

Abstract
As a continuation of our studies aimed at the development of a new cytostatic agent derived from an Amaryllidaceae alkaloid lycorine, we synthesized 32 analogues of this natural product. This set of synthetic analogues included compounds incorporating selective derivatization of the C1 versus C2 hydroxyl groups, aromatized ring C, lactamized N6 nitrogen, dihydroxylated C3-C3a olefin functionality, transposed olefin from C3-C3a to C2-C3 or C3a-C4, and C1 long-chain fatty esters. All synthesized compounds were evaluated for antiproliferative activities in vitro in a panel of tumor cell lines including those exhibiting resistance to proapoptotic stimuli and representing solid cancers associated with dismal prognoses, such as melanoma, glioblastoma, and non-small-cell lung cancer. Most active analogues were not discriminatory between cancer cells displaying resistance or sensitivity to apoptosis, indicating that these compounds are thus able to overcome the intrinsic resistance of cancer cells to pro-apoptotic stimuli. 1,2-Di-O-allyllycorine was identified as a lycorine analogue, which is 100 times more potent against a U373 human glioblastoma model than the parent natural product. Furthermore, a number of synthetic analogues were identified as promising for the forthcoming in vivo studies.
Keywords: cancer, apoptosis resistance, lycorine, melanoma, glioblastoma, alkaloid
1. Introduction
Glioma, melanoma, oesophageal, non-small-cell lung, and a number of other types of cancer known to be associated with dismal prognoses, all have the characteristic of being intrinsically resistant to proapoptotic stimuli.1–4 Moreover, more than 90% of cancer patients die from tumor metastases, which are incompetent in initiating the apoptotic program as well. Nonetheless, a large majority of therapeutic agents used by oncologists to treat cancer patients are pro-apoptotic.1,2 Thus, the rise in incidence of gliomas and melanomas has not been paralleled by improved therapeutic options over the years. New types of drugs are, therefore, urgently needed to combat cancers that are resistant to proapoptotic stimuli and thus poorly responsive to current therapies.
Lycorine, a phenanthridine alkaloid isolated from various species of the Amaryllidaceae plant family, has attracted considerable attention due to its promising biological activities.5 Specifically, its anticancer properties have been evaluated in vitro and in vivo in various preclinical models of human cancers.6 These promising biological results have led to some exploration of structure-activity relationships (SAR) through isolation of lycorine's natural congeners and synthesis of its analogues.6e,f,h,7 Recently, we reported biochemical experiments providing a mechanistic insight into lycorine's antiproliferative effects.7a We showed that at therapeutic concentrations, lycorine does not induce apoptosis in cancer cells, but rather exhibits cytostatic effects through the targeting of the actin cytoskeleton. This mode of action accounts for its promising activity in vitro against a panel of cancer cell lines, resistant to proapoptotic stimuli, as well as in vivo in a mouse melanoma model. Additionally, we provided considerable evidence supporting the eEF1A elongation factor as a likely intracellular target for the structurally related Amaryllidaceae isocarbostyrils, identifying this protein as a potential target for lycorine as well.8 These results make lycorine an excellent lead for the generation of compounds able to combat cancers, which are naturally resistant to pro-apoptotic stimuli, such as glioblastoma, melanoma, non-small-cell-lung and metastatic tumors, among others. In this article, we describe the synthesis of 32 lycorine analogues and their evaluation against a panel of cancer cell lines diversely representing these challenging types of human malignancy.
2. Results and discussion
2.1. Chemistry
Because lycorine is poorly soluble in most organic solvents, the initial derivatization efforts centered on the use of its diacetate 2 (Scheme 1). For the purposes of a systematic SAR study, it was decided to prepare some of the previously reported lycorine analogues together with the new derivatives. Thus, diol 3 had been synthesized previously on two occasions by a low-yielding reaction of diacetate 2 with KMnO4.5b,9 In our hands, the synthetically useful yield of 3 was achieved through the use of the OsO4/NMO system. The aromatized derivative 4, obtained as a minor byproduct, was further transformed to phenol 5. The known reaction of 2 with PhI(OAc)2 provided lactam 6,10 which was hydrolyzed to lactam diol 7 and additionally converted to a new derivative, thiolactam 8. Furthermore, diol 3 was used to synthesize new polyhydroxylated derivatives 9, 10 and 13 through the application of basic hydrolysis (9), LiAlH4 reduction (10) and secondary hydroxyl acetylation/elimination/hydrolysis sequence9 (11→12→13), respectively (Scheme 2).
Scheme 1.

Synthesis of known and new derivatives of lycorine starting with diacetate 2
Scheme 2.

Synthesis of a new polyhydroxylated analogue 13
Next, we investigated various methods to selectively functionalize either the C1- or C2-hydroxyl groups. We expected the C2-hydroxyl to be more reactive both due to its allylic nature and steric accessibility. Indeed, it was previously reported that the C2-position could be selectively oxidized with either Jones reagent5b or the DCC/DMSO system,11 albeit in rather low yields. We found that the Dess-Martin reagent afforded the desired ketone 14 in a useful 66% yield and exclusive C2-regioselectivity (Scheme 3). In addition, although selective silylation of the C2-hydroxyl with t-BuMe2SiCl described by McNulty and coworkers5a did not work in our hands (possibly due to differences in solubility of different crystalline forms of 1), we were able to prepare C2-pivaloate 15, C2-tri-iso-propylsilyl ether 16 and C2-benzoate 19 with exclusive regioselectivity. We also found that the C1- and C2-hydroxyls could be differentiated by a controlled hydrolysis of dibenzoate 17 to give corresponding monobenzoates 18 and 19 (Scheme 4).
Scheme 3.
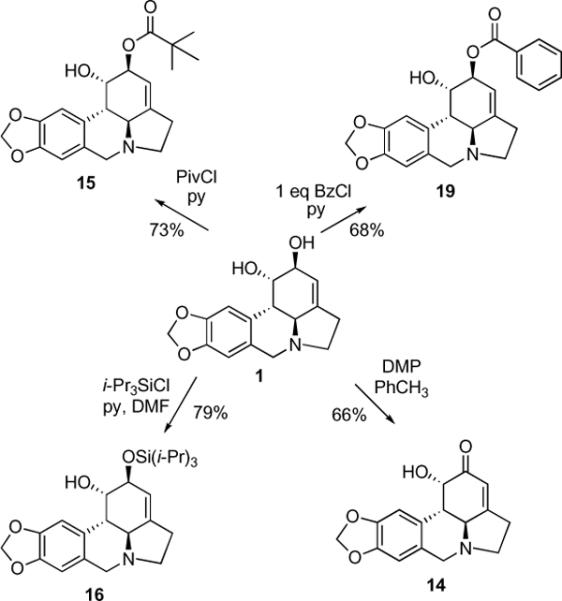
Selective functionalization of the C1 and C2-hydroxyl functionality
Scheme 4.
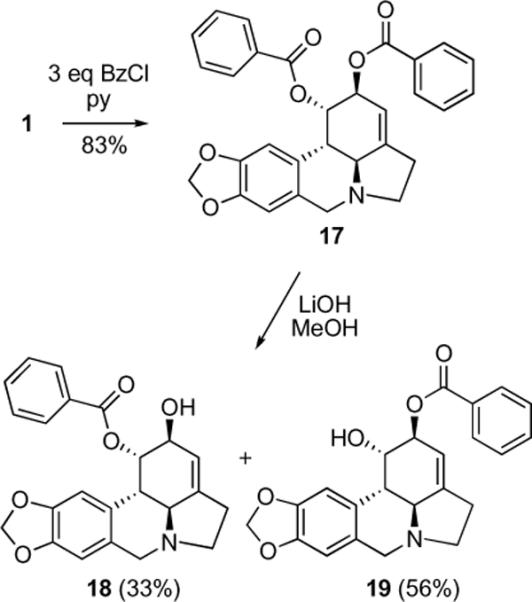
Preparation of C1- and C2-benzoates
Utilizing the facile selective protection of the C2-hydroxyl as the tri-iso-propylsilyl ether, a number of C1-derivatives were obtained. To this end, lycorine (1) was treated with i-Pr3SiCl and pyridine in DMF and the crude silyl ether was acylated or alkylated. Without purification this material was then desilylated to produce palmitate 22, acetate 23, stearate 24, oleate 25, docosahexaenoate 26, and benzyl ether 27 (Figure 1). Intermediate silyl ethers 20 and 21 were also isolated and purified for biological testing.
Figure 1.
