

Abstract
Although PDK1 regulates several signaling pathways that respond to neurotrophins, direct evidence for its involvement in neurotrophin-mediated survival has not yet been reported. Here we show high neuronal expression of active PDK1 in the rat cortex and hippocampus at the developmental stages with pronounced dependence on extracellular survival signals. Also, in cultured cortical neurons from newborn rats, BDNF resulted in PDK1- and extracellular signal-regulated kinase-1/2 (ERK1/2)-mediated activation of their direct target, the p90 ribosomal S6 kinase 1/2 (RSK1/2). In trophic-deprived cortical neurons, knockdown of endogenous PDK1 attenuated the antiapoptotic survival response to 10 ng/ml BDNF, whereas an overexpressed active mutant form of PDK1 reduced apoptosis. The neuroprotection by BDNF or active PDK1 required RSK1/2. Conversely, PDK1 knockdown reversed the survival effects of combining the overexpressed RSK1 with a low, subprotective BDNF concentration of 2 ng/ml. Likewise, the protection by the overexpressed, active PDK1 was enhanced by coexpression of an active RSK1 mutant. Consistent with the observations that in BDNF-stimulated neurons RSK1/2 activation required both PDK1 and ERK1/2, ERK1/2 knockdown removed BDNF-mediated survival. Selective activation of ERK1/2 with an overexpressed active mutant form of MKK1 resulted in RSK1/2- and PDK1-dependent neuroprotection. Finally, at subprotective plasmid DNA dosage, overexpression of the active MKK1 and PDK1 mutants produced synergistic effect on survival. Our findings indicate a critical role for PDK1-RSK1/2 signaling in BDNF-mediated neuronal survival. Thus, the PDK1 is indispensable for the antiapoptotic effects of the ERK1/2 pathway offering previously unrecognized layer of survival signal processing and integration.
Keywords: neurotrophins, TrkB, p90RSK, MAP-kinase, protein kinase B, Akt, development, survival
Introduction
During neuronal development, avoidance of programmed cell death depends on extracellular signals provided by neurotrophins such as brain-derived neurotrophic factor (BDNF), and the neurotransmitter glutamate, acting at the N-methyl-d-aspartate receptors (NMDAR) (Alcántara et al., 1997; Olney, 2004; Buss et al., 2006). In differentiating postmitotic rodent neurons that undergo synaptogenesis, these signals prevent the default apoptosis matching neuronal connectivity with their number. The signal transduction pathways that mediate neuronal survival responses include phosphatidyloinositol-3-kinase (PI3K)-Akt and ERK1/2-RSK1/2 (Dudek et al., 1997; Bonni et al., 1999).
PDK1 is a Ser/Thr kinase that regulates several AGC kinases including ERK1/2-dependent activation of RSK1/2 and PI3K-dependent activation of Akt (Mora et al., 2004). The antiapoptotic effects of PDK1 have been reported in non-neuronal cells (Flynn et al., 2000; Cho et al., 2001; Sato et al., 2002; Liang et al., 2006; Hayashi et al., 2007). The involvement of PDK1 in neuronal development has been indicated by the phenotype of PDK1-deficient mice, which includes the lack of a forebrain and dorsal root ganglia at embryonic day 9.5 and ultimately results in death in utero (Lawlor et al., 2002). Although it has been proposed that these developmental disturbances may be attributable to excessive neuronal death, the PDK1 contribution to neuronal survival has not been directly verified.
The survival effects of PDK1 have been attributed to its role as a PI3K-dependent activator of Akt (Flynn et al., 2000; Cho et al., 2001; Sato et al., 2002; Liang et al., 2006; Hayashi et al., 2007); however, the prosurvival effects of PDK1 may also involve other PDK1 targets. For instance, survival of cancer cells was affected relatively more by manipulating PDK1 rather than Akt (Flynn et al., 2000; Liang et al., 2006). In addition, forebrain development remained disturbed in mice whose PDK1 was replaced by the mutants with normal ability to regulate Akt but not the PI3K-independent targets, including RSK1/2 (Collins et al., 2003; McManus et al., 2004; Collins et al., 2005). Conversely, no neurodevelopmental abnormalities were reported in mice whose PDK1 was selectively impaired in PI3K-dependent interactions with Akt (Bayascas et al., 2008). The identity of non-Akt targets that contribute to PDK1-mediated cell survival remains to be determined.
PDK1-mediated phosphorylation of RSK1/2 Ser-221/227 activates the RSK1/2 N-terminal kinase domain (NTK) in a PI3K-independent manner (Jensen et al., 1999; Hauge and Frödin, 2006). PDK1 is recruited to RSK1/2 after ERK1/2-dependent activation of the RSK1/2 C-terminal kinase domain (CTK) which autophosphorylates RSK1/2 Ser-380/386 thereby creating a PDK1-binding site to phosphorylate Ser-221/227 of NTK. Then, the active RSK NTK phosphorylates its substrates including the proapoptotic protein Bad or the transcription factor CREB that contribute to neuronal survival and/or synaptic plasticity (Bonni et al., 1999; Adams and Sweatt, 2002; Hauge and Frödin, 2006). However, the physiological significance of PDK1-RSK1/2 interactions has not been yet directly tested in neurons.
