

Abstract
Regulatory T cells (Tregs) migrate into peripheral sites of inflammation such as allografts undergoing rejection, where they serve to suppress the immune response. In this study, we find that ~30–40% of human CD25hi FOXP3+ CD4+ Tregs express the peripheral CXC chemokine receptor 3 (CXCR3) and that this subset has potent immunoregulatory properties. Consistently, we observed that proliferative responses as well as IFN-γ production were significantly higher using CXCR3-depleted versus undepleted responders in the mixed lymphocyte reaction, as well as following mitogen-dependent activation of T cells. Using microfluidics, we also found that CXCR3 was functional on CXCR3pos Tregs, in as much as chemotaxis and directional persistence towards interferon-c-inducible protein of 10 kDa (IP-10) was significantly greater for CXCR3pos than CXCR3neg Tregs. Following activation, CXCR3-expressing CD4+ Tregs were maintained in vitro in cell culture in the presence of the mammalian target of rapamycin (mTOR) inhibitor rapamycin, and we detected higher numbers of circulating CXCR3+ FOXP3+ T cells in adult and pediatric recipients of renal transplants who were treated with mTOR-inhibitor immunosuppressive therapy. Collectively, these results demonstrate that the peripheral homing receptor CXCR3 is expressed on subset(s) of circulating human Tregs and suggest a role for CXCR3 in their recruitment into peripheral sites of inflammation.
Keywords: Chemokine receptors, Chemokines, CXCR3, Immunoregulation, Tregs
Introduction
Regulatory T cells (Tregs) are essential for the suppression of immune responses to foreign antigens, including alloantigens, and they are well established to function in the development and maintenance of self-tolerance [1, 2]. Forkhead box P3 (FOXP3) has emerged as the master regulator of the development and function of Tregs in both mice and humans [3–5]. Furthermore, expansion of CD4+FOXP3+ T-cell subsets is generally considered to be critical for tolerance induction and for the suppression of a wide range of immune-mediated diseases [6].
Tregs utilize multiple mechanisms to suppress effector cell expansion and to mediate immunoregulation [1, 7]. These include cell–cell contact-dependent suppression [8], secretion of immunosuppressive cytokines including IL-10 [9, 10], TGF-β [11, 12] and IL-35 [13], and the consumption of IL-2 produced by responder T cells [14]. Furthermore, Tregs may function via the generation of pericellular adenosine catalyzed by CD39 and CD73 [15] and/or the modification of APCs via the delivery of negative/apoptotic signals [7]. In addition, several studies have indicated that the in vivo function of Tregs is dependent on their migration into sites of inflammation [16–19]. Although compartmentalization of Tregs is not a new phenomenon [20], the concept that Tregs migrate into allografts and inhibit rejection is a very recent observation [16–18, 21]. An emerging model is that tolerance to alloantigens can only be achieved if Tregs are allowed to migrate in an appropriate pattern within allografts and within lymph nodes [16, 18].
It has been reported that Tregs express multiple chemokine receptors [22]; some studies have identified that the majority of human Tregs express CD62L [23], CCR4 [22, 24] and CCR7 [25]. These combinations should allow Tregs to migrate into lymph nodes and into the periphery. Nevertheless, most studies have been performed in rodents [18, 20, 24], and few studies have evaluated expression of these receptors in human Treg subsets.
The CXC chemokine receptor 3 (CXCR3) is classically expressed on activated human CD4+ T cells, and is well established to mediate effector cell trafficking [26–28]. Consistent with these findings, the expression of CXCR3 [28–30] and its chemokine ligands, monokine induced by IFN-γ (Mig or CXCL9), IFN-γ-inducible protein of 10 kDa (IP-10 or CXCL10) and IFN-γ-inducible T-cell α-chemoattractant (or CXCL11) have been reported to be associated with both cardiac and renal allograft rejection [28, 30–37]. However, paradoxically, some recent studies have suggested that CXCR3 may also be expressed on Tregs [22, 38–41], and blockade of CXCR3 is reported to have variable functional effects in different animal models [32, 42, 43]. Nevertheless, little is known about its expression pattern or its association with Treg subsets and their immunoregulatory function(s).
In this study, we characterized the expression of CXCR3 on human CD25hi FOXP3+ CD4+ T regulatory cells, and we demonstrate that CXCR3hi Tregs are functional to suppress effector alloimmune responses. Furthermore, we demonstrate that levels of CXCR3 increase on Tregs following activation, and that CXCR3hi Tregs are enriched in cell culture in the presence of rapamycin.
Results
Human CD4+CD25hiFOXP3+ T cells express CXCR3
We initially analyzed the co-expression of CXCR3 and CD25 on CD4+ T cells by four color flow cytometry. Consistently, we observed two subpopulations of CD25hi cells that were either CXCR3hi or CXCR3lo/neg (Fig. 1A). As illustrated in Fig. 1B and C, we also found that FOXP3 was expressed within both populations and, further, that the level of FOXP3 expression in each subset was similar. We gated on CD25hi, CD25int/lo and CD25neg CD4+ T-cell subsets, and we assessed the relative expression of CXCR3 on each population. As illustrated in Fig. 2A, we found that CXCR3 is expressed by all subsets, irrespective of CD25 expression; but notably, double positive CXCR3+CD25hi populations co-express significant levels of FOXP3.
Figure 1.
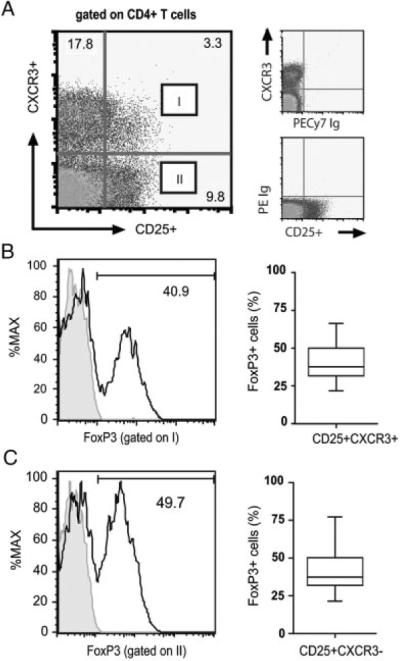
A subset of CXCR3+ CD4+ T cells express FOXP3. Mono-nuclear cells were isolated from healthy volunteers and were stained with antibodies to CD4, CD25, CXCR3 and FOXP3, or with isotype control mAbs. (A) Representative dot plot showing CD25 and CXCR3 expression, after gating on CD4+ T cells. (B) Representative FOXP3 expression (open histograms, left panel) after gating on the CD41 CD25+CXCR3+ subset (labeled I) in (A). (C) FOXP3 expression after gating on the CD4+CD25+CXCR3− subset (labeled II) in (A). In both (B) and (C), the box plots on the right illustrate the median, 25th and 75th percentile and range of FOXP3 expression in (B) CXCR3pos and (C) CXCR3neg Tregs from n = 20 subjects.
Figure 2.