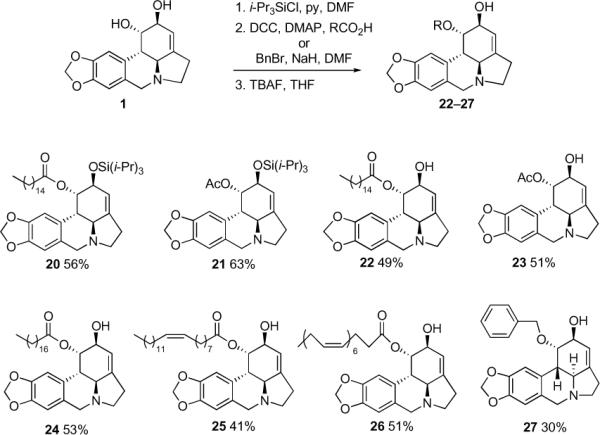
Lycorine derivatives obtained through selective protection of the C2-hydroxyl [% yields are from lycorine (1)]
In an attempt to prepare aromatic lycorine derivatives, we found that a simple treatment of lactam 7 with thionyl chloride in pyridine produced indoline lactam 28 (Scheme 5), which was previously synthesized by total synthesis efforts and converted to indole lactam 29 by oxidation with DDQ.12 This oxidation reproduced well in our hands and was applied to prepare indole lactam 30 with equal success.
Scheme 5.
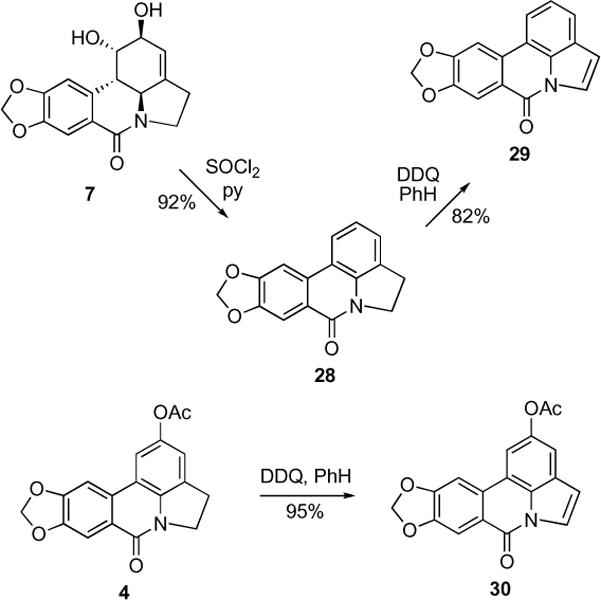
Synthesis of aromatic derivatives
Lastly, conversion of the C1- and C2-hydroxyl functionality into lithium and sodium bis-alkoxides allowed us to synthesize di-Boc carbonate 31 and diallyl ether 32 without the competing quaternization of the basic nitrogen (Scheme 6). The attempted Claisen rearrangement using ethyl orthoacetate and propionic acid did not lead to the desired product containing a new quaternary center at C3a and a transposed olefin at C2-C3. Instead, dipropionate 33 was isolated from this reaction mixture as the only product. We wondered whether this unsuccessful outcome was caused by a competing formation of the cyclic orthoester at the C1 and C2-hydroxyl functionality. Therefore, this reaction was performed on C1-protected acetate 23 and, gratifyingly gave the desired rearranged product 34 under slightly modified conditions in a reasonable yield of 38%.
Scheme 6.
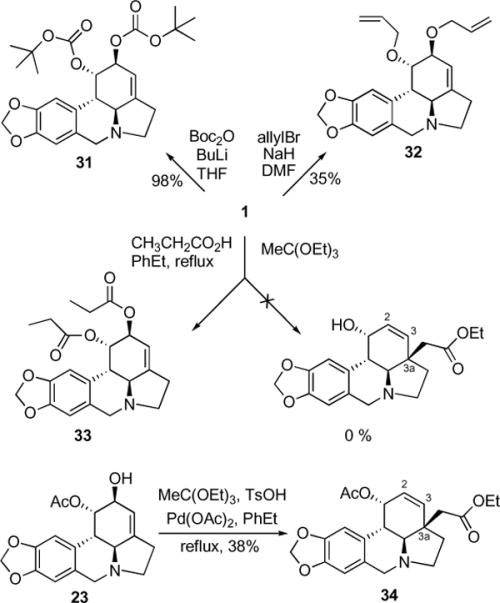
Synthesis of dicarbonate and diether derivatives and C3a-quaternization/olefin transposition using Claisen rearrangement
2.2. In vitro growth inhibitory properties
The synthesized compounds were evaluated for in vitro growth inhibition using the MTT colorimetric assay13 against a panel of eight cancer cell lines including those resistant to proapoptotic stimuli [human U373 and T98G glioblastoma (GBM, from astroglial origin14),13b,15a,15b human A549 non-small-cell-lung cancer (NSCLC),13b,16 and human SKMEL-28 melanoma17] as well as apoptosis-sensitive tumor models (human Hs683 anaplastic oligodendroglioma,13b,15a human LoVo colon cancer15c,15d and mouse B16F10 melanoma).17,18 Analysis of these data reveals that lycorine, as well as most of its active derivatives, does not discriminate between the cancer cell lines based on the apoptosis sensitivity criterion and displays comparable potencies in both cell types, further corroborating our conclusion that apoptosis induction is not the primary mechanism responsible for antiproliferative activity in this series of compounds, at least in solid cancers (Table, Figure 2).7a Although the large variations in cancer cell line sensitivities preclude reliable statistical analyses to be performed, compound 23 stands out as uniformly potent in inhibiting the growth of apoptosis-resistant cancer cells (Figure 2).
Figure 2.
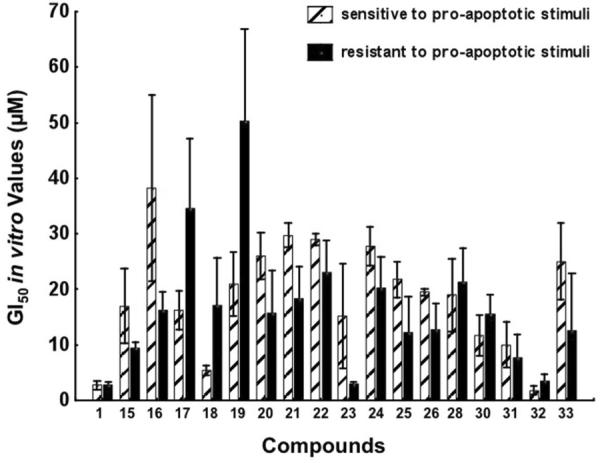
Illustration of the in vitro growth inhibitory effects of lycorine and 17 of its derivatives in four cancer cell lines that are sensitive to proapoptotic stimuli (the hatched bars) versus four cancer cell lines that display resistance to pro-apoptotic stimuli (the black bars). The data are presented as mean ± SEM values. We included in the present analysis all the compounds displaying GI50 values < 100 μM in each of the eight cancer cell lines under study (see Table).
The data also indicate the absolute requirement for the presence of a basic nitrogen at N6, as the compounds oxidized at C7, and thus containing amidic nitrogen at this position, are inactive (3, 6, 7, 9, 11, 12 and 13). In the same vein, thiolactam 8 also displays reduced activity. Exceptions to this rule constitute compounds with an aromatic ring C (4, 5, 28, 29 and 30) and the resultant planarity of the pentacyclic skeleton.
The sterically bulky substituents at the C1- and C2-positions appear to be tolerated well, provided they can be removed by intracellular hydrolysis. Such compounds include dipropionate 33, dicarbonate 31 and the benzoate substituted analogues (17, 18 and 19). Of interest is the comparison of ester 17 with ether 27, where the significantly higher potency of the hydrolyzable analogue 17 leads to a proposal that these large hydrophobic elements are not part of the pharmacophore but only assist in cell penetration. This is also consistent with the observation that tetraol 7, lying at the other end of the polarity spectrum, is inactive. A possible exception is represented by moderately active C2-silyl ethers (16, 20 and 21), which could be resistant to intracellular hydrolysis.
The idea behind synthesizing the long-chain fatty acid derivatives was to make these molecules selectively toxic to tumors as opposed to normal tissues. Certain fatty acids have shown in vitro and in vivo growth inhibitory effect against cancer cells19 and, more importantly, many natural fatty acids are selectively taken up by tumors, presumably for use as biochemical precursors and energy sources.20 This phenomenon has been utilized by Bradley and co-workers to prepare a paclitaxel-docosahexanoic acid conjugate, which showed enhanced antitumor activity in mice compared to that of paclitaxel itself.21 Furthermore, Ahn and co-workers have synthesized a series of saturated and unsaturated fatty acid esters of 4'-demethyldeoxypodophyllotoxin and shown that many of such derivatives possessed enhanced anticancer activities.22 Although analysis of the GI50 values associated with palmitate 22, stearate 24, oleate 25 and docosahexaenoate 26 reveals that these compounds do not display elevated potencies compared with those of lycorine (1) in vitro, the low micromolar potencies associated with these compounds and their chemical stability in the range of pH 5–7.4 for at least 3 days are encouraging and bode well for the forthcoming in vivo tests where their propensity for selective uptake into tumors can be properly evaluated.
Importantly, diallyllycorine 32 is equipotent to lycorine (1) across the entire panel of cell lines and as much as 100 times more potent against the apoptosis-resistant U373 glioblastoma. Because it is unlikely that the allyl ether functionality is removed by intracellular hydrolysis, this structural element must be part of the lycorine pharmacophore and a successful computer-based theoretical model must be able to account for this result.
