high frequency rotamer for Lys258. The closest remaining contact was 2.4 Å. No attempt was made at the subtle adjustments needed to resolve contacts that were suboptimal by only ~ 0.5 Å given the resolution of this study. The need for at most subtle adjustment of side chains is consistent with the absence of features in the EM density indicative of significant conformational changes.
Density-based footprint
AAV amino acids contacted by Fab A20 were identified as those with any atom within 4 Å of any homology model atom. An alternative model-independent identification was performed as follows: Between density of the Fab′ and AAV-2, a clear constriction at the periphery of the interface could be used to demark an outline. The contour level of 2.2 σ was used, because it corresponded to the calculated Fab′ solvent-excluded volume in a map of the complex segmented in Chimera (Pettersen et al., 2004). On the 2.2 σ contour of the non-segmented map, the boundary between Fab′ and AAV-2 was outlined in 3D by connecting saddle points with markers placed interactively using Coot (Emsley et al., 2010). The outline was projected onto the surface of the AAV-2 model using Rivem (Xiao and Rossmann, 2006), identifying AAV-2 amino acids whose surfaces were > 25% covered. A solvent-accessible outline was obtained by extending the markers outwards by the 1.4 Å radius of a solvent probe, and additional potential contact regions were determined using the corresponding 1.1 σ contour level.
Supplementary Material
Acknowledgments
We acknowledge the Florida State University shared High-Performance Computing facility and staff for contributions to results presented in this paper. The research was supported by grants from the National Institutes of Health (R01GM66875 & R01GM78538 to MSC), the Office of Naval Research (N000141010082 to John Carruthers at Portland State University), and a grant partially supporting the purchase of the microscope (S10-RR025080 to KAT).
ABBREVIATIONS AND SYMBOLS
- AAV
adeno-associated Virus
- CPV
canine parvovirus
- CTF
contrast transfer function
- EM
electron microscopy
- FPV
feline panleukopenia virus
- HSPG
heparan sulfate proteoglycan
- MVM
minute virus of mouse
- mAb
monoclonal antibody
- NCS
non crystallographic symmetry
- PCR
polymerase chain reaction
- rAAV
recombinant AAV
- RMS(D)
root mean square (deviation)
- VP
viral protein
- VR
variable region
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agbandje-McKenna M, Chapman MS. Correlating structure with function in the viral capsid. In: Kerr JR, Cotmore SF, Bloom ME, Linden RM, Parrish CR, editors. Parvoviruses. Hodder Arnold, Ltd; London: 2006. pp. 124–139. [Google Scholar]
- Agbandje-McKenna M, Kleinschmidt J. AAV capsid structure and cell interactions. Methods Mol Biol. 2011;807:47–92. doi: 10.1007/978-1-61779-370-7_3. [DOI] [PubMed] [Google Scholar]
- Blacklow NR, Hoggan MD, Rowe WP. Serologic evidence for human infection with adenovirus-associated viruses. J Natl Cancer Inst. 1968;40(2):319–27. [PubMed] [Google Scholar]
- Boutin S, Monteilhet V, Veron P, Leborgne C, Benveniste O, Montus MF, Masurier C. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther. 2010;21(6):704–12. doi: 10.1089/hum.2009.182. [DOI] [PubMed] [Google Scholar]
- Brockstedt DG, Podsakoff GM, Fong L, Kurtzman G, Mueller-Ruchholtz W, Engleman EG. Induction of immunity to antigens expressed by recombinant adeno-associated virus depends on the route of administration. Clin Immunol. 1999;92(1):67–75. doi: 10.1006/clim.1999.4724. [DOI] [PubMed] [Google Scholar]
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system: A new software system for macromolecular structure determination. Acta Crystallographica. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Bueler H. Adeno-associated viral vectors for gene transfer and gene therapy. Biol Chem. 1999;380(6):613–22. doi: 10.1515/BC.1999.078. [DOI] [PubMed] [Google Scholar]