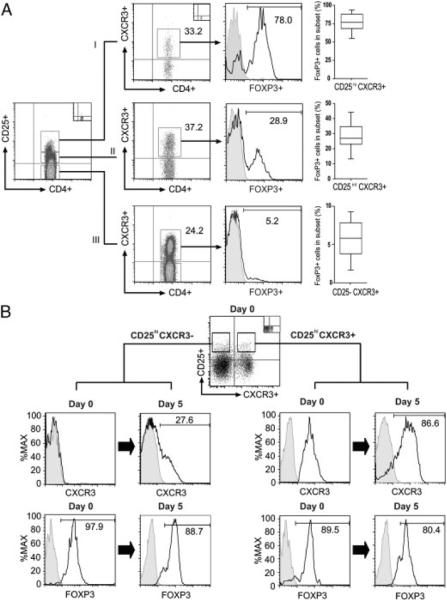
Expression of CXCR3 on CD4+ Tregs. Freshly isolated mononuclear cells were stained with antibodies to CD4, CD25, CXCR3 and FOXP3. (A) After gating on lymphocytes, the expression of CXCR3 was evaluated on CD25hi (I), CD25int/lo (II), and CD25neg (III) CD4+ T cells, and FOXP3 expression (histograms) was evaluated in each subset. The box plots on the right illustrate the median, 25th and 75th percentile and range of FOXP3 expression among the CXCR3-expressing CD4+ T-cell subsets in n = 20 subjects. (B) FACS-sorted populations of CD25hiCXCR3neg or CD25hiCXCR3pos CD4+ T cells (as illustrated) were stimulated with immobilized anti-CD3 and soluble anti-CD28 in the presence of IL-2 (100 IU/mL) for 5 days. The expression of CXCR3 and FOXP3 was evaluated by flow cytometry on day 0 and on day 5 following activation. The histograms on the left illustrate representative expression of CXCR3 and FOXP3 on sorted CXCR3neg cells before and after activation, as indicated. The histograms on the right illustrate expression of CXCR3 and FOXP3 on sorted CXCR3pos cells. Representative of n = 3 experiments.
To determine whether CXCR3 expression is representative of Tregs in different states of activation, we sorted CD25hiCXCR3neg cells and CD25hiCXCR3pos cells, and we stimulated each subset for 5 days with immobilized anti-CD3 and soluble anti-CD28 in the presence of IL-2 (Fig. 2B). Prior to activation, both subsets were found to express high levels of FOXP3 at the mRNA and protein levels (Fig. 2B, and data not shown). As illustrated, we found that the expression of CXCR3 was maintained on activated CXCR3pos Tregs (Fig. 2B). Furthermore, following activation, we found that CXCR3 was induced in expression on a subset of CXCR3neg cells, suggesting that differences in CXCR3 expression on each Treg subset may in part relate to their state of activation.
We also performed additional phenotypic profiling of CXCR3pos Tregs by evaluating co-expression of CXCR3 with cytotoxic T-lymphocyte antigen 4 (CTLA-4) and CD39, well-established markers of Tregs [15, 44]. As summarized in Fig. 3A–C, we found that CXCR3 is expressed at similar levels on both FOXP3+ and CTLA-4+ CD4+ T-cell subsets. In addition, we observed that up to half of FOXP3+CTLA-4+ or FOXP3+CD39+ double positive Tregs co-express CXCR3 (Fig. 3D and E). Since these markers tend to be expressed on activated cells, this finding is again consistent with the interpretation that levels of CXCR3 expression on Tregs are in part reflective of their state of activation.
Figure 3.

Co-expression of CXCR3 with FOXP3, CTLA-4 and CD39 on CD4+ T cells. (A and B) Representative flow cytometry plots illustrating co-expression of CXCR3 and CD25 on (A) the CD4+FOXP3+ T-cell subset and (B) the CD4+CTLA-4+ subset. Staining with isotype control antibodies is shown on the right of each dot plot. (C) Median, 25th and 75th percentile and range of CXCR3 expression on CD4+FOXP3+ or CD4+CTLA-4+ subsets. (D and E) Representative flow cytometry plots of CXCR3 expression on (D) double positive FOXP3+CTLA-4+ cells and (E) FOXP3+CD39+ cells after gating on CD4+ cells. The box plot on the right of each panel illustrates median, 25th and 75th percentile and range of expression in multiple experiments. (F) Fold difference (mean1SEM) in mRNA expression for Tbet in sorted populations of CD4+CD25hiCXCR3pos cells (white bars) or CD4+CD25hiCXCR3neg Tregs (black bars). Expression was evaluated in unactivated Tregs (left panel) or in Tregs following 6h activation with anti-CD3/anti-CD28 (right panel). Representative of n = 3 experiments with similar results.
Finally, we compared the expression of Tbet in CXCR3pos and CXCR3neg Tregs. Tbet is reported to identify a subset of Tregs that control Th1-type inflammation in murine models [45]. As illustrated in Fig. 3F, we found that Tbet mRNA expression was higher in CXCR3pos Tregs as compared with CXCR3neg subsets, regardless of their state of activation. Collectively, these observations indicate that CXCR3 is expressed on subsets of Tregs, most notably on recently activated cells.
Subsets of CXCR3-expressing CD4+ T cells are immunoregulatory
To next determine the immunoregulatory function(s) of CXCR3-expressing CD4+ T cells, pooled populations or CXCR3-depleted populations of CD4+ T cells were used as responders in alloantigen- (Fig. 4A and B) and mitogen- (Fig. 4C and D) induced assays. CD25-depleted CD4+ T-cell responders were used as a control. As illustrated in Fig. 4A and B, we found that proliferation and IFN-γ production (as assessed by ELISPOT) was greater (p<0.01) in CXCR3-depleted responders, compared with undepleted cells, in the mixed lymphocyte reaction. Also, following mitogen-dependent activation, proliferation (Fig. 4C) and IFN-γ production (Fig. 4D) was significantly greater (p<0.001 and p<0.05 respectively) in cultures using CXCR3-depleted responders. The increased proliferation and production of IFN-γ in CXCR3-depleted responder cultures was similar to that observed in control cultures when CD25-depleted CD4+ cells were used as responders (Fig. 4A–D). IL-2 production was also increased when CXCR3-depleted responders were used in mitogen-induced assays (p<0.05, data not shown). Finally, to confirm the suppressive function of CXCR3-expressing T cells, we repleted responders with sorted populations of CXCR3+ CD25hi T cells in a standard in vitro suppression assay. As illustrated in Fig. 4E, the addition of CXCR3+ CD25hi cells into the cultures in increasing ratios suppressed proliferative responses to baseline. Taken together, these observations indicate that subset(s) of CXCR3-expressing T cells have potent immunoregulatory properties.
Figure 4.