The calculated log P values for each synthesized analogue are also listed in the Table. Although a direct correlation between the lipophilicity and anticancer activity cannot be easily seen, it is noteworthy that compounds with clog P values lower than those of lycorine are all inactive (see 3, 7, 9, 10, 11, 13). It is likely that this observation reflects the difficulty of cell penetration for highly hydrophilic lycorine analogues and it provides useful guidelines for future analogue synthesis.
3. Conclusions
The synthesis and evaluation of 32 analogues of lycorine revealed structural elements necessary for antiproliferative activity in this series of compounds. 1,2-Di-O-allyllycorine was identified as a lycorine analogue, which is 100 times more potent against an U373 human glioblastoma model in vitro. Because active lycorine derivatives are equally potent toward the cell lines exhibiting resistance to proapoptotic stimuli, this finding further corroborates our conclusion that apoptosis induction is not the primary mechanism responsible for the antiproliferative activity in this series of compounds and bodes well for the discovery of a lycorine-derived agent able to combat such malignancies as glioblastoma, melanoma, non-small-cell-lung, metastatic cancers, among others. The synthetic chemistry findings and the new SAR insights described in this article are a valuable step toward this ambitious goal.
4. Experimental
4.1. Chemistry
All reagents, solvents and catalysts were purchased from commercial sources (Acros Organics and Sigma-Aldrich) and used without purification. CH2Cl2 was distilled from CaH2 and kept under argon; THF and toluene were distilled from sodium/benzophenone prior to use. Lycorine was isolated from dried bulbs of Sterbergia lutea Ker Gawl according to a published procedure23 and its purity was confirmed by NMR and HRMS. All reactions were performed in oven-dried flasks under nitrogen or argon and monitored by thin layer chromatography on TLC precoated (250 μm) silica gel 60 F254 glass-backed plates (EMD Chemicals Inc.). Visualization was accomplished with UV light. Flash column chromatography was performed on silica gel (32–63 μm, 60 Å pore size). 1H and 13C NMR spectra were recorded on Jeol Eclipse 300 or Bruker Avance III 400 spectrometers. Chemical shifts (δ) are reported in ppm relative to the TMS internal standard. HRMS analyses were performed at the Mass Spectrometry Facility, University of New Mexico. Samples were run on LCT Premier TOF mass spec.
4.1.1. (1S,2S,3a1S,12bS)-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridine-1,2-diyl diacetate (2)
This compound was prepared using a published procedure.24
4.1.2. Dihydroxylation of the C3-C3a olefin
Diacetyl lycorine (2) (0.096 g, 0.258 mmol) was dissolved in acetone (6 mL) and to this solution of OsO4 (0.25 mL, 2.5 % wt solution in t-BuOH), 0.038 g of NMO (0.284 mmol) of water (1.5 mL) were added over 5 min. The solution was stirred at room temperature for 14 hrs, and then the volatiles were evaporated under reduced pressure. To the obtained slurry were added EtOAc (20 mL) and saturated NH4Cl (5 mL), followed by washing with saturated NaCl (10 mL). The extraction was repeated twice with EtOAc (15 mL) and the combined organic phases were dried with MgSO4. The residue after evaporation of the solvent was subjected to silica gel column chromatography (CH2Cl2, then CH2Cl2/MeOH = 96/4) yielded 3 and 4.
(1S,2S,3S,3aR,3a1S,12bS)-3,3a-dihydroxy-7-oxo-2,3,3a,3a1,4,5,7,12b-octahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridine-1,2-diyl diacetate (3)
53.2% (0.0577 g) as light yellow viscous oil. 1H and 13C NMR were identical to those published in the literature.5b
7-Oxo-5,7-dihydro-4H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridin-2-yl acetate (4)
12.9% as light yellow viscous oil. 1H and 13C NMR were identical to those published in the literature.5b
4.1.3. General procedure for acetate hydrolysis to obtain 5, 7, 9, 13
A fresh 0.5 M solution of LiOH in THF/H2O (1/1) was used and its volume was adjusted to 1.05 equivalents of LiOH per acetate residue in the starting material (4, 6, 3, 12). The acetates were dissolved in the required volume of LiOH solution and stirred overnight at 18 °C. After that time the two-layered solution became uniform and 0.05 g of Aberlyst 15(dry) ion-exchange resin (ACROS) were added and stirred for 1 h. The resin was filtered (glass filter) and washed on filter with a mixture of MeOH/CH2Cl2 (1/1, 5 mL). Toluene (15 mL) was added to the obtained mother liquor and evaporated to dryness to obtain pure deacetylated products 5, 7, 9, and 13.
2-Hydroxy-4H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridin-7(5H)-one (5)
93%. 1H and 13C NMR were identical to those published in the literature.25
(1S,2S,3a1S,12bS)-1,2-dihydroxy-3a1,4,5,12b-tetrahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridin-7(2H)-one (7)
95%. 1H and 13C NMR were identical to those published in the literature.5b
(1S,2R,3S,3aR,3a1S,12bS)-1,2,3,3a-tetrahydroxy-3,3a,3a1,4,5,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridin-7(2H)-one (9)
93%. 1H NMR (300 MHz, CD3OD) δ: 7.31 (s, 1H), 6.89 (s, 1H), 6.01 (s, 2H), 4.39 (d, J = 3.0 Hz, 1H), 4.08 (dd, J = 7.4 Hz, 1H), 3.90 (dd, J = 9.1 Hz, 2H), 3.62 (d, J = 9.4 Hz, 1H), 3.48 (quintet, J = 5.8 Hz, 1H), 3.08 (d, J = 14.0 Hz, 1H), 2.14 (quintet, J = 6.0 Hz, 2H). 13C NMR (75 MHz, CD3OD) δ: 162.5, 151.0, 146.6, 134.1, 124.1, 107.1, 105.9, 101.8, 79.2, 77.0, 75.1, 71.4, 63.8, 43.6, 38.5, 22.8. HRMS m/z (ESI+) calc'd for C16H17NO7 (M+Na+) 358.0903, found 358.0905.
(1S,2R,3R,3a1S,12bS)-1,2,3-trihydroxy-2,3,5,12b-tetrahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridin-7(3a1H)-one (13)
90%. 1H NMR (300 MHz, CD3OD) δ: 7.59 (s, 1H), 6.84 (s, 1H), 6.00 (s, 2H), 5.87 (s, 1H), 4.80 (d, J = 15.6 Hz, 1H), 4.60 (d, J = 13.6 Hz, 1H), 4.50 (d, J = 12.4 Hz, 1H), 4.45 (s, 1H), 4.34 (d, J = 15.8 Hz, 1H), 4.18 (s, 1H), 3.21 (d, J = 12.9 Hz, 1H). 13C NMR (100 MHz, CD3OD) δ: 163.2, 151.1, 146.6, 139.4, 135.0, 125.2, 121.4, 107.6, 104.1, 72.4, 69.8, 68.8, 59.7, 52.0, 42.8. HRMS m/z (ESI+) calc'd for C16H15NO6 (M+Na+) 340.0797, found 340.0807.
(1S,2S,3a1S,12bS)-7-oxo-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrole[3,2,1-de]phenanthridine-1,2-diyl diacetate (6)
This compound was prepared using a published procedure.10
4.1.4. Preparation of thiolactam 8
Compound 6 (0.0385g, 0.1 mmol) was suspended in dry benzene (10 mL), then phosphorous pentasulfide (0.02 g, 0.06 mmol) and hexamethyldisiloxane (0.02 g, 0.16 mmol) was added. The mixture was refluxed under N2 for 2 hours, after that solvent was removed under reduced pressure. The crude residue was purified by PTLC (CH2Cl2:MeOH=99/1, Rf = 0.45) to give 0.035 g (88%) of 8 as a yellow solid.
(1S,2S,3a1S,12bS)-7-thioxo-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridine-1,2-diyl diacetate (8)
1H NMR (300 MHz, CDCl3) δ: 8.01 (s, 1H), 6.60 (s, 1H), 5.98 (d, J = 8.0 Hz, 2H), 5.75 (s, 1H), 5.66 (s, 1H), 5.27 (d, J = 1.4 Hz, 1H), 4.24-3.95 (m, 3H), 3.06 (dd, J = 1.9 Hz, J = 12.1 Hz, 1H), 2.89 (t, J = 8.6 Hz, 2H), 2.08 (s, 3H), 2.03 (s, 3H). 13C NMR (75 MHz, CDCl3) δ: 194.7, 169.9, 151.1, 147.0, 142.8, 131.1, 127.7, 118.9, 116.4, 112.5, 102.7, 74.7, 69.8, 67.2, 56.9, 51.1, 39.5, 28.6, 21.1. HRMS m/z (ESI+) calc'd for C20H19NO6S (M+H+) 402.1011, found 402.1025.