- Calcedo R, Vandenberghe LH, Gao G, Lin J, Wilson JM. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J Infect Dis. 2009;199(3):381–90. doi: 10.1086/595830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter B, Flotte T. Development of adeno-associated virus vectors for gene therapy of cystic fibrosis. Current Topics in Microbiology and Immunology. 1996;218:119–144. doi: 10.1007/978-3-642-80207-2_8. [DOI] [PubMed] [Google Scholar]
- Carter BJ. Adeno-associated virus vectors in clinical trials. Hum Gene Ther. 2005;16(5):541–50. doi: 10.1089/hum.2005.16.541. [DOI] [PubMed] [Google Scholar]
- Carter BJ, Burstein H, Peluso RW. Adeno-associated Virus and AAV Vectors for Gene delivery. In: Templeton NS, editor. Gene and cell therapy: therapeutic mechanisms and strategies. CRC Press; Boca Raton: 2008. pp. 115–156. [Google Scholar]
- Chapman MS. Restrained Real-Space Macromolecular Atomic Refinement using a New Resolution-Dependent Electron Density Function. Acta Crystallographica. 1995;A51:69–80. [Google Scholar]
- Chirmule N, Propert K, Magosin S, Qian Y, Qian R, Wilson J. Immune responses to adenovirus and adeno-associated virus in humans. Gene Ther. 1999;6(9):1574–1583. doi: 10.1038/sj.gt.3300994. [DOI] [PubMed] [Google Scholar]
- Chirmule N, Xiao W, Truneh A, Schnell MA, Hughes JV, Zoltick P, Wilson JM. Humoral immunity to adeno-associated virus type 2 vectors following administration to murine and nonhuman primate muscle. J Virol. 2000;74(5):2420–5. doi: 10.1128/jvi.74.5.2420-2425.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidoff AM, Gray JT, Ng CY, Zhang Y, Zhou J, Spence Y, Bakar Y, Nathwani AC. Comparison of the ability of adeno-associated viral vectors pseudotyped with serotype 2, 5, and 8 capsid proteins to mediate efficient transduction of the liver in murine and nonhuman primate models. Mol Ther. 2005;11(6):875–88. doi: 10.1016/j.ymthe.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Dong JY, Fan PD, Frizzell RA. Quantitative analysis of the packaging capacity of recombinant adeno-associated virus. Human Gene Therapy. 1996;7(17):2101–2112. doi: 10.1089/hum.1996.7.17-2101. [DOI] [PubMed] [Google Scholar]
- Dunbrack RL., Jr Rotamer libraries in the 21st century. Curr Opin Struct Biol. 2002;12(4):431–40. doi: 10.1016/s0959-440x(02)00344-5. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N, Eramian D, Webb B, Shen M, Sali A. Protein Structure Modeling With MODELLER. In: Baxevanis AD, Stein LD, Stormo GD, Yates JR III, editors. Current Protocols in Bioinformatics. John Wiley & Sons, Inc; 2006. pp. 5.6.1–5.6.30. Vol. Supplement 15. [Google Scholar]
- Fernandez JJ, Luque D, Caston JR, Carrascosa JL. Sharpening high resolution information in single particle electron cryomicroscopy. J Struct Biol. 2008;164(1):170–5. doi: 10.1016/j.jsb.2008.05.010. [DOI] [PubMed] [Google Scholar]
- Flotte TR. Recent developments in recombinant AAV-mediated gene therapy for lung diseases. Curr Gene Ther. 2005;5(3):361–6. doi: 10.2174/1566523054064986. [DOI] [PubMed] [Google Scholar]
- Flotte TR, Carter BJ. Adeno-associated virus vectors for gene therapy of cystic fibrosis. Methods Enzymol. 1998;292:717–32. doi: 10.1016/s0076-6879(98)92055-9. [DOI] [PubMed] [Google Scholar]
- Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116(1):190–9. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- Gerstein M, Richards F, Chapman MS, Connolly M. Protein surfaces and volumes: measurement and use. In: Rossmann MG, Arnold E, editors. International Tables for Crystallography. Crystallography of Biological Molecules. F. Kluwer Academic Publishers; Dortrecht, Netherlands: 2001. pp. 531–45. (Cpt. 22.1) [Google Scholar]
- Girod A, Ried M, Wobus C, Lahm H, Leike K, Kleinschmidt J, Deleage G, Hallek M. Genetic capsid modifications allow efficient re-targeting of adeno-associated virus type 2. Nat Med. 1999;5(9):1052–1056. doi: 10.1038/12491. [DOI] [PubMed] [Google Scholar]