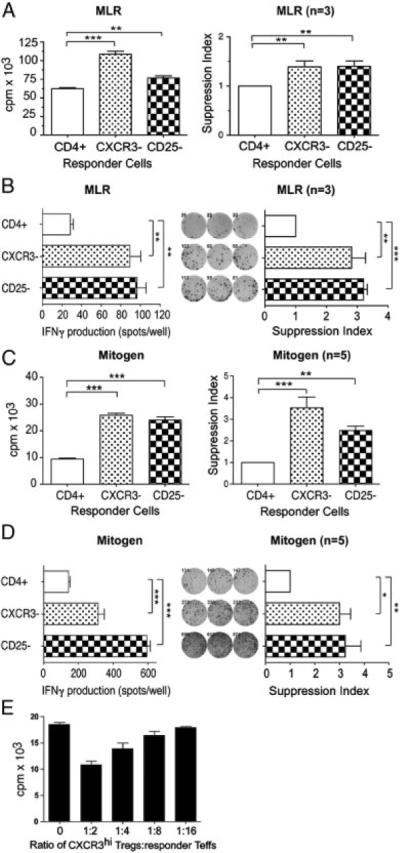
Immunoregulation by CD4+CXCR3+ T cells. CD4+ T cells were positively isolated from PBMCs, and were subsequently depleted of CXCR3-expressing cells (labeled CXCR3−) or CD25-expressing cells (labeled CD25−) using magnetic beads, as described in Materials and methods. Proliferation was assessed by standard 3H thymidine incorporation, and IFN-γ production was assessed by ELISPOT, (A and B) after allogeneic stimulation in a standard mixed lymphocyte reaction (MLR) or (C and D) after mitogen-dependent activation with anti-CD3/anti-CD28. In each panel, a representative experiment is illustrated in the left graph, and the average suppression from multiple experiments (mean1SEM) is illustrated in the graph on the right. (E) Mitogen-induced proliferation of CXCR3-depleted responders, repleted with increasing ratio's of sorted CXCR3hi CD25+ T cells (mean+SEM, representative of n = 3 experiments). Statistical significance was determined by ANOVA and Bonferroni's post test, *p<0.05, **p<0.01 and ***p<0.001.
Migration of CXCR3+ Treg subsets to IP-10
We next evaluated the functional implications of CXCR3 expression on Tregs for IP-10-dependent chemotaxis. Leukocyte migration was measured using a microfluidic technique that allows for precise and robust measurements of leukocyte migration at single-cell resolution [46]. Purified CD4+CD25+CD127dim/− Tregs were FACS-sorted into CXCR3pos or CXCR3neg subsets and were introduced into the main channel of the microfluidic device (Fig. 5A). Subsequently, images of live-time cell migration toward the chemokine IP-10 were captured using time-lapse imaging, as described in Materials and methods. In the absence of a chemoattractant stimulus, we found minimal migration of T cells into the 6 × 6 μm side channels, and cells that entered the channels appeared to move at random. However, as illustrated in Fig. 5B and C, we found that CXCR3+ Tregs had a marked chemotactic response toward IP-10, and their directional persistence was significantly greater (p<0.01) than that observed for CXCR3neg Tregs (Fig. 5D). CXCR3neg subsets were found to move in a random manner, some cells entered the channel and returned to baseline, and some migrated toward IP-10. In general, the directional persistence of CXCR3neg subsets was limited (Fig. 5D). We also observed that the velocity of CXCR3pos cells during persistent directional migration was consistently slower than the velocity of random migrating CXCR3neg Tregs (but this difference did not reach statistical significance, data not shown). Collectively, these studies demonstrate that CXCR3 is functional to elicit chemotaxis in CXCR3-expressing Tregs.
Figure 5.

Migration of CXCR3+ Tregs to the chemokine IP-10. CD4+CD25+CD127dim/− Tregs were isolated from PBMCs using magnetic beads and were sorted into populations of CXCR3neg or CXCR3pos cells. (A and B) Each Treg subset was introduced into the main channel of microfluidic devices, as described in Materials and methods, and migration to IP-10 was evaluated as a time course for 3 h. Illustrated are images of the T cells (A) following introduction into the device and (B) at sequential times over the initial course of a representative experiment. (C) Representative analysis of single-cell migration of seven individual CXCR3neg cells (left) or CXCR3pos cells (right). Time-lapse microscopy images were captured every 1.5 min, and migration was measured in μm of integrated distance traveled in successive images (representative of a mean of 60 cells evaluated/experiment). (D) Total displacement/integrated distance was calculated for each experiment. Directional chemotaxis was subsequently evaluated based on parameters ranging from 0 (for random motion) to 1 (for directed chemotaxis), as described in Materials and methods. The box plot illustrates the median, 25th and 75th percentile and range of the chemotaxis response for a total of 20 cells examined in the experiment illustrated in (C). As illustrated, while CXCR3neg Tregs have a variable/random pattern of migration, CXCR3pos Tregs migrate efficiently toward IP-10. p-Values were calculated using Student's t-test. Representative of n = 3 experiments.
Co-expression of additional trafficking receptors on CXCR3-expressing Tregs
We next wished to evaluate the co-expression of CXCR3 with well-established lymphoid and peripheral homing receptors on FOXP3+ Tregs. We stained PBMC for CD4, CD25, FOXP3 and either CXCR3, CD62L, CCR4, CCR5 and CCR7, established to be expressed on Tregs [22–26]. We also evaluated the co-expression of CXCR3 with Treg-associated homing receptors. Illustrated in Fig. 6A and B, we found comparable levels of CXCR3 and CD62L expression on both CD25hiFOXP3+ Tregs and CD25loFOXP3− Teff subsets. However, among chemokine receptors, we found lower levels of expression of CCR7 and higher levels of CCR4 and CCR5 on FOXP3+ Tregs versus Teff subsets. Also, we observed that CXCR3 is co-expressed with CD62L on ~30% of FOXP3+ Tregs, while only ~12% Tregs co-express CCR7 and CXCR3; and ~20% CXCR3pos Tregs co-express CCR4 or CCR5 (Fig. 6B). These observations indicate that lymphoid and peripheral homing receptors are co-expressed by human FOXP3+ Tregs and further that CXCR3 is a major peripheral homing receptor expressed on high numbers of human Tregs.
Figure 6.
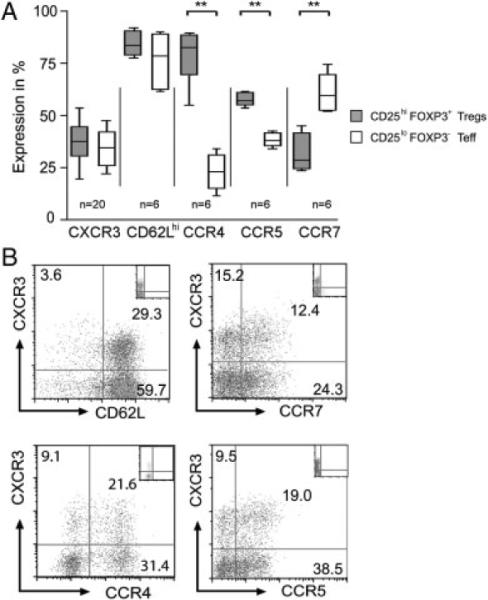
Co-expression of lymphoid and peripheral homing receptors on CXCR3+ Tregs. (A) PBMCs were stained with antibodies to CD4, CD25, FOXP3 and either CXCR3, CD62L, CCR4, CCR5 or CCR7. Gating was performed either on the CD4+CD25hiFOXP3+ Treg subset (grey box plots) or on the CD25loFOXP3− T effector subset (white box plots). Illustrated is the median, 25th and 75th percentile and range of expression of each homing receptor in n≥6 donors, as indicated. (B) Purified populations of CD4+ T cells were stained with antibodies to CD25, FOXP3 and CXCR3, and either the lymphoid homing receptors CD62L and CCR7 or the peripheral homing receptors CCR4 or CCR5. After gating on CD4+CD25hiFOXP3+ cells, co-expression of CXCR3 with CD62L, CCR7, CCR4 and CCR5 is illustrated (representative of n = 4 with similar results).