This study was initiated to directly evaluate the role of PDK1 in the BDNF-dependent survival of developing cortical neurons. In addition we set out to determine whether RSK1/2 is a relevant antiapoptotic target of neuronal PDK1.
Materials and Methods
Materials.
The following plasmids have been described previously: pCEP4-HA-MKK1 wild-type, constitutively active and dominant negative (Mansour et al., 1994), rat wild-type pCMV26-FLAG-RSK1, rat pCMV26-FLAG-RSK1 constitutively active, mouse wild-type pCMV26-FLAG-RSK2 (Silverman et al., 2004); rat wild-type pCDNA3-HA-ERK1 and -ERK2 (Dimitri et al., 2005); pON260 (Cherrington and Mocarski, 1989); pcDNA3-FLAG-BCL-2 (del Peso et al., 1997); CMV-p53-DD (Shaulian et al., 1992); mouse pcDNA-PDK1A280V (Wick et al., 2000); the TCF/Elk luciferase reporter construct pE74-Luc (Vickers et al., 2004); pSUPER vector (Brummelkamp et al., 2002); EF1αLacZ and pSuper-based small hairpin RNA (shRNA) construct targeting GFP (Kalita et al., 2006). The following antibodies and reagents were obtained from commercial sources: goat anti-RSK1, rabbit anti-phospho-Ser380/386-RSK1/2, rabbit anti-phospho-Ser221/227-RSK1/2 (Santa Cruz Biotechnology), rabbit anti-phospho(Thr183/Tyr185)-ERK1/2 antibody and mouse anti-β-galactosidase (Promega), rabbit anti-PDK1 (Genscript), rabbit anti-phospho(Ser241)-PDK1 and mouse anti-ERK1/2 antibodies (Cell Signaling Technology), mouse anti-HA antibody (Roche), rabbit anti-β-galactosidase (MP Biomedicals), monoclonal mouse anti-FLAG M2 and mouse anti-β-Actin antibodies (Sigma), BDNF (Alomone Labs), dizocilpine maleate (MK-801), N-α-tosyl-l-phenylalanyl chloromethyl ketone (TPCK), 2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one (LY294002), U0126, CREBtide, Hoechst 33258 and protein G-Sepharose (Sigma or Calbiochem).
Generation of shRNA expression constructs.
To design shRNA constructs the respective human, rat and mouse mRNA sequences were analyzed using small interfering RNA design software (http://sonnhammer.cgb.ki.SE/). For inhibition of PDK1, we selected two target sequences that were conserved between human, mouse and rat: shPDK1–1, GCACATCCAGATCACAGA; shPDK1–2, GGGTTTATTTGCAAGACGA. For RSK silencing 2 appropriate target sequences were selected that were conserved between mouse and rat: shRSK1–1, GGATCCTTTGGCAAAGTCT; shRSK1–2, GGAAACTGGAACACGGTTT; shRSK2–1, GGATGAGTCTGGTAATCCA; shRSK2–2, CTGTTTCAGACACAGCAAA. For ERK1/2 shRNAs, we used modified target sequences that were identified by others (Yun et al., 2005; Vantaggiato et al., 2006): shERK1–1, GACCGGATGTTAACCTTTA; shERK1–2, ATGTCATAGGCATCCGAGA; shERK2–1, GTACAGAGCTCCAGAAATT; shERK2–2, AGTTCGAGTTGCTATCAAG. Based on the target sequences, the appropriate complementary oligonucleotides were designed, synthesized, annealed and cloned into pSUPER vector according to the manufacturer's recommendations (Oligoengine).
Cell culture and transfection.
Cortical neurons were prepared from newborn Sprague Dawley rats at postnatal day 0 (P0) as described previously (Habas et al., 2006). Briefly, culture medium was Basal Medium Eagle (BME) supplemented with 10% heat-inactivated bovine calf serum (Hyclone), 35 mm glucose, 1 mm l-glutamine, 100 U/ml of penicillin and 0.1 mg/ml streptomycin. Cytosine arabinoside (2.5 μm) was added to cultures on the second day after seeding [day in vitro 2 (DIV 2)] to inhibit the proliferation of non-neuronal cells. Cells were used for experiments on DIV 4–6 unless indicated otherwise. Transient transfections were performed on DIV 3–4 using the Lipofectamine 2000 reagent (Invitrogen) as described previously (Hetman et al., 2002). Electroporation of freshly dissociated newborn rat cortical neurons was conducted using a rat neuron nucleofection reagent kit (Amaxa).
Trophic deprivation.