4.1.5. Synthesis of Compound 10
LiAlH4 (0.01 g, 0.26 mmol) was dissolved in 2 mL of THF and compound 3 (0.011 g, 0.026 mmol) was added to this solution. The mixture was refluxed for 5 hours under argon, after that quenched with brine (6 mL) and extracted with EtOAc (3 × 10 mL). The solvent was evaporated under the reduced pressure to afford 0.0056 g (66%) of derivative 10 as white flakes.
(1S,2R,3S,3aR,3a1S,12bS)-2,3,3a,3a1,4,5,7,12b-octahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridine-1,2,3,3a-tetraol (10)
1H NMR (300 MHz, CD3OD) δ: 6.95 (s, 1H), 6.76 (s, 1H), 5.94 (s, 2H), 4.51 (d, J = 13.3 Hz, 1H), 4.21 (d, J = 15.1 Hz, 1H), 4.11 (m, 1H), 3.75 (d, J = 5.2 Hz, 1H), 3.57 (m, 1H), 3.15 (d, J = 8.7 Hz, 1H), 3.09 (d, J = 8.7 Hz, 1H). 13C NMR (100 MHz, CD3OD) δ: 151.9, 150.9, 148.4, 126.6, 107.4, 105.8, 101.6, 78.6, 69.7, 69.0, 61.4, 54.5, 52.5, 39.1, 35.3, 28.8. HRMS m/z (ESI+) calc'd for C16H19NO6 (M+H+) 322.1281, found 322.1291.
(1S,2R,3S,3aR,3a1S,12bS)-3a-hydroxy-7-oxo-2,3,3a,3a1,4,5,7,12b-octahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridine-1,2,3-triyl triacetate (11)
This compound was prepared using a published procedure.9
(1S,2R,3R,3a1S,12bS)-7-oxo-2,3,3a1,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrole[3,2,1-de]phenanthridine-1,2,3-triyl triacetate (12)
This compound was prepared using a published procedure.9
4.1.6. Synthesis of Compound 14
An oven-dry flask was charged with lycorine (1) (0.0143 g, 0.049 mmol) and Dess-Martin periodinane (0.044 g, 0.103 mmol) and then purged with argon. Dry toluene (2 mL) was then added and the reaction suspension was heated for 2 hours (complete consumption of 1 by TLC). The mixture was cooled to rt and diluted with EtOAc (10 mL), followed by washing with saturated NaHCO3 (15 mL) and brine, drying over MgSO4, and concentration under reduced pressure. The residue was purified on silica gel column with CH2Cl2:MeOH=9/1 as an eluent to isolate 0.0093 g (66%) of 14 (Rf = 0.63).
(1S,3a1S,12bS)-1-hydroxy-4,5,7,12b-tetrahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridin-2(3a1H)-one (14)
Pink amorphous solid. 1H and 13C NMR were identical to those published in the literature.7a
4.1.7. Discrimination of the C1 and C2-hydroxyls, synthesis of compounds 15 and 19
Lycorine (1) (0.014 g, 0.049 mmol) was mixed with pyridine (0.5 mL) and DMAP (0.0001 g). To the obtained mixture pivaloyl chloride (0.0066 mL, 0.054 mmol) or benzoyl chloride (0.0063 mL, 0.054 mmol) was added in one portion at rt and then stirred for 12 h under the argon. The residue obtained after rotary evaporation was separated by PTLC to afford 15 (CH2Cl2:MeOH=49/1; Rf = 0.49) or 19 (CH2Cl2:MeOH=97/3; Rf = 0.40).
(1S,2S,3a1S,12bS)-1-hydroxy-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridin-2-yl pivalate (15)
0.013 g (73%) as a yellow oil. 1H NMR (300 MHz, CDCl3) δ: 6.79 (s, 1H), 6.60 (s, 1H), 5.93 (d, J = 6.3 Hz, 2H), 5.46 (s, 1H), 5.43 (s, 1H), 4.45 (s, 1H), 4.14 (d, J = 14.3 Hz, 1H), 3.56 (d, J = 14.3 Hz, 1H), 3.38 (q, 1H), 2.81 (d, J = 9.1 Hz, 1H), 2.69 (m, 3H), 1.19 (s, 9H). 13C NMR (100 MHz, CDCl3) δ: 178.3, 146.6, 146.4, 145.8, 129.9, 127.1, 113.7, 107.8, 104.5, 101.1, 73.2, 69.4, 60.7, 57.7, 53.7, 41.0, 39.0, 29.0, 27.9. HRMS m/z (ESI+) calc'd for C21H25NO5 (M+H+) 372.1811, found 372.1812.
(1S,2S,3a1S,12bS)-1-hydroxy-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridin-2-yl benzoate (19)
0.0136 g (68%) as a yellow oil. 1H NMR (300 MHz, CDCl3) δ: 8.05 (d, J = 7.4 Hz, 2H), 7.54 (q, J = 7.4 Hz, 1H), 7.43 (t, J = 7.7 Hz, J = 15.1 Hz, 2H), 6.84 (s, 1H), 6.63 (s, 1H), 5.92 (s, 2H), 5.65 (s, 1H), 5.59 (s, 1H), 4.70 (s, 1H), 4.21 (t, J = 3.6 Hz, J = 12.4 Hz, 1H), 3.66 (d, J = 3.4 Hz, 1H), 2.76 (s, 1H), 2.30 (d, J = 7.4 Hz, 1H), 2.02 (s, 1H), 1.40 (d, J = 7.0 Hz, 2H), 1.21 (d, J = 7.4 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 166.1, 149.2, 147.5, 147.3, 133.8, 129.7, 128.8, 128.5, 118.0, 108.8, 105.8, 101.2, 73.3, 68.2, 53.6, 53.3, 40.5, 29.7. HRMS m/z (ESI+) calc'd for C23H21NO5 (M+H+) 392.1498, found 392.1486.
4.1.8. Synthesis of Compound 16
To a solution of lycorine (1) (0.0287 g, 0.1 mmol) in DMF (0.22 mL) under argon, pyridine (0.025 mL, 0.31 mmol), AgNO3 (0.0685 g, 0.4 mmol) and tri-iso-propysilyl chloride (0.042 mL, 0.2 mmol) were added at 20 °C and the reaction was kept at this temperature in a dark place for 4 hours. Then saturated NH4Cl solution (5 mL) was added and organic phase was extracted with Et2O (3 × 10), washed with brine (10 mL). The ether was then removed under reduced pressure and crude was separated by flash column chromatography (CHCl3:MeOH:NH4OH=95/5/0.5) to give 0.0351 g (79%) of compound 16 (Rf = 0.48).
(1S,2S,3a1S,12bS)-2-(triisopropylsilyloxy)-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridin-1-ol (16)
1H NMR (300 MHz, CDCl3) δ: 6.81 (s, 1H), 6.59 (s, 1H), 5.92 (s, 2H), 5.50 (s, 1H), 4.50 (s, 1H), 4.39 (s, 1H), 4.16 (d, J = 13.8 Hz, 1H), 3.51 (d, J = 13.6 Hz, 1H), 3.34 (m, 1H), 2.86 (d, J = 12.9 Hz, 1H), 2.73 (d, J = 12.7 Hz, 1H), 2.61 (m, 2H), 2.35 (t, J = 9.0 Hz, J = 17.0 Hz, 1H), 1.09 (s, 21H). 13C NMR (75 MHz, CDCl3) δ: 146.7, 146.3, 141.8, 130.5, 128.0, 118.5, 107.9, 104.5, 101.1, 72.6, 72.3, 61.1, 57.3, 54.0, 41.1, 28.7, 18.2. HRMS m/z (ESI+) calc'd for C25H37NO4Si (M+H+) 444.2570, found 444.2576.
4.1.9. Synthesis of Compound 17
To a suspension of lycorine (1) (0.0287 g, 0.1 mmol) and DMAP (0.0005 g) in dry pyridine (1.5 mL) was added benzoyl chloride (0.038 mL, 0.33 mmol) at rt. Reaction was stirred at 40 °C for 5 hours, after that time it was diluted with toluene (20 mL) and evaporated on the rotary evaporator. The residue was dissolved in CH2Cl2 (20 mL) and washed with brine (10 mL) and dried over MgSO4. The solution was concentrated and purified by column chromatography (CH2Cl2) yielding 0.04 g (83%) of compound 17.
(1S,2S,3a1S,12bS)-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridine-1,2-diyl dibenzoate (17)
White amorphous material. 1H NMR (300 MHz, CDCl3) δ: 8.08 (d, J = 8.0 Hz, 2H), 7.91 (d, J = 8.0 Hz, 2H), 7.56-7.37 (m, 6H), 6.86 (s, 1H), 6.58 (s, 1H), 6.15 (s, 1H), 5.87 (s, 1H), 5.84 (s, 1H), 5.69 (s, 2H), 4.24 (d, J = 14.0 Hz, 1H), 3.67 (d, J = 14.1 Hz, 1H), 3.46 (s, 1H), 3.18 (d, J = 9.9 Hz, 1H), 3.12 (d, J = 9.8 Hz, 1H), 2.75 (s, 2H), 2.58 (s, 1H). 13C NMR (75 MHz, CDCl3) δ: 165.5, 165.3, 146.8, 146.5, 146.1, 133.3, 129.8, 128.4, 126.4, 113.9, 107.4, 105.1, 101.2, 70.9, 69.4, 61.7, 56.9, 53.5, 40.8, 28.9. HRMS m/z (ESI+) calc'd for C30H25NO6 (M+H+) 496.1760, found 496.1775.