Effect of rapamycin on CD25+FOXP3+CXCR3hi T-cell subsets
We also examined the effect of immunosuppressants on the survival and expansion of CXCR3-expressing Tregs. Inactivation of the mammalian target of rapamycin (mTOR) kinase and its signaling pathway in T cells has been reported to inhibit activation-induced expansion of CD4+CD25lo effector T cells in vitro and in vivo, while enabling the preferential expansion of Tregs [47, 48]. Furthermore, Tregs that expand in the presence of mTOR inhibitors have been found to possess immunoregulatory activity [48]. We stimulated purified populations of CD4+ T cells with immobilized anti-human CD3, soluble anti-human CD28 and IL-2 in the presence of rapamycin or cyclosporine. As expected [47, 48], CD4+CD25+FOXP3+ Tregs expanded after 5 days of culture in the presence of rapamycin (10 ng/mL). In contrast, culture in the presence of cyclosporine A (CsA) (0.1 μg/mL) inhibited Treg cell expansion (Fig. 7A). By FACS, CXCR3 was expressed at high levels on FOXP3+ Tregs following mitogen-dependent activation both in the absence and in the presence of rapamycin (1 and 10 ng/mL, Fig. 7B and C). However, culture in the presence of CsA (0.1 and 1 μg/mL) inhibited CXCR3 expression on surviving CD25+FOXP3+ cells (p<0.01, Fig. 7B and C). We interpret these observations to indicate that FOXP3+ T cells that expand in the presence of mTOR inhibitors express CXCR3.
Figure 7.
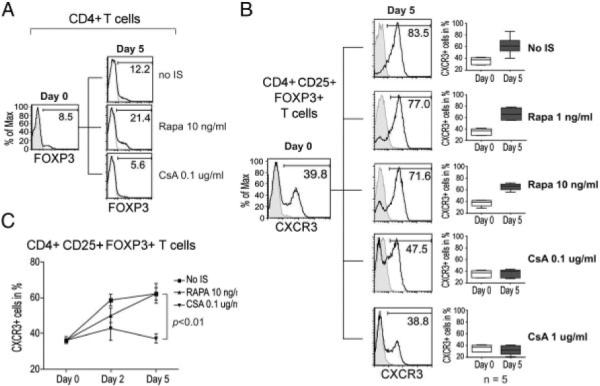
Culture of mitogen-activated CD4+ T cells with rapamycin results in the expansion of CXCR3-expressing Tregs. Purified populations of CD4+ T cells were stimulated with plate-bound anti-CD3/soluble anti-CD28 and IL-2 (100 IU/mL) in the absence or presence of rapamycin (Rapa) or CsA, as indicated, and were stained for CD25, FOXP3 and CXCR3 or isotype controls (shaded histograms). (A) Expression of FOXP3 (open histograms) on CD4+ T cells on day 0 and day 5 following activation (representative of three independent experiments). (B) Representative expression of CXCR3 (open histograms) on CD4+CD25hiFOXP3+ T cells on day 0 and day 5 following activation. The box plots on the right depict summarized data from n = 5 subjects, and illustrate the median, 25th and 75th percentile and range of expression. (C) Expansion of CXCR3+ cells (mean±SEM) within the CD4+ CD25+FOXP3+ population of T cells, plotted over time (n = 5).
CXCR3 expressing CD4+FOXP3+ Tregs in renal transplant recipients treated with rapamycin
Finally, to investigate the pathophysiological significance of our observations, we isolated PBMCs from renal transplant recipients who were treated with mTOR-inhibitor therapy. Two groups of patients were evaluated. The first group consisted of 18 adult recipients of deceased donor transplants, eight of whom were converted to mTOR-inhibitor-based immunosuppression after 3 months of therapy with cyclosporine. The other ten patients were maintained on cyclosporine for the first post transplant year. The second group was pediatric recipients of living related donor transplants who received mTOR-inhibitor therapy de novo, and were enrolled in an NIH-sponsored calcineurin inhibitor avoidance therapy study. These patients received an immunosuppression protocol consisting of induction therapy with an IL-2R antagonist, and maintenance with sirolimus, mycophenolate mofetil and steroids [49]. As illustrated in Fig. 8A, at 1 year post transplantation, we found that adult recipients treated with an mTOR inhibitor had higher levels of circulating FOXP3+ Tregs than patients treated with cyclosporine. In addition, there was an overall increase in numbers of FOXP3+CXCR3+ cells (p<0.01) in recipients treated with mTOR inhibitors as compared with those treated with cyclosporine (Fig. 8B). We noted a trend for association between Treg expression of CXCR3 and better GFRs at year 2 post transplantation in this small cohort of patients (data not shown), but this trend did not reach statistical significance. We also evaluated CXCR3 expression on Tregs in sequential blood samples collected from four pediatric recipients over the first post transplant year. We found that the numbers of CXCR3-expressing FOXP3+ Tregs increased over the first 6–12 months post transplantation in two of these patients, (Fig. 8C). In the other two, expression remained at low levels. Although these results are observational, the two patients with higher numbers of CXCR3-expressing Tregs had excellent renal function at 24 and 36 months post transplantation. In contrast, both patients with low levels of CXCR3 on circulating Tregs had acute rejection episodes within the first post transplant year, and one patient developed graft failure by 24 months post transplantation. Thus, kidney transplant recipients treated with mTOR-inhibitor therapy have circulating CXCR3-expressing Tregs. It will be intriguing to determine whether the patterns of expression seen in this small cohort of patients are associated with differences in long-term graft outcome.
Figure 8.
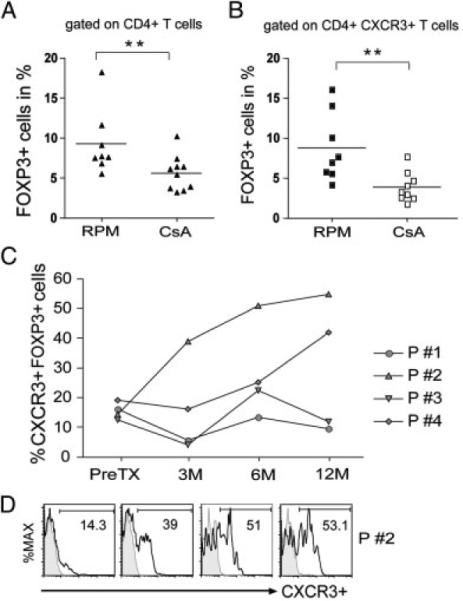
CXCR3-expression on Tregs in renal transplant recipients. (A) Expression of FOXP3 within the CD4+ population of T cells in adult recipients of deceased donor renal transplants who were switched either to an mTOR inhibitor-based protocol (RPM, n = 8) or were maintained on CsA (n = 10). (B) Expression of FOXP3 within the CXCR3+CD4+ T-cell population in the patients identified in (A). In (A) and in (B) expression was assessed at >6 but <12 months post transplantation. The solid line represents the mean level of expression for each cohort (**p<0.01 was determined using the Mann-Whitney U-test). (C) Expansion of CXCR3+ among CD25hiFOXP3+ CD4+ T cells in pediatric recipients of living donor renal transplants enrolled in the CNO1 study [49]. All these patients were treated with an mTOR inhibitor-based immunosuppressive regimen de novo and they did not receive calcineurin inhibitor therapy. (D) Representative histograms showing the expression of CXCR3 pretransplant and at 3, 6 and 12 months post transplantation in one of the patients illustrated in (C).