- Govindasamy L, Padron E, McKenna R, Muzyczka N, Kaludov N, Chiorini JA, Agbandje-McKenna M. Structurally mapping the diverse phenotype of adeno-associated virus serotype 4. J Virol. 2006;80(23):11556–70. doi: 10.1128/JVI.01536-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafenstein S, Bowman VD, Sun T, Nelson CD, Palermo LM, Chipman PR, Battisti AJ, Parrish CR, Rossmann MG. Structural comparison of different antibodies interacting with parvovirus capsids. J Virol. 2009;83:5556–5566. doi: 10.1128/JVI.02532-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafenstein S, Palermo LM, Kostyuchenko VA, Xiao C, Morais MC, Nelson CD, Bowman VD, Battisti AJ, Chipman PR, Parrish CR, Rossmann MG. Asymmetric binding of transferrin receptor to parvovirus capsids. Proc Natl Acad Sci U S A. 2007;104(16):6585–9. doi: 10.1073/pnas.0701574104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbert CL, Rutledge EA, Allen JM, Russell DW, Miller AD. Repeat transduction in the mouse lung by using adeno-associated virus vectors with different serotypes. J Virol. 2000;74(3):1524–32. doi: 10.1128/jvi.74.3.1524-1532.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbert CL, Standaert TA, Aitken ML, Alexander IE, Russell DW, Miller AD. Transduction by adeno-associated virus vectors in the rabbit airway: efficiency, persistence, and readministration. J Virol. 1997;71(8):5932–41. doi: 10.1128/jvi.71.8.5932-5941.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauck B, Xiao W. Characterization of tissue tropism determinants of adeno-associated virus type 1. J Virol. 2003;77(4):2768–74. doi: 10.1128/JVI.77.4.2768-2774.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez YJ, Wang J, Kearns WG, Loiler S, Poirier A, Flotte TR. Latent adeno-associated virus infection elicits humoral but not cell-mediated immune responses in a nonhuman primate model. J Virol. 1999;73(10):8549–58. doi: 10.1128/jvi.73.10.8549-8558.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewat EA, Blaas D. Structure of a neutralizing antibody bound bivalently to human rhinovirus 2. EMBO J. 1996;15(7):1515–23. [PMC free article] [PubMed] [Google Scholar]
- Hewat EA, Marlovits TC, Blaas D. Structure of a neutralizing antibody bound monovalently to human rhinovirus 2. J Virol. 1998;72(5):4396–402. doi: 10.1128/jvi.72.5.4396-4402.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann B, Lopez-Bueno A, Mateu MG, Chipman PR, Nelson CD, Parrish CR, Almendral JM, Rossmann MG. Minute virus of mice, a parvovirus, in complex with the Fab fragment of a neutralizing monoclonal antibody. J Virol. 2007;81(18):9851–8. doi: 10.1128/JVI.00775-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay MA, Manno CS, Ragni MV, Larson PJ, Couto LB, McClelland A, Glader B, Chew AJ, Tai SJ, Herzog RW, Arruda V, Johnson F, Scallan C, Skarsgard E, Flake AW, High KA. Evidence for gene transfer and expression of factor IX in haemophilia B patients treated with an AAV vector. Nat Genet. 2000;24(3):257–61. doi: 10.1038/73464. [DOI] [PubMed] [Google Scholar]
- Lander GC, Stagg SM, Voss NR, Cheng A, Fellmann D, Pulokas J, Yoshioka C, Irving C, Mulder A, Lau PW, Lyumkis D, Potter CS, Carragher B. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J Struct Biol. 2009;166(1):95–102. doi: 10.1016/j.jsb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Dunn JJ, Luft BJ, Lawson CL. Crystal structure of Lyme disease antigen outer surface protein A complexed with an Fab. Proc Natl Acad Sci U S A. 1997;94(8):3584–9. doi: 10.1073/pnas.94.8.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochrie MA, Tatsuno GP, Christie B, McDonnell JW, Zhou S, Surosky R, Pierce GF, Colosi P. Mutations on the external surfaces of adeno-associated virus type 2 capsids that affect transduction and neutralization. J Virol. 2006;80(2):821–34. doi: 10.1128/JVI.80.2.821-834.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallick SP, Carragher B, Potter CS, Kriegman DJ. ACE: automated CTF estimation. Ultramicroscopy. 2005;104(1):8–29. doi: 10.1016/j.ultramic.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Manning WC, Zhou S, Bland MP, Escobedo JA, Dwarki V. Transient immunosuppression allows transgene expression following readministration of adeno-associated viral vectors. Hum Gene Ther. 1998;9(4):477–85. doi: 10.1089/hum.1998.9.4-477. [DOI] [PubMed] [Google Scholar]
- Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, Ozelo MC, Hoots K, Blatt P, Konkle B, Dake M, Kaye R, Razavi M, Zajko A, Zehnder J, Rustagi PK, Nakai H, Chew A, Leonard D, Wright JF, Lessard RR, Sommer JM, Tigges M, Sabatino D, Luk A, Jiang H, Mingozzi F, Couto L, Ertl HC, High KA, Kay MA. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12(3):342–7. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- Moskalenko M, Chen L, van Roey M, Donahue BA, Snyder RO, McArthur JG, Patel SD. Epitope mapping of human anti-adeno-associated virus type 2 neutralizing antibodies: implications for gene therapy and virus structure. J Virol. 2000;74(4):1761–6. doi: 10.1128/jvi.74.4.1761-1766.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy SL, Bhagwat A, Edmonson S, Zhou S, High KA. High-throughput Screening and Biophysical Interrogation of Hepatotropic AAV. Mol Ther. 2008;16(12):1960–1967. doi: 10.1038/mt.2008.210. [DOI] [PubMed] [Google Scholar]
- North B, Lehmann A, Dunbrack RL., Jr A new clustering of antibody CDR loop conformations. J Mol Biol. 2011;406(2):228–56. doi: 10.1016/j.jmb.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell J, Taylor KA, Chapman MS. Adeno-associated virus-2 and its primary cellular receptor-Cryo-EM structure of a heparin complex. Virology. 2009;385:434–443. doi: 10.1016/j.virol.2008.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peden CS, Burger C, Muzyczka N, Mandel RJ. Circulating anti-wild-type adeno-associated virus type 2 (AAV2) antibodies inhibit recombinant AAV2 (rAAV2)-mediated, but not rAAV5-mediated, gene transfer in the brain. J Virol. 2004;78(12):6344–59. doi: 10.1128/JVI.78.12.6344-6359.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Rossmann MG. Fitting atomic models into electron-microscopy maps. Acta Crystallogr D Biol Crystallogr. 2000;56(Pt 10):1341–1349. doi: 10.1107/s0907444900009562. [DOI] [PubMed] [Google Scholar]
- Rossmann MG, Arnold E, Erickson JW, Frankenberger EA, Griffith JP, Hecht H, Johnson JE, Kamer G, Luo M, Mosser A, Rueckert R, Sherry B, Vriend G. Structure of human common cold virus and functional relationship to other picornaviruses. Nature. 1985;317:145–153. doi: 10.1038/317145a0. [DOI] [PubMed] [Google Scholar]
- Shi W, Arnold GS, Bartlett JS. Insertional mutagenesis of the adeno-associated virus type 2 (AAV2) capsid gene and generation of AAV2 vectors targeted to alternative cell-surface receptors. Hum Gene Ther. 2001;12(14):1697–711. doi: 10.1089/104303401750476212. [DOI] [PubMed] [Google Scholar]
- Smith TJ, Chase ES, Schmidt TJ, Olson NH, Baker TS. Neutralizing antibody to human rhinovirus 14 penetrates the receptor-binding canyon. Nature. 1996;383(6598):350–4. doi: 10.1038/383350a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, Carragher B. Automated molecular microscopy: the new Leginon system. J Struct Biol. 2005;151(1):41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Summerford C, Samulski RJ. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J Virol. 1998;72(2):1438–45. doi: 10.1128/jvi.72.2.1438-1445.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol. 2007;157(1):38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Thouvenin E, Laurent S, Madelaine MF, Rasschaert D, Vautherot JF, Hewat EA. Bivalent binding of a neutralising antibody to a calicivirus involves the torsional flexibility of the antibody hinge. J Mol Biol. 1997;270(2):238–46. doi: 10.1006/jmbi.1997.1095. [DOI] [PubMed] [Google Scholar]