4.1.10. Synthesis of Compound 18
To a solution of compound 17 (0.03 g, 0.06 mmol) in MeOH (3 mL) was added LiOH monohydrate (0.0025 g, 0.06 mmol) at rt. After 3 hours the reaction mixture was subjected to PTLC (CH2Cl2:MeOH=48/3) to isolate compound 18 (33%, Rf = 0.71) and compound 19 (56%, Rf = 0.56).
(1S,2S,3a1S,12bS)-2-hydroxy-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridin-1-yl benzoate (18)
0.007 g (33%) white amorphous solid. 1H and 13C NMR were identical to those published in the literature.5a
4.1.11. General procedure for acylation of compound 16. Synthesis of compounds 20 and 21
Compound 16 (0.032 g, 0.072 mmol) was co-evaporated with toluene (5 mL) and dissolved in CH2Cl2 (10 mL). Selected carboxylic acid (0.08 mmol), DCC (0.018 g, 0.085 mmol) and DMAP (0.0005 g) were added at 0 °C. The mixture was kept under argon at rt for 12 hours. In the case of compounds 20 and 21 the mixture was subjected to column chromatography using CHCl3 as a solvent to obtain pure 20 and 21. In the other cases, the crude material was used in the next step.
(1S,2S,3a1S,12bS)-2-(triisopropylsilyloxy)-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo [4,5-j]pyrrolo[3,2,1-de]phenanthridin-1-yl palmitate (20)
0.035 g (71%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ: 6.75 (s, 1H), 6.55 (s, 1H), 5.90 (s, 2H), 5.69 (s, 1H), 5.50 (s, 1H), 4.30 (s, 1H), 4.12 (d, J = 14.3 Hz, 1H), 3.52 (d, J = 14.1 Hz, 1H), 3.36 (m, 1H), 2.97 (d, J = 10.4 Hz, 1H), 2.76 (d, J = 10.5 Hz, 1H), 2.63 (m, 2H), 2.28 (t, J = 8.9 Hz, J = 16.8 Hz, 1H), 2.17 (t, J = 5.8 Hz, J = 14.6 Hz, 2H), 1.60 (q, 2H), 1.44 (q, J = 7.7 Hz, 2H), 1.25 (s, 21H), 1.08 (d, J = 5.5 Hz, 24H), 0.87 (t, J = 6.6 Hz, J = 13.2 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ: 173.2, 146.5, 146.2, 142.2, 129.5, 127.8, 118.4, 107.3, 105.1, 101.0, 72.0, 69.9, 61.9, 57.0, 53.9, 39.5, 34.5, 32.0, 29.8, 29.5, 28.9, 28.7, 25.1, 22.8, 18.2, 14.2. HRMS m/z (ESI+) calc'd for C41H67NO5Si (M+H+) 682.4867, found 682.4873.
(1S,2S,3a1S,12bS)-2-(triisopropylsilyloxy)-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo [4,5-j]pyrrolo[3,2,1-de]phenanthridin-1-yl acetate (21)
0.028 g (80%) as an oil. 1H NMR (300 MHz, CDCl3) δ: 6.75 (s, 1H), 6.56 (s, 1H), 5.91 (d, J = 1.4 Hz, 2H), 5.66 (s, 1H), 5.50 (s, 1H), 4.32 (s, 1H), 4.17 (d, J = 14.0 Hz, 1H), 3.54 (d, J = 14.0 Hz, 1H), 3.36 (m, 2H), 3.01 (d, J = 10.2 Hz, 1H), 2.77 (d, J = 10.2 Hz, 1H), 2.63 (m, 2H), 2.39 (t, J = 9.1 Hz, 1H), 1.93 (s, 3H), 1.08 (m, 21H). 13C NMR (75 MHz, CDCl3) δ: 170.5, 146.2, 142.3, 129.5, 127.8, 118.2, 107.4, 105.0, 100.7, 72.4, 69.8, 61.7, 57.2, 53.9, 39.4, 28.7, 21.2, 18.2. HRMS m/z (ESI+) calc'd for C27H39NO5Si (M+H+) 486.2676, found 486.2664.
4.1.12. General procedure for the removal of the TIPS-protecting group. Synthesis of compounds 22, 23, 24, 25, 26
Pure TIPS-protected 20, 21 or crude mixtures for the other C1-acylated lycorines from the procedure above (0.06 mmol) were dissolved in THF (3 mL) and TBAF (0.066 mL, 0.066 mmol, 1.0 M solution in THF) was added to the obtained solution dropwise at rt. The reaction was left for 4 hours at this temperature, then quenched with saturated solution of NH4Cl (5 mL) and extracted with EtOAc (3 × 5 mL). Column chromatography (CH2Cl2:MeOH=48/2) afforded pure C1-acylated lycorines 22, 23, 24, 25, 26.
(1S,2S,3a1S,12bS)-2-hydroxy-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrole [3,2,1-de]phenanthridin-1-yl palmitate (22). 88% as a yellow oil. 1H and 13C NMR were identical to those published in the literature.26
(1S,2S,3a1S,12bS)-2-hydroxy-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrole [3,2,1-de]phenanthridin-1-yl acetate (23). 81% as a yellow oil. 1H and 13C NMR were identical to those published in the literature.5b
(1S,2S,3a1S,12bS)-2-hydroxy-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridin-1-yl stearate (24). 67% as a viscous oil. 1H NMR (300 MHz, CDCl3) δ: 6.69 (s, 1H), 6.56 (s, 1H), 5.91 (d, J = 2.5 Hz, 2H), 5.65 (s, 1H), 5.55 (s, 1H), 4.21 (s, 1H), 4.17 (d, J = 14 Hz, 1H), 3.54 (d, J = 14 Hz, 1H), 3.36 (q, 1H), 2.89 (d, J = 10.4 Hz, 1H), 2.75 (d, J = 10.4 Hz, 1H), 2.64 (m, 2H), 2.41 (t, J = 6.8 Hz, 1H), 2.18 (t, J = 7.4 Hz, 1H), 1.43 (t, J = 6.6 Hz, 2H), 1.25 (s, 32H), 1.14 (s, 2H), 0.88 (t, J = 6.6 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ: 173.6, 146.6, 146.3, 141.8, 129.2, 127.2, 117.2, 107.4, 105.1, 101.0, 72.4, 70.0, 61.7, 57.0, 53.8, 39.6, 34.4, 32.0, 29.8, 29.5, 29.3, 28.8, 25.1, 22.8, 14.2. HRMS m/z (ESI+) calc'd for C34H51NO5 (M+H+) 554.3845, found 554.3854.
(1S,2S,3a1S,12bS)-2-hydroxy-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridin-1-yl oleate (25). 52% as a colorless oil. 1H NMR (300 MHz, CDCl3) δ: 6.68 (s, 1H), 6.56 (s, 1H), 5.90 (s, 2H), 5.64 (s, 1H), 5.55 (s, 1H), 5.34 (d, J = 3.84 Hz, 2H), 4.19 (s, 1H), 4.12 (d, J = 14.5 Hz, 1H), 3.53 (d, J = 14.5 Hz, 1H), 3.34 (q, J = 9.1 Hz, 1H), 2.85 (d, J = 9.6 Hz, 1H), 2.76 (d, J = 9.6 Hz, 1H), 2.63 (m, 2H), 2.40 (t, J = 8.8 Hz, 1H), 2.17 (t, J = 7.1 Hz, J = 14.5 Hz, 2H), 1.99 (m, 4H), 1.45 (t, J = 7.1 Hz, J = 13.5 Hz, 2H), 1.26 (s, 20H), 1,15 (s, 2H), 0.87 (t, J = 5.5 Hz, J = 12.1 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ: 173.6, 146.5, 144.5, 130.1, 129.8, 129.5, 127.2, 117.2, 107.3, 105.1, 101.0, 72.4, 69.9, 61.7, 57.0, 53.8, 39.6, 34.4, 32.0, 29.9, 29.6, 29.4, 29.2, 27.3, 25.0, 22.8, 14.2. HRMS m/z (ESI+) calc'd for C34H49NO5 (M+H+) 552.3689, found 552.3691.