Discussion
In this report, we demonstrate that CXCR3 is expressed on human FOXP3+CD4+ T-cell subsets, and that CXCR3hiCD4+ Treg subsets function as potent immunoregulatory cells to suppress allogeneic and mitogen-induced effector T-cell activation in vitro. We also find that CXCR3+ Tregs migrate toward their chemokine ligand IP-10, and their directional persistence and chemotaxis response is significantly greater than that of CXCR3neg Tregs. We interpret these observations to suggest that the expression of CXCR3 on Tregs may facilitate their accumulation at sites of inflammation including allografts undergoing rejection.
Understanding the compartmentalization and migration of Tregs is an area of intense study, and is likely of great importance for tolerance induction following solid organ transplantation [16–18]. Tregs are well established to express both adhesion and chemokine receptors [20, 22, 23], and they have potential to suppress anti-donor immune responses following transplantation [16–18]. The trafficking of Tregs into secondary lymphoid organs as well as into the periphery has been proposed to be important for alloimmune tolerance induction [16, 18], and for the prevention of chronic rejection [17]. Indeed, recently, it was observed that effective immunoregulation in vivo was not achieved in the absence of defined patterns of migration [18]. In these studies, we found that greater than 80% of human Tregs express the lymph node homing receptor CD62L. Also, consistent with others [22, 24, 25], we find that CXCR3+FOXP3+ Tregs co-express the peripheral homing receptors CCR4 and CCR5. However, we also find notable differences in the expression of additional homing receptors on Tregs versus T effector cells including α-integrins, β-integrins and PSGL-1 (p<0.01, p<0.05 and p<0.01 respectively, data not shown), further indicating the potential that human Tregs have potential to traffic to lymph nodes as well as to peripheral sites of inflammation, as observed in mouse models [16–18].
CXCR3-expressing T cells, as well as the CXCR3 chemokine ligand IP-10, have been reported to be found within human renal and cardiac allografts in association with rejection [28, 33–36, 50]. Our findings outlined in these studies support the possibility that local intragraft expression of IP-10 facilitates the migration of expanded Tregs into the graft. Consistent with our observations, CXCR3+ cells isolated from inflamed livers were found to have suppressive function [40, 41]. Also, FOXP3+ T cells have been observed within renal allografts in association with rejection [50]. These findings as well as others [16, 17, 51] strongly suggest that alloactivated Tregs migrate into allografts where they have the potential to suppress the local inflammatory response. Our observations are suggestive that CXCR3 faciltates the peripheral migration of Tregs into allografts and that this subset has the potential to suppress ongoing rejection.
It is well established that mTOR inhibitors augment the expansion of Tregs [47, 48] and promote tolerance induction in vivo [48, 52, 53]. We find that the mTOR inhibitor rapamycin also permits the expansion of CXCR3hi Tregs in vitro, and we found higher numbers of circulating FOXP3+CXCR3+ Tregs in transplant recipients treated with mTOR inhibitors versus those treated with calcineurin inhibitors as part of their maintenance immunosuppressive therapy. Our studies involved small numbers of patients, but they are suggestive that the use of mTOR-inhibitor therapy may enable the expansion of CXCR3+ Tregs in vivo, and may have an impact on long-term graft survival. Further evaluation of this observation in a larger cohort of patients may identify if expansion of this subset, for instance in association with the use of mTOR inhibitors, may serve as a biomarker and/or predict long-term graft survival.
In summary, although CXCR3 is classically reported to be expressed on T effector cells, these new findings demonstrate that it is also expressed on populations of immunoregulatory T cells. Our findings explain the variable effects of CXCR3 blockade in allograft rejection [32, 42], in as much as it was not previously known that CXCR3 may mediate the local trafficking of Tregs. Thus, an important implication of our observations is that the activation and expansion of CXCR3-expressing Tregs in vivo will facilitate the compartmentalization of T-cell regulatory subsets within allografts.
Materials and methods
Reagents and Abs
Mouse anti-human CD4-FITC (RPA-T4), anti-human CD4-PE (RPA-T4), anti-human CD4-PECy7 (RPA-T4), anti-human CD39-FITC (A1), anti-human CCR7-PE (3D12), anti-human CCR5-FITC (HEK/1/85a) and anti-human FOXP3-FITC (206D) were obtained from Biolegend (San Diego, CA). Mouse anti-human FOXP3-APC (3G3), mouse anti-human CD62L-APC (DREG-56) and mouse anti-human CCR4-FITC were purchased from Miltenyi Biotec (Auburn, CA), eBioscience (San Diego, CA) and R&D Systems (Minneapolis, MN) respectively. Mouse anti-human CXCR3-PE or CXCR3-APC (1C6), anti-human CD25-FITC or CD25-PECy7 (M-A251), anti-human CD45RO (UCHL1), anti-human CTLA-4-APC (BNI3) and murine monoclonal subclass-specific isotype controls were purchased from BD Biosciences (San Diego, CA). For functional assays, mouse anti-human CD3 (HIT3a) and anti-human CD28 (CD28.2) were purchased from BD Biosciences. For ELISPOT assays, mouse anti-human IFN-γ capture mAbs and a biotinylated anti-human IFN-γ mAbs were purchased from Fisher Scientific (Pierce Biotechnology, Rockford, IL); mouse anti-human IL-2 capture mAbs, biotinylated anti-human IL-2 mAbs and recombinant IL-2 were purchased from R&D Systems. Pooled human AB serum was purchased from Pel Freeze Biologicals (Rogers, AR). Rapamycin was gifted to the laboratory by Wyeth-Ayest Research (Princeton, NJ) and CsA was purchased from Novartis Pharmaceuticals (East Hanover, NJ).
Isolation of T-cell subsets
Human peripheral blood was obtained from healthy volunteers consented in accordance with IRB approval by Children's Hospital Boston. CD4+ T cells were isolated from PMBCs using magnetic beads (Dynal CD4 Positive Isolation Kit, Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. The purity of isolated CD4+ cells was found to be >97% by FACS. For depletion studies, purified CD4+ T cells were incubated for 20 min with 1 μg per 106 target cells of anti-CXCR3 mAbs (1C6, BD Biosciences) or anti-CD25 mAbs (M-A251, BD Biosciences) at 4°C, and were washed in PBS/0.5% BSA. The cells were subsequently incubated with Pan mouse IgG magnetic beads (Dynal Cellection Kit, Invitrogen) and CXCR3+ or CD25+ cells were removed by magnetic separation. The purity of the depleted populations was >92% as assessed by flow cytometry. For migration assays, CD4+CD25+CD127dim/− cells were isolated from PBMCs using magnetic beads (Miltenyi Biotec) and were FACS-sorted (using 1C6, BD Biosciences) into CXCR3+ and CXCR3neg populations.
Cell culture
Cell culture was performed at 37°C in 5% CO2 in RPMI 1640 media (Cambrex, Charles City, IA) containing 10% human AB serum, 2 mM L-glutamine, 100 U/mL penicillin/streptomycin (Gibco-Invitrogen), 1% sodium bicarbonate and 1% sodium pyruvate (Cambrex) in 96-well, round-bottom plates (Corning Life Sciences, Lowell, MA).