- Topf M, Lasker K, Webb B, Wolfson H, Chiu W, Sali A. Protein Structure Fitting and Refinement Guided by Cryo-EM Density. Structure. 2008;16(2):295–307. doi: 10.1016/j.str.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Regenmortel MHV. Structure of antigens. CRC Press; Boca Raton, Fla: 1992. [Google Scholar]
- Voss NR, Yoshioka CK, Radermacher M, Potter CS, Carragher B. DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. J Struct Biol. 2009;166(2):205–13. doi: 10.1016/j.jsb.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Calcedo R, Bell P, Lin J, Grant RL, Siegel DL, Wilson JM. Impact of pre-existing immunity on gene transfer to nonhuman primate liver with adeno-associated virus 8 vectors. Hum Gene Ther. 2011;22(11):1389–401. doi: 10.1089/hum.2011.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Calcedo R, Wang H, Bell P, Grant R, Vandenberghe LH, Sanmiguel J, Morizono H, Batshaw ML, Wilson JM. The pleiotropic effects of natural AAV infections on liver-directed gene transfer in macaques. Mol Ther. 2010;18(1):126–34. doi: 10.1038/mt.2009.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikoff WR, Wang G, Parrish CR, Cheng RH, Strassheim ML, Baker TS, Rossmann MG. The structure of a neutralized virus: canine parvovirus complexed with neutralizing antibody fragment. Structure. 1994;2(7):595–607. doi: 10.1016/s0969-2126(00)00062-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wistuba A, Weger S, Kern A, Kleinschmidt JA. Intermediate of Adeno-Associated Virus Type 2 Assembly: Identification of Soluble Complexes Containing Rep and Cap Proteins. Journal of Virology. 1995;69:5311–5319. doi: 10.1128/jvi.69.9.5311-5319.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wobus CE, Hugle-Dorr B, Girod A, Petersen G, Hallek M, Kleinschmidt JA. Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: epitope mapping and identification of capsid domains involved in AAV-2-cell interaction and neutralization of AAV-2 infection. J Virol. 2000;74(19):9281–93. doi: 10.1128/jvi.74.19.9281-9293.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, Xiao W, Conlon T, Hughes J, Agbandje-McKenna M, Ferkol T, Flotte T, Muzyczka N. Mutational analysis of the adeno-associated virus type 2 (AAV2) capsid gene and construction of AAV2 vectors with altered tropism. J Virol. 2000;74(18):8635–47. doi: 10.1128/jvi.74.18.8635-8647.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Asokan A, Samulski RJ. Adeno-associated virus serotypes: vector toolkit for human gene therapy. Mol Ther. 2006;14(3):316–27. doi: 10.1016/j.ymthe.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Xiao C, Rossmann MG. Interpretation of electron density with stereographic roadmap projections. J Struct Biol. 2006 doi: 10.1016/j.jsb.2006.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, Chirmule N, Berta SC, McCullough B, Gao G, Wilson JM. Gene therapy vectors based on adeno-associated virus type 1. J Virol. 1999;73(5):3994–4003. doi: 10.1128/jvi.73.5.3994-4003.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Li J, McCown TJ, Samulski RJ. Gene transfer by adeno-associated virus vector into the central nervous system. Experimental Neurology. 1997;144(1):113–124. doi: 10.1006/exnr.1996.6396. [DOI] [PubMed] [Google Scholar]
- Xie Q, Bu W, Bhatia S, Hare J, Somasundaram T, Azzi A, Chapman MS. The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc Natl Acad Sci U S A. 2002;99(16):10405–10. doi: 10.1073/pnas.162250899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Q, Hare J, Turnigan J, Chapman MS. Large-scale production, purification and crystallization of wild-type adeno-associated virus-2. J Virol Methods. 2004;122(1):17–27. doi: 10.1016/j.jviromet.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Xie Q, Lerch TF, Meyer NL, Chapman MS. Structure-function analysis of receptor-binding in adeno-associated virus serotype 6 (AAV-6) Virology. 2011;420(1):10–9. doi: 10.1016/j.virol.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.