(4Z,7Z,10Z,13Z,16Z,19Z)-((1S,2S,3a1S,12bS)-2-hydroxy-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridin-1-yl) docosa-4,7,10,13,16,19-hexaenoate (26). 65% as an oil. 1H NMR (300 MHz, CDCl3) δ: 6.66 (s, 1H), 6.55 (s, 1H), 5.90 (s, 2H), 5.63 (s, 1H), 5.53 (s, 1H), 5.37 (m, 12H), 4.18 (s, 1H), 4.11 (d, J = 14.0 Hz, 1H), 3.52 (d, J = 14.0 Hz, 1H), 3.36 (q, J = 4.1 Hz, 1H), 2.84–2.62 (m, 16H), 2.24 (m, 3H), 2.74 (t, J = 7.4 Hz, 1H), 0.96 (t, J = 7.4 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ: 172.8, 146.6, 146.3, 143.8, 129.3, 128.7, 128.3, 128.2, 127.1, 117.5, 107.4, 105.1, 101.0, 72.7, 69.6, 61.7, 56.9, 53.8, 39.4, 34.2, 28.7, 25.7, 25.6, 22.8, 20.6, 14.4. HRMS m/z (ESI+) calc'd for C34H47NO5 (M+H+) 598.3532, found 598.3538.
4.1.13. Synthesis of compound 27
Compound 16 (0.032 g, 0.072 mmol) was dissolved in dry DMF (0.5 mL). To the obtained solution under argon at 20 °C were added NaH (0.008 g, 0.2 mmol, 60% suspension in mineral oil) and after 10 min benzyl bromide (0.012 mL, 0.1 mmol). The reaction was left stirring for 20 hours at rt. The mixture was diluted with 20 ml of 2-propanol and solvents were removed at reduced pressure. Remaining residue was dissolved in THF (3 mL) and TBAF (0.066 mL, 0.066 mmol, 1.0 M solution in THF) was added to the obtained solution dropwise at rt. The reaction was left for 4 hours at this temperature, then quenched with saturated solution of NH4Cl (5 mL) and extracted with EtOAc (3 × 5 mL). Column chromatography (CH2Cl2:MeOH=48/2) afforded pure 27.
(1S,2S,3a1S,12bS)-1-(benzyloxy)-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridin-2-ol (27). 38% as a yellow oil. 1H NMR (300 MHz, CDCl3) δ: 7.28–7.25 (m, 5H), 6.57 (s, 1H), 6.49 (s, 1H), 5.92 (s, 2H), 5.63 (s, 1H), 4.66 (d, J = 11.8 Hz, 1H), 4.42 (s, 1H), 4.27 (s, 1H), 4.13 (d, J = 14.3 Hz, 1H), 3.41 (m, 1H), 4.63 (d, J = 13.9 Hz, 1H), 2.67 (s, 2H). 13C NMR (100 MHz, CDCl3) δ: 149.8, 146.0, 144.4, 137.9, 135.4, 128.3, 128.2, 128.1, 128.0, 127.7, 125.6, 117.9, 107.2, 105.3, 101.0, 72.2, 72.0, 67.9, 53.5, 52.6, 50.4, 29.4. HRMS m/z (ESI+) calc'd for C23H23NO4 (M+H+) 378.1705, found 378.1713.
4.1.14. Synthesis of compound 28
To a solution of compound 7 (0.0141 g, 0.0468 mmol) in 0.5 mL of dry pyridine at 20 °C was added thionyl chloride (0.1 mL). After 4 hours of stirring at 20 °C the mixture was separated by PTLC (CH2Cl2:MeOH=49/1) give 0.011 g (92%) of compound 28 (Rf = 0.51) as a white solid.
4H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridin-7(5H)-one (28). 92%; 1H and 13C NMR were identical to those published in the literature.12
4.1.15. Synthesis of compounds 29 and 30
Selected compound 28 (0.0048 g, 0.018 mmol) or 4 (0.0058 g, 0.018 mmol) was refluxed under N2 in dry benzene (1 mL) with DDQ (0.126 mmol) for 24 hours. After that time the reaction mixtures were diluted with EtOAc (7 mL) and washed with saturated aqueous solution of Na2SO3 (6 mL), then saturated solution of NaHCO3 (6 mL) followed by brine (3 mL). The organic phase was concentrated in vacuum and subjected to PTLC (CH2Cl2:MeOH=99/1) to afford pure compounds 29 and 30 respectively.
7H-[1,3]Dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridin-7-one (29). 0.0039 g (82%) as a white solid. 1H and 13C NMR were identical to those published in the literature.12
7-Oxo-7H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridin-2-yl acetate (30). 0.0054 g (95%) as a white solid. 1H NMR (300 MHz, CDCl3) δ: 8.06 (d, J = 3.6 Hz, 1H), 7.97 (s, 1H), 7.62 (d, J = 1.7 Hz, 1H), 7.55 (s, 1H), 7.46 (d, J = 1.7 Hz, 1H), 6.87 (d, J = 3.6 Hz, 1H), 6.17 (s, 2H), 2.39 (s, 3H). 13C NMR (75 MHz, CDCl3) δ: 194.4, 170.0, 152.9, 148.8, 147.4, 131.0, 128.9, 124.7, 122.8, 122.6, 115.9, 112.3, 110.8, 108.3, 102.5, 102.1, 21.3. HRMS m/z (ESI+) calc'd for C18H11NO5 (M+H+) 322.0715, found 322.0726.
4.1.16. Synthesis of compound 31
n-Butyllithium (0.09 mL, 0.22 mmol, 2.5 M solution in hexanes) was added under argon to a cold (0 °C) suspension of lycorine (1) (0.0287 g, 0.1 mmol) in THF (3 mL). The resulting solution was stirred at 0 °C for 20 min and then transferred to a solution of BOC anhydride (0.048g, 0.21 mmol) in THF (5 mL) at 20 °C, then the mixture was stirred at this temperature for 5 h. The reaction was quenched with brine (2 mL), and extracted with EtOAc followed by washing with brine (3 × 10 mL), then dried (MgSO4). Rotary evaporation of the volatiles provided 0.048 g (98%) of compound 31 as a light yellow oil.
tert-Butyl (1S,2S,3a1S,12bS)-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrole[3,2,1-de]phenanthridine-1,2-diyl dicarbonate (31). 1H NMR (300 MHz, CDCl3) δ: 6.78 (s, 1H), 6.55 (s, 1H), 5.89 (d, J = 1.92 Hz, 2H), 5.60 (s, 1H), 5.57 (s, 1H), 5.15 (s, 1H), 4.09 (d, J = 13.5 Hz, 1H), 3.54 (d, J = 13.5 Hz, 1H), 3.32 (m, 1H), 2.87 (d, J = 10.4 Hz, 1H), 2.80 (d, J = 10.5 Hz, 1H), 2.61 (m, 2H), 2.38 (t, J = 8.52 Hz, 1H), 1.50 (s, 9H), 1.41 (s, 9H). 13C NMR (75 MHz, CDCl3) δ: 152.9, 152.6, 146.7, 146.4, 129.7, 126.7, 113.5, 107.4, 105.3, 101.0, 82.9, 82.8, 73.6, 72.5, 61.1, 57.0, 53.7, 40.6, 28.9, 27.9. HRMS m/z (ESI+) calc'd for C26H33NO8 (M+H+) 488.2284, found 488.2294.
4.1.17. Synthesis of compound 32
To a solution of lycorine (1) (0.0287 g, 0.1 mmol) in 0.5 mL of dry DMF under argon were added at 20 °C NaH (0.016 g, 0.4 mmol, 60% suspension in mineral oil) and after 10 min allyl bromide (0.02 mL, 0.2 mmol). The reaction was left stirring for 20 hours at rt. The mixture was diluted with 20 mL of 2-propanol and the solvents were removed at reduced pressure. PTLC (CH2Cl2:MeOH=48/2, Rf = 0.25) afforded 0.013 g (35%) of compound 32 as a yellow oil.
(1S,2S,3a1R,12bS)-1,2-bis(allyloxy)-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridine (32). 1H NMR (300 MHz, CDCl3) δ: 6.78 (s, 1H), 6.57 (s, 1H), 5.98–5.75 (m, 4H), 5.56 (s, 1H), 5.35–5.17 (m, 4H), 4.23–4.03 (m, 7H), 3.56 (d, J = 11.6 Hz, 1H), 3.31 (dd, J = 6.6 Hz, 1H), 2.82 (d, J = 9.6 Hz, 1H), 2.75 (t, J = 9.9 Hz, 1H), 2.62 (m, 3H), 2.40 (s, 1H). 13C NMR (100 MHz, CDCl3) δ: 146.2, 145.9, 135.1, 135.0, 128.6, 117.7, 117.1, 115.6, 105.1, 100.8, 75.4, 75.1, 71.1, 70.5, 61.4, 53.7, 41.0, 28.7. HRMS m/z (ESI+) calc'd for C22H25NO4 (M+H+) 368.1862, found 368.1869.
(1S,2S,3a1S,12bS)-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridine-1,2-diyl dipropionate (33). This compound was obtained as a side product during the attempted Claisen rearrangement with propionic acid as an additive. 1H and 13C NMR were identical to those published in the literature.5b
4.1.17. Claisen rearrangement to synthesize compound 34
Compound 23 (0.01 g, 0.03 mmol), triethyl orthoacetate (0.19 mL, 0.76 mmol) and p-TsOH (0.001 g, 0.005 mmol) were suspended in toluene (2 mL) and refluxed under N2 for 60 h. After that time the volatiles were removed under the reduced pressure and crude was purified by preparative TLC with CH3Cl:MeOH=95/5 mixture as an eluent to isolate 0.0046 g (38%) of compound 34 (Rf = 0.71).