Proliferation assays
Mitogen-dependent assays were performed in 96-well round bottom cell culture plates in triplicate wells (1 × 105 T cells/well) in a final volume of 200 μL. The cells were stimulated with either immobilized anti-CD3 mAbs (5 μg/mL) alone or with immobilized anti-CD3 mAbs in combination with soluble anti-CD28 mAbw (1 μg/mL) for 3 days. Mixed lymphocyte reactions were performed using 2 × 105 responders and γ-irradiated PBMCs (1700 rad) as stimulators in a ratio of 1:1. Cells were cultured in triplicate wells using either allogeneic or autologous stimulators. Proliferation was assessed after 5 days by 3H-Thymidine (Perkin Elmer, Boston, MA; 1 μCi/well) incorporation for the final 18 h of culture, and data were analyzed using suppression ratios. Cells were harvested using an automated cell harvester (Tomtec, Hamden, CT) and incorporated radioactivity was determined by liquid scintillation (MicroBeta Trilux Liquid Scintillation Counter, Wallac, Gaithersburg, MD). In selected experiments, rapamycin 1 or 10 ng/mL or CsA 0.1 or 1.0 mcg/mL was added into cultures containing 100 IU/mL human recombinant IL-2.
ELISPOT assays
Multiscreen-IP 96-well microtiter plates (Millipore, Bedford, MA) were coated with a mouse anti-human CD3 mAbs (2 μg/mL) and mouse anti-human IFN-γ capture mAbs (4 μg/mL). Freshly isolated T cells (1 × 105 cells/well in 200 μL) were cultured for 36 h, isolated, washed and incubated with a biotinylated mouse anti-human IFN-γ mAbs (2 μg/mL). After washing, HRP-labeled streptavidin (DAKO, Carpinteria, CA) was added for 1 h and subsequently the spots were developed with AEC substrate (Sigma-Aldrich, St. Louis, MO) and analyzed in an ImmunoSpot analyzer (Cellular Technology, Shaker Heights, OH). Cytokine secretion is expressed as spots/well.
Flow cytometry and cell sorting
CD41 T cells were stained with up to four directly conjugated fluorescent antibodies or control antibodies for 30 min at 4°C. After extensive washing the cells were fixed and permeabilized using the Fixation & Permeabilization kit (eBioscience), and intracellular staining of FOXP3 and CTLA-4 was performed according to the manufacturer's recommendations. Data were acquired on a FACsCalibur (BD Biosciences, San Jose, CA) and analyzed using FlowJo software (Tree Star, Ashland, OR). For cell sorting experiments, CD4+ cells stained for desired cell surface markers were isolated using a FACSAria or FACSVantage (BD Biosciences) apparatus.
Real-time PCR
PCR was performed using the TaqMan Gene Expression Assay Kit (TaqMan, MGC probes, Applied Biosystems, Foster City, CA) and the 7300 real-time PCR system. Gene-specific primers for the analysis of human Tbet and GAPDH by real-time PCR were obtained from Applied Biosystems.
Lymphocyte migration assays
Migration of lymphocyte subpopulations in response to IP-10 (CXCL10) was quantified at single-cell resolution using microfluidic devices and time-lapse microscopy, as described previously [46]. Briefly, photoresist (SU8, Microchem, Newton, MA), was patterned within silicon wafers, which were used as a mold to produce a PDMS (Fisher Scientific, Fair Lawn, NJ) device, which was then bonded onto standard 1 × 3 in. glass slides (Fisher Scientific). The microfluidic network inside each device consisted of an array of up to 450 parallel channels (6 × 6 μm cross-section and 800 μm long) connected to one main channel, (50 μm tall, 400 μm wide and 10 mm long) with inlets and outlets. The devices were first primed with a solution of IP-10 (100 nM) and fibronectin (250 nM) for 15 min. After priming, sorted populations of either CXCR3+ or CXCR3− CD4+CD25+CD127dim/−Tregs (~1 × 105/condition) suspended in 15 μL of media were introduced into the main channel through tubing connected to the main inlet. The cells were flushed through the main channel until media was seen to emerge from the main outlet. Immediately after loading, the inlet and outlet were sealed and the device was placed on a Nikon Ti microscope fitted with an environmental chamber, motorized stage and Nikon Elements software for time-lapse imaging. At least 20 fields were imaged every 90 s, such that one frame equals 1.5 min for each condition.
Cell migration was manually tracked from time-lapse microscopy images using ImageJ (NIH). One-dimensional trajectories were analyzed for the following quantitative metrics: (i) total displacement (the difference between the initial and the final cell position within the device), (ii) total integrated distance (the sum of the distances traveled in successive images), and (iii) directional persistence (the nondimensional ratio of total displacement to total integrated distance. This parameter was designated as equal to zero for completely random motion, where the cell travels distances but ultimately returns to its initial position. Conversely, the parameter was designated as equal to one for directed motion where the cell travels toward its final position along the chemokine gradient and ends up at its destination. Thus, if cell motion is directed toward a chemokine, but then reverses toward its initial position the final designation will be less than one.) Average velocity (the total integrated distance divided by the duration of the trajectory) was also calculated as a quantitative metric.
Patient samples for in vivo studies
PBMCs were obtained from pediatric recipients of living-donor kidney transplants (n 5 = 4) and adult recipients of cadaveric kidney transplants, who received long-term immunosuppression with prednisone, mycophenolate mofetil and rapamycin [49]. Adult recipients received CsA in the initial post-transplantation period and were converted into an everolimus-based regimen at 2 or 3 months post transplantation (n = 8) or maintained on the calcineurin inhibitor-based regimen (CsA, n = 10). Human peripheral blood was obtained in accordance with IRB approval at Children's Hospital Boston and the University of Dvisberg Essen. Patient blood samples collected during the first 12 months post transplantation were cryopreserved in cell culture medium (above) containing 10% DMSO (Sigma-Aldrich) until analysis. Cells were carefully thawed and washed and cultured for 3 h before flow cytometric analysis.
Statistical analysis
Statistical analyses were performed, using the Wilcoxon matched pair test, Mann-Whitney U-test test and/or Student's t test, as indicated, for comparison of multiple groups. p-Values<0.05 were considered statistically significant.
Acknowledgements
This work was supported by National Institutes of Health Grants U01 AI46135 (To W.E.H and D.M.B) and PO1 AI50157 (to D. M. B.), R01 GM092804 (to D. I.), and by research grants from the Deutsche Forschungsgemeinschaft (HO2581/3-1 to A. H.) and the Damon Runyon Cancer Research Foundation (to I. W.). Adult patients evaluated in this study were managed by Dr. Oliver Witzke, Department of Nephrology, University Hospital Essen, Essen, Germany. Pediatric patients evaluated in this study were under the auspices of the Cooperative Clinical Trials in Pediatric Transplantation (CCTPT) of the National Institute of Allergy and Infectious Diseases, National Institutes of Health. The authors thank Leslie Spaneas RN for help with pediatric patient data analysis.
Abbreviations
- CTLA-4
cytotoxic T-lymphocyte antigen 4;
- CsA
Cyclosporine A;
- CXCR3
CXC chemokine receptor 3;
- FOXP3
forkhead box P3;
- IP-10
inducible protein of 10 kDa;
- mTOR
mammalian target of rapamycin
Footnotes
Conflict of interest: The authors declare no financial or commercial conflicts of interest.
References
- 1.Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+regulatory T cells in the human immune system. Nat. Rev. Immunol. 2010;10:490–500. doi: 10.1038/nri2785. [DOI] [PubMed] [Google Scholar]
- 2.Shevach EM. Mechanisms of foxp31T regulatory cell-mediated suppression. Immunity. 2009;30:636–645. doi: 10.1016/j.immuni.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 3.Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 2001;27:20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 4.Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, Levy-Lahad E, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat. Genet. 2001;27:18–20. doi: 10.1038/83707. [DOI] [PubMed] [Google Scholar]
- 5.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+regulatory T cells. Nat. Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 6.Shevach EM, DiPaolo RA, Andersson J, Zhao DM, Stephens GL, Thornton AM. The lifestyle of naturally occurring CD4+CD25+Foxp3+regulatory T cells. Immunol. Rev. 2006;212:60–73. doi: 10.1111/j.0105-2896.2006.00415.x. [DOI] [PubMed] [Google Scholar]
- 7.Tang Q, Bluestone JA. The Foxp3+regulatory T cell: a jack of all trades, master of regulation. Nat. Immunol. 2008;9:239–244. doi: 10.1038/ni1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thornton AM, Shevach EM. CD4+CD25+immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med. 1998;188:287–296. doi: 10.1084/jem.188.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hara M, Kingsley CI, Niimi M, Read S, Turvey SE, Bushell AR, Morris PJ, et al. IL-10 is required for regulatory T cells to mediate tolerance to alloantigens in vivo. J. Immunol. 2001;166:3789–3796. doi: 10.4049/jimmunol.166.6.3789. [DOI] [PubMed] [Google Scholar]
- 10.Asseman C, Mauze S, Leach MW, Coffman RL, Powrie F. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J. Exp. Med. 1999;190:995–1004. doi: 10.1084/jem.190.7.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Green EA, Gorelik L, McGregor CM, Tran EH, Flavell RA. CD4+CD25+T regulatory cells control anti-islet CD8+ T cells through TGF-beta-TGF-beta receptor interactions in type 1 diabetes. Proc. Natl. Acad. Sci. USA. 2003;100:10878–10883. doi: 10.1073/pnas.1834400100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fahlen L, Read S, Gorelik L, Hurst SD, Coffman RL, Flavell RA, Powrie F. T cells that cannot respond to TGF-beta escape control by CD4(+)CD25(+) regulatory T cells. J. Exp. Med. 2005;201:737–746. doi: 10.1084/jem.20040685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, Cross R, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450:566–569. doi: 10.1038/nature06306. [DOI] [PubMed] [Google Scholar]
- 14.Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+T cells. Nat. Immunol. 2007;8:1353–1362. doi: 10.1038/ni1536. [DOI] [PubMed] [Google Scholar]
- 15.Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, Chen JF, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007;204:1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Long ET, Baker S, Oliveira V, Sawitzki B, Wood KJ. Alpha-1,2-mannosidase and hence N-glycosylation are required for regulatory T cell migration and allograft tolerance in mice. PLoS One. 2010;5:e8894. doi: 10.1371/journal.pone.0008894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nadig SN, Wieckiewicz J, Wu DC, Warnecke G, Zhang W, Luo S, Schiopu A, et al. In vivo prevention of transplant arteriosclerosis by ex vivo-expanded human regulatory T cells. Nat. Med. 2010;16:809–813. doi: 10.1038/nm.2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang N, Schroppel B, Lal G, Jakubzick C, Mao X, Chen D, Yin N, et al. Regulatory T cells sequentially migrate from inflamed tissues to draining lymph nodes to suppress the alloimmune response. Immunity. 2009;30:458–469. doi: 10.1016/j.immuni.2008.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huehn J, Siegmund K, Lehmann JC, Siewert C, Haubold U, Feuerer M, Debes GF, et al. Developmental stage, phenotype, and migration distinguish naive- and effector/memory-like CD4+regulatory T cells. J. Exp. Med. 2004;199:303–313. doi: 10.1084/jem.20031562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huehn J, Hamann A. Homing to suppress: address codes for Treg migration. Trends Immunol. 2005;26:632–636. doi: 10.1016/j.it.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 21.Roncarolo MG, Battaglia M. Regulatory T-cell immunotherapy for tolerance to self antigens and alloantigens in humans. Nat. Rev. Immunol. 2007;7:585–598. doi: 10.1038/nri2138. [DOI] [PubMed] [Google Scholar]
- 22.Hirahara K, Liu L, Clark RA, Yamanaka K, Fuhlbrigge RC, Kupper TS. The majority of human peripheral blood CD4+CD25high-Foxp3+regulatory T cells bear functional skin-homing receptors. J. Immunol. 2006;177:4488–4494. doi: 10.4049/jimmunol.177.7.4488. [DOI] [PubMed] [Google Scholar]
- 23.Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+CD25high regulatory cells in human peripheral blood. J. Immunol. 2001;167:1245–1253. doi: 10.4049/jimmunol.167.3.1245. [DOI] [PubMed] [Google Scholar]
- 24.Lee I, Wang L, Wells AD, Dorf ME, Ozkaynak E, Hancock WW. Recruitment of Foxp3+T regulatory cells mediating allograft tolerance depends on the CCR4 chemokine receptor. J. Exp. Med. 2005;201:1037–1044. doi: 10.1084/jem.20041709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoffmann P, Eder R, Kunz-Schughart LA, Andreesen R, Edinger M. Large-scale in vitro expansion of polyclonal human CD4(+)CD25high regulatory T cells. Blood. 2004;104:895–903. doi: 10.1182/blood-2004-01-0086. [DOI] [PubMed] [Google Scholar]
- 26.Baggiolini M. Chemokines and leukocyte traffic. Nature. 1998;392:565–568. doi: 10.1038/33340. [DOI] [PubMed] [Google Scholar]
- 27.Lacotte S, Brun S, Muller S, Dumortier H. CXCR3, inflammation, and autoimmune diseases. Ann. N. Y. Acad. Sci. 2009;1173:310–317. doi: 10.1111/j.1749-6632.2009.04813.x. [DOI] [PubMed] [Google Scholar]
- 28.Melter M, Exeni A, Reinders ME, Fang JC, McMahon G, Ganz Hancock, W. W., Briscoe DM. Expression of the chemokine receptor CXCR3 and its ligand IP-10 during human cardiac allograft rejection. Circulation. 2001;104:2558–2564. doi: 10.1161/hc4601.098010. [DOI] [PubMed] [Google Scholar]
- 29.Hoffmann U, Segerer S, Rummele P, Kruger B, Pietrzyk M, Hofstadter F, Banas B, Kramer BK. Expression of the chemokine receptor CXCR3 in human renal allografts - a prospective study. Nephrol. Dial. Transplant. 2006;21:1373–1381. doi: 10.1093/ndt/gfk075. [DOI] [PubMed] [Google Scholar]