Ethyl 2-((1R,3aR,3a1S,12bS)-1-acetoxy-3a,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridin-3a-yl)acetate (34). White amorphous solid. 1H NMR (300 MHz, CDCl3) δ: 6.56 (s, 1H), 6.52 (s, 1H), 6.50 (d, J = 11.1 Hz, 1H), 5.91 (s, 2H), 5.89 (s, 1H), 5.75 (dd, J = 4.1 Hz, J = 11.1 Hz, 1H), 4.13 (q, J = 7.1 Hz, J = 14.3 Hz, 2H), 4.05 (d, J = 14.5 Hz, 1H), 3.44 (m, 2H), 3.18 (dd, J = 7.1 Hz, 1H), 2.70 (d, J = 14.6 Hz, 1H), 2.61 (s, 1H), 2.57 (m, 2H), 2.28 (m, 2H), 1.82 (s, 3H), 1.24 (t, J = 7.1 Hz, J = 14.3 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ: 172.5, 170.7, 146.9, 146.1, 138.6, 136.3, 132.3, 125.8, 107.2, 106.7, 101.0, 67.4, 67.2, 60.4, 57.2, 53.3, 43.0, 40.0, 37.5, 31.7, 21.1, 14.3. HRMS m/z (ESI+) calc'd for C22H25NO6 (M+H+) 400.1760, found 400.1769.
4.2. Determination of GI50 Growth Inhibitory Values
The overall growth levels of each human cancer cell line was determined using the colorimetric MTT (3-[4,5-dimethylthiazol-2yl])-diphenyl tetrazolium bromide, Sigma, Belgium) assay.6h,8,13a,17 Briefly, the cell lines were incubated for 24 h in 96-microwell plates (at a concentration of 10,000 to 40,000 cells per 1 mL of culture medium depending on the cell type) to ensure adequate plating prior to cell growth determination. The assessment of cell population growth by means of the MTT assay is based on the capability of living cells to reduce the yellow MTT reagent to the blue product, formazan, by a reduction reaction occurring in the mitochondria. The number of living cells after 72 h of culture in the presence (or absence: control) of the various compounds is directly proportional to the intensity of the blue staining, which is quantitatively measured by spectrophotometry – in our case using a Biorad Model 680XR (Biorad, Nazareth, Belgium) – at a 570 nm wavelength (with a reference of 630 nm). Each set of experimental conditions was evaluated in sextuplicate. 6h,8,13a,17
Supplementary Material
Table. In Vitro Growth Inhibitory Effects of Lycorine Derivatives.
| GI50 in vitro values (μM) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| lycorine derivatives | clog P | glioma | carcinoma | melanoma | m ± SEM | |||||
| Hs683 | T98G | U373 | A549 | PC3 | MCF7 | SKMEL −28 | B16 | |||
|
| ||||||||||
| lycorine (1) | 0.5 | 0.9 | 3 | 3 | 0.9 | 4 | 4 | 4 | 2 | 3 ± 1 |
| 3 | 0.3 | * | * | * | * | * | * | * | 86 | > 98 |
| 4 | 2.5 | 20 | 29 | 21 | 23 | 33 | 50 | * | 20 | > 37 ± 10 |
| 5 | 2.4 | 40 | 41 | 49 | 44 | 48 | 9 | * | 38 | > 46 ± 9 |
| 6 | 1.1 | 49 | 32 | 39 | 36 | * | * | 99 | 75 | > 66 ± 11 |
| 7 | −0.3 | * | * | * | * | * | * | * | * | > 100 |
| 8 | 2.6 | 52 | 57 | 46 | 57 | * | 35 | 47 | 30 | > 53 ± 8 |
| 9 | −1.8 | * | * | * | * | * | 96 | * | * | > 99 |
| 10 | −0.9 | * | * | * | * | * | * | * | 83 | > 98 |
| 11 | 0.1 | * | * | * | * | * | * | * | * | > 100 |
| 12 | 0.9 | * | * | * | * | * | * | * | 83 | > 98 |
| 13 | −1.0 | * | * | * | * | * | * | * | * | > 100 |
| 14 | 0.4 | 3 | 38 | 45 | 4 | 92 | 35 | * | 16 | > 42 ± 13 |
| 15 | 2.7 | 15 | 12 | 9 | 7 | 12 | 36 | 10 | 5 | 15 ± 4 |
| 16 | 3.4 | 22 | 11 | 13 | 15 | 18 | 24 | 26 | 22 | 19 ± 2 |
| 17 | 5.4 | 10 | 54 | 1 | 54 | 11 | 20 | 29 | 24 | 25 ± 7 |
| 18 | 3.0 | 4 | 32 | 0.6 | 32 | 5 | 5 | 4 | 8 | 11 ± 5 |
| 19 | 3.0 | 6 | 70 | 1 | 70 | 34 | 23 | 60 | 21 | 36 ± 10 |
| 20 | 9.4 | 21 | 3 | 13 | 9 | 20 | 25 | 38 | 38 | 21 ± 5 |
| 21 | 4.1 | 36 | 6 | 15 | 18 | 26 | 27 | 34 | 30 | 24 ± 4 |
| 22 | 8.2 | 27 | 7 | 24 | 26 | 29 | 32 | 35 | 28 | 26 ± 3 |
| 23 | 1.2 | 11 | 2 | 4 | 3 | 3 | 4 | 3 | 43 | 9 ± 5 |
| 24 | 8.8 | 24 | 16 | 8 | 23 | 26 | 38 | 34 | 23 | 24 ± 3 |
| 25 | 9.3 | 14 | 0.8 | 4 | 14 | 22 | 30 | 30 | 21 | 17 ± 4 |
| 26 | 6.9 | 20 | 3 | 8 | 15 | 19 | 21 | 25 | 18 | 16 ± 3 |
| 27 | 2.8 | 80 | 34 | 50 | 67 | 97 | 74 | * | 57 | > 70 ± 8 |
| 28 | 2.9 | 14 | 24 | 4 | 24 | 38 | 16 | 33 | 8 | 20 ± 4 |
| 29 | 3.3 | 19 | 96 | 27 | 96 | * | 27 | * | 34 | > 62 ± 14 |
| 30 | 2.8 | 14 | 18 | 5 | 18 | 21 | 7 | 21 | 5 | 14 ± 3 |
| 31 | 3.8 | 9 | 4 | 4 | 3 | 22 | 5 | 20 | 4 | 9 ± 3 |
| 32 | 3.1 | 2 | 4 | 0.03 | 4 | 1 | 0.2 | 6 | 4 | 3 ± 1 |
| 33 | 2.7 | 8 | 0.6 | 3 | 4 | 41 | 22 | 43 | 29 | 19 ± 6 |
| 34 | 2.2 | * | * | * | * | * | * | * | 79 | >97 |
> 100 μM
Acknowledgment
This work is supported by the US National Institutes of Health (RR-16480 and CA-135579) under the BRIN/INBRE and AREA programs as well as by the Fonds Yvonne Boël (Brussels, Belgium). V. M. and R. K. are a post-doctoral fellow and the director of research with the Fonds National de la Recherche Scientifique (FNRS, Belgium). In addition, grants from the Italian Ministry of University and Research are gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- (1).Lefranc F, Brotchi J, Kiss R. J. Clin. Oncol. 2005;23:2411–2422. doi: 10.1200/JCO.2005.03.089. [DOI] [PubMed] [Google Scholar]
- (2).La Porta CA. Curr. Med. Chem. 2007;14:387–391. doi: 10.2174/092986707779941078. [DOI] [PubMed] [Google Scholar]
- (3).D'Amico TA, Harpole DH., Jr. Chest Surg. Clin. N. Am. 2000;10:451–469. [PubMed] [Google Scholar]
- (4).Fenell DA. Clin. Cancer Res. 2005;11:2097–2105. doi: 10.1158/1078-0432.CCR-04-1482. [DOI] [PubMed] [Google Scholar]
- (5).For some recent reports, see: McNulty J, Nair JJ, Little JRL, Brennan JD, Bastida J. Bioorg. Med. Chem. Lett. 2010;20:5290–5294. doi: 10.1016/j.bmcl.2010.06.130.. Cedron JC, Gutierrez D, Flores N, Ravelo AG, Estevez-Braun A. Bioorg. Med. Chem. 2010;18:4694–4701. doi: 10.1016/j.bmc.2010.05.023..