- 30.Panzer U, Reinking RR, Steinmetz OM, Zahner G, Sudbeck U, Fehr S, Pfalzer B, et al. CXCR3 and CCR5 positive T-cell recruitment in acute human renal allograft rejection. Transplantation. 2004;78:1341–1350. doi: 10.1097/01.tp.0000140483.59664.64. [DOI] [PubMed] [Google Scholar]
- 31.Hancock WW, Gao W, Csizmadia V, Faia KL, Shemmeri N, Luster AD. Donor-derived IP-10 initiates development of acute allograft rejection. J. Exp. Med. 2001;193:975–980. doi: 10.1084/jem.193.8.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hancock WW, Lu B, Gao W, Csizmadia V, Faia K, King JA, Smiley ST, et al. Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J. Exp. Med. 2000;192:1515–1520. doi: 10.1084/jem.192.10.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tatapudi RR, Muthukumar T, Dadhania D, Ding R, Li B, Sharma VK, Lozada-Pastorio E, et al. Noninvasive detection of renal allograft inflammation by measurements of mRNA for IP-10 and CXCR3 in urine. Kidney Int. 2004;65:2390–2397. doi: 10.1111/j.1523-1755.2004.00663.x. [DOI] [PubMed] [Google Scholar]
- 34.Zhao DX, Hu Y, Miller GG, Luster AD, Mitchell RN, Libby P. Differential expression of the IFN-gamma-inducible CXCR3-binding chemokines, IFN-inducible protein 10, monokine induced by IFN, and IFN-inducible T cell alpha chemoattractant in human cardiac allografts: association with cardiac allograft vasculopathy and acute rejection. J. Immunol. 2002;169:1556–1560. doi: 10.4049/jimmunol.169.3.1556. [DOI] [PubMed] [Google Scholar]
- 35.Fahmy NM, Yamani MH, Starling RC, Ratliff NB, Young JB, McCarthy PM, Feng J, et al. Chemokine and receptor-gene expression during early and late acute rejection episodes in human cardiac allografts. Transplantation. 2003;75:2044–2047. doi: 10.1097/01.TP.0000069601.73079.94. [DOI] [PubMed] [Google Scholar]
- 36.Hu H, Aizenstein BD, Puchalski A, Burmania JA, Hamawy MM, Knechtle SJ. Elevation of CXCR3-binding chemokines in urine indicates acute renal-allograft dysfunction. Am. J. Transplant. 2004;4:432–437. doi: 10.1111/j.1600-6143.2004.00354.x. [DOI] [PubMed] [Google Scholar]
- 37.Hu H, Kwun J, Aizenstein BD, Knechtle SJ. Noninvasive detection of acute and chronic injuries in human renal transplant by elevation of multiple cytokines/chemokines in urine. Transplantation. 2009;87:1814–1820. doi: 10.1097/TP.0b013e3181a66b3e. [DOI] [PubMed] [Google Scholar]
- 38.Muller M, Carter SL, Hofer MJ, Manders P, Getts DR, Getts MT, Dreykluft A, et al. CXCR3 signaling reduces the severity of experimental autoimmune encephalomyelitis by controlling the parenchymal distribution of effector and regulatory T cells in the central nervous system. J. Immunol. 2007;179:2774–2786. doi: 10.4049/jimmunol.179.5.2774. [DOI] [PubMed] [Google Scholar]
- 39.Hasegawa H, Inoue A, Kohno M, Lei J, Miyazaki T, Yoshie O, Nose M, Yasukawa M. Therapeutic effect of CXCR3-expressing regulatory T cells on liver, lung and intestinal damages in a murine acute GVHD model. Gene Ther. 2008;15:171–182. doi: 10.1038/sj.gt.3303051. [DOI] [PubMed] [Google Scholar]
- 40.Oo YH, Weston CJ, Lalor PF, Curbishley SM, Withers DR, Reynolds GM, Shetty S, et al. Distinct roles for CCR4 and CXCR3 in the recruitment and positioning of regulatory T cells in the inflamed human liver. J. Immunol. 2010;184:2886–2898. doi: 10.4049/jimmunol.0901216. [DOI] [PubMed] [Google Scholar]
- 41.Eksteen B, Miles A, Curbishley SM, Tselepis C, Grant AJ, Walker LS, Adams DH. Epithelial inflammation is associated with CCL28 production and the recruitment of regulatory T cells expressing CCR10. J. Immunol. 2006;177:593–603. doi: 10.4049/jimmunol.177.1.593. [DOI] [PubMed] [Google Scholar]
- 42.Kwun J, Hazinedaroglu SM, Schadde E, Kayaoglu HA, Fechner J, Hu HZ, Roenneburg D, et al. Unaltered graft survival and intragraft lymphocytes infiltration in the cardiac allograft of Cxcr3−/− mouse recipients. Am. J. Transplant. 2008;8:1593–1603. doi: 10.1111/j.1600-6143.2008.02250.x. [DOI] [PubMed] [Google Scholar]
- 43.Sporici R, Issekutz TB. CXCR3 blockade inhibits T-cell migration into the CNS during EAE and prevents development of adoptively transferred, but not actively induced, disease. Eur. J. Immunol. 2010;40:2751–2761. doi: 10.1002/eji.200939975. [DOI] [PubMed] [Google Scholar]
- 44.Read S, Malmstrom V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+) CD4(+) regulatory cells that control intestinal inflammation. J. Exp. Med. 2000;192:295–302. doi: 10.1084/jem.192.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat. Immunol. 2009;10:595–602. doi: 10.1038/ni.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Butler KL, Ambravaneswaran V, Agrawal N, Bilodeau M, Toner M, Tompkins RG, Fagan S, Irimia D. Burn injury reduces neutrophil directional migration speed in microfluidic devices. PLoS One. 2010;5:e11921. doi: 10.1371/journal.pone.0011921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zeiser R, Leveson-Gower DB, Zambricki EA, Kambham N, Beilhack A, Loh J, Hou JZ, Negrin RS. Differential impact of mammalian target of rapamycin inhibition on CD4+CD25+Foxp31 regulatory T cells compared with conventional CD4+T cells. Blood. 2008;111:453–462. doi: 10.1182/blood-2007-06-094482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, Worley PF, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harmon W, Meyers K, Ingelfinger J, McDonald R, McIntosh M, Ho M, Spaneas L, et al. Safety and efficacy of a calcineurin inhibitor avoidance regimen in pediatric renal transplantation. J. Am. Soc. Nephrol. 2006;17:1735–1745. doi: 10.1681/ASN.2006010049. [DOI] [PubMed] [Google Scholar]
- 50.Muthukumar T, Dadhania D, Ding R, Snopkowski C, Naqvi R, Lee JB, Hartono C, et al. Messenger RNA for FOXP3 in the urine of renal-allograft recipients. N. Engl. J. Med. 2005;353:2342–2351. doi: 10.1056/NEJMoa051907. [DOI] [PubMed] [Google Scholar]
- 51.Warnecke G, Feng G, Goto R, Nadig SN, Francis R, Wood KJ, Bushell A. CD4+ regulatory T cells generated in vitro with IFN-{gamma} and allogeneic APC inhibit transplant arteriosclerosis. Am. J. Pathol. 2010;177:464–472. doi: 10.2353/ajpath.2010.090292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gao W, Lu Y, El Essawy B, Oukka M, Kuchroo VK, Strom TB. Contrasting effects of cyclosporine and rapamycin in de novo generation of alloantigen-specific regulatory T cells. Am. J. Transplant. 2007;7:1722–1732. doi: 10.1111/j.1600-6143.2007.01842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bestard O, Cruzado JM, Grinyo JM. Inhibitors of the mammalian target of rapamycin and transplant tolerance. Transplantation. 2009;87:S27–S29. doi: 10.1097/TP.0b013e3181a07b08. [DOI] [PubMed] [Google Scholar]