- (6).Liu J, Hu WX, He LF, Ye M, Li Y. FEBS Letters. 2004;578:245–250. doi: 10.1016/j.febslet.2004.10.095.. Liu J, Li Y, Tang LJ, Zhang GP, Hu WX. BioMed. Pharmacother. 2007;61:229–234. doi: 10.1016/j.biopha.2007.01.003.. Liu XS, Jiang J, Jiao XY, Wu YE, Lin JH, Cai YM. Cancer Letters. 2009;274:16–24. doi: 10.1016/j.canlet.2008.08.029.. Kornienko A, Evidente A. Chem. Rev. 2008;108:1982–2014. doi: 10.1021/cr078198u.. Evidente A, Kireev AS, Jenkis AR, Romero AE, Steelant WFA, Van Slambrouck S, Kornienko A. Planta Med. 2009;75:501–507. doi: 10.1055/s-0029-1185340.. Evidente A, Kornienko A. Phytochem. Rev. 2009;8:449–459.. Liu J, Hu JL, Shi BW, He Y, Hu WX. Cancer Cell Internat. 2010;10:25. doi: 10.1186/1475-2867-10-25.. Van Goietsenoven G, Andolfi A, Lallemand B, Cimmino A, Lamoral-Theys D, Gras T, Abou-Donia A, Dubois J, Lefranc F, Mathieu V, Kornienko A, Kiss R, Evidente A. J. Nat. Prod. 2010;73:1223–1227. doi: 10.1021/np9008255..
- (7).(a) Lamoral-Theys D, Andolfi A, Van Goietsenoven G, Cimmino A, Le Calvé B, Wauthoz N, Mégalizzi V, Gras T, Bruyère C, Jacques Dubois J, Véronique Mathieu V, Kornienko A, Kiss R, Evidente A. J. Med. Chem. 2009;52:6244–6256. doi: 10.1021/jm901031h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lamoral-Theys D, Decaestecker C, Mathieu V, Dubois J, Kornienko A, Kiss R, Evidente A, Pottier L. Mini- Rev. Med. Chem. 2010;10:41–50. doi: 10.2174/138955710791112604. [DOI] [PubMed] [Google Scholar]; (c) McNulty J, Nair JJ, Singh M, Crankshaw DJ, Holloway AC, Bastida J. Bioorg. Med. Chem. Lett. 2009;19:3233–3237. doi: 10.1016/j.bmcl.2009.04.086. [DOI] [PubMed] [Google Scholar]; (d) McNulty J, Nair JJ, Bastida J, Pandey S, Griffin C. Phytochem. 2009;70:913–919. doi: 10.1016/j.phytochem.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Van Goietsenoven G, Hutton J, Becker J-P, Lallemand B, Robert F, Lefranc F, Pirker C, Vandenbussche G, Van Antwerpen P, Evidente A, Berger W, Prévost M, Pelletier J, Kiss R, Kinzy TG, Kornienko A, Mathieu V. FASEB J. 2010;24:4575–4584. doi: 10.1096/fj.10-162263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Toda J, Sano T, Tsuda Y, Itatani Y. Chem. Pharm. Bull. 1982;30:1322–1332. [Google Scholar]
- (10).Huang WJ, Singh OV, Chen CH, Chiou SY, Lee SS. Helv. Chim. Acta. 2002;85:1069–1078. [Google Scholar]
- (11).Evidente A, Cicala MR, Randazzo G, Riccio R, Calabrese G, Liso R, Arrigoni O. Phytochemistry. 1983;22:2193–2196. [Google Scholar]
- (12).Ganton MD, Kerr MA. Org. Lett. 2005;7:4777–4779. doi: 10.1021/ol052086c. [DOI] [PubMed] [Google Scholar]
- (13).(a) Dumont P, Ingrassia L, Rouzeau S, Ribaucour F, Thomas S, Roland I, Darro F, Lefranc F, Kiss R. Neoplasia. 2007;9:766–776. doi: 10.1593/neo.07535. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ingrassia L, Lefranc F, Dewelle J, Pottier L, Mathieu V, Spiegl-Kreinecker S, Sauvage S, El Yazidi M, Dehoux M, Berger W, Van Quaquebeke E, Kiss R. J. Med. Chem. 2009;52:1100–1114. doi: 10.1021/jm8013585. [DOI] [PubMed] [Google Scholar]; (c) Lefranc F, Sauvage S, Van Goietsenoven G, Mégalizzi V, Lamoral-Theys D, Debeir O, Spiegl-Kreinecker S, Berger W, Mathieu V, Decaestecker C, Kiss R. Mol. Cancer Ther. 2009;8:1–12. doi: 10.1158/1535-7163.MCT-08-0932. [DOI] [PubMed] [Google Scholar]
- (14).Belot N, Rorive S, Doyen I, Lefranc F, Bruyneel E, Dedecker R, Micik S, Brotchi J, Decaestecker C, Salmon I, Kiss R, Camby I. Glia. 2001;36:375–390. doi: 10.1002/glia.1124. [DOI] [PubMed] [Google Scholar]
- (15).(a) Branle F, Lefranc F, Camby I, Jeuken J, Geurts-Moespot A, Sprenger S, Sweep F, Kiss R. Cancer. 2002;95:641–655. doi: 10.1002/cncr.10710. [DOI] [PubMed] [Google Scholar]; (b) Le Calvé B, Rynkowski M, Le Mercier M, Bruyère C, Lonez C, Gras T, Haibe-Kains B, Bontempi G, Decaestecker C, Ruysschaert JM, Kiss R, Lefranc F. Neoplasia. 2010;12:727–739. doi: 10.1593/neo.10526. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Peng Q, Zhou J, Zhou Q, Pan F, Zhong D, Liang H. Hepatogastroenterology. 2009;56:355–360. [PubMed] [Google Scholar]; (d) Yun HJ, Kim SY, Kwon YY, Kim CH, Kang CM, Kim EJ. Cancer Biol. Ther. 2010;10:354–361. doi: 10.4161/cbt.10.4.12382. [DOI] [PubMed] [Google Scholar]
- (16).Mijatovic T, Mathieu V, Gaussin JF, De Neve N, Ribaucour F, Van Quaquebeke E, Dumont P, Darro F, Kiss R. Neoplasia. 2006;8:402–412. doi: 10.1593/neo.05850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Mathieu V, Pirker C, Martin de Lasalle E, Vernier M, Mijatovic T, De Neve N, Gaussin J.F. ; Dehoux, M., Lefranc F, Berger W, Kiss R. J. Cell. Mol. Med. 2009;13:3960–3972. doi: 10.1111/j.1582-4934.2009.00708.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Mathieu V, Le Mercier M, De Neve N, Sauvage S, Gras T, Roland I, Lefranc F, Kiss R. J. Invest. Dermatol. 2007;127:2399–2410. doi: 10.1038/sj.jid.5700869. [DOI] [PubMed] [Google Scholar]
- (19).(a) Igarashi M, Miyazawa T. Cancer Lett. 2000;148:173–179. doi: 10.1016/s0304-3835(99)00332-8. [DOI] [PubMed] [Google Scholar]; (b) Zhu YP, Su ZW, Li CH. J. Natl. Cancer Inst. 1989;81:1302–1306. doi: 10.1093/jnci/81.17.1302. [DOI] [PubMed] [Google Scholar]; (c) Ito H, Kasama K, Naruse S, Shimura K. Cancer Lett. 1982;17:197–203. doi: 10.1016/0304-3835(82)90032-5. [DOI] [PubMed] [Google Scholar]
- (20).Sauer LA, Dauchy RT. Brit. J. Cancer. 1992;66:297–303. doi: 10.1038/bjc.1992.260.; Jaracz S, Chen J, Kuznetsova LV, Ojima I. Bioorg. Med. Chem. 2005;13:5043–5054. doi: 10.1016/j.bmc.2005.04.084..
- (21).Bradley MO, Webb NL, Anthony FH, Devanesan P, Witman PA, Hemamalini S, Chander MC, Baker SD, He L, Horwitz SB, Swindell CS. Clinical Cancer Res. 2001;7:3229–3238. [PubMed] [Google Scholar]
- (22).(a) You YJ, Kim Y; Nam, N. H., Bang SC, Ahn BZ. Eur. J. Med. Chem. 2004;39:189–193. doi: 10.1016/j.ejmech.2003.10.002. [DOI] [PubMed] [Google Scholar]; (b) You YJ, Kim Y, Nam NH, Ahn BZ. Bioorg. Med. Chem. Lett. 2003;13:2629–2632. doi: 10.1016/s0960-894x(03)00558-4. [DOI] [PubMed] [Google Scholar]
- (23).Evidente A, Iasiello I, Randazzo G. Chem. Ind. 1984:348–349. [Google Scholar]
- (24).Lee S-S, Venkatesham U, Rao CP, Lam S-H, Lin J-H. Bioorg. Med. Chem. 2007;15:1034–1043. doi: 10.1016/j.bmc.2006.10.026. [DOI] [PubMed] [Google Scholar]
- (25).Lauk U, Dürstand D, Fischer W. Tetrahedron Lett. 1991;32:65–68. [Google Scholar]
- (26).Ghosal S, Shanthy A, Kumar A, Kumar Y. Phytochemistry. 1985;24:2703–2706. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.