Trophic deprivation (TD) that included serum withdrawal and NMDAR block was performed as described (Hetman et al., 2000). Briefly, cells were washed twice with serum-free BME and then incubated 24 h in serum-free BME supplemented with 35 mm glucose, 1 mm l-glutamine, 100 units/ml penicillin, 0.1 mg/ml streptomycin, 10 μm MK-801 and 2.5 μm cytosine arabinoside. The experimental controls included sham treatments consisting of the removal of growth media (conditioned media), washes with serum-free BME and replacement of the conditioned media.
Drug treatment.
BDNF was diluted in PBS containing 0.1% bovine serum albumin before addition to the cells. TPCK, U0126, and LY294002 were dissolved in dimethyl sulfoxide (DMSO). The final concentration of DMSO in the medium was 0.2–0.5%. For Western blot analysis and immunocomplex kinase assays, inhibitors were added 3 h before BDNF stimulation. For apoptosis analysis, BDNF was added at the onset of TD.
Immunofluorescence and immunohistochemistry.
Transfected cells were detected by immunostaining with a rabbit antibody against β-galactosidase (MP Biomedicals) and Texas Red-conjugated goat antibody against rabbit Ig (Calbiochem) using standard immunofluorescence methodology. For pERK and pRSK immunostaining, mouse anti-pERK (Cell Signaling Technology) and rabbit anti-phospho-Ser221/227-RSK1/2 (Santa Cruz Biotechnology) were used. Cells were fixed with 4% paraformaldehyde in PBS and incubated in methanol for 10 min at −20°C. Primary antibodies were diluted in PBS/0.3% TritonX100 at the following concentrations: pRSK 1:100 and pERK 1:200 and incubated overnight at 4°C. Secondary biotin anti-rabbit antibody was added to enhance the pRSK signal followed by streptavidine-Alexa 555 and anti-mouse Alexa 488 for pERK (Invitrogen). Nuclei were counterstained with 2.5 μg/ml Hoechst 33258.
For immunohistochemical analysis of PDK1 expression, naive P7 (n = 4) Sprague Dawley rats were housed with their siblings under a 12 h light/dark cycle and with ad libitum access to water and food. All animals were treated in accordance with the guidelines of the National Institutes of Health and the University of Louisville Guidelines for the Care and Use of Laboratory Animals. Anesthetized animals were perfused transcardially, and free-floating 30 μm-thick coronal brain sections were prepared after postfixation (Yang et al., 2005). For staining, sections were washed in Tris-buffered saline (TBS; 0.05 mm Tris and 0.9% NaCl, pH 8.0), the endogenous peroxidase was quenched for 5 min (10% methanol and 1% H2O2 in TBS), and the cell membrane was permeabilized (15 min in 0.5% Triton X-100 in TBS). A rabbit polyclonal anti-PDK1 antibody (1:1000; anti-PDK1 Ab-241; Genscript) was applied in a blocking buffer (10% normal goat serum, 3% BSA in TBS) overnight at 4°C. The sections were then washed with TBS and incubated with secondary antibody (biotinylated anti-rabbit IgG; 1:1000 dilution; Invitrogen) for 2 h at room temperature, followed by a standard ABC/DAB histochemistry (Vector Laboratories).
Quantification of apoptosis.
Cell nuclei were visualized with Hoechst 33258. The transfected, β-gal-positive cells with uniformly stained nuclei were scored as viable. The transfected cells displaying condensed or fragmented nuclei were scored as apoptotic. At least 200 cells were evaluated for each condition in each independent experiment.
Western analysis and RSK1/2 immunocomplex kinase assays.
For Western analysis, cortical neurons were washed twice with PBS and lysed in SDS-PAGE sample buffer. Rat brain tissue samples were grinded and homogenized in Triton lysis buffer (TLB: 20 mm Tris-HCl pH 7.4, 140 mm NaCl, 10 mm NaF, 25 mm β-glycerolphosphate, 2 mm EDTA, 2 mm NaPPi, 1 mm Na3VO4, 1% Triton X-100, 10% glycerol, 1 mm DTT and Calbiochem set III protease inhibitors) and centrifuged at 10,000 g for 15 min. Supernatant protein concentrations were determined by the Bradford method. Standard SDS-PAGE and immunoblotting techniques were applied.
For RSK1/2 kinase assays neurons were lysed in TLB. RSK1/2 was immunoprecipitated using 1 μg anti-RSK1 antibody (Santa Cruz Biotechnology) or monoclonal mouse anti-FLAG M2 antibody (Sigma) and 20 μl protein G-Sepharose/2 × 106 neurons. The anti-RSK1 antibody detected overexpressed RSK1 or RSK2 with similar potencies (data not shown). Kinase reactions were performed in 25 mm Hepes, pH 7.4, 25 mm β-glycerolphosphate, 25 mm MgCl2, 1 mm Na3VO4, 100 μM ATP, 0.025 μCi/μl γ32P-ATP, and 200 μm CREBtide as a substrate at 30°C for 30 min. The reactions were terminated by spotting on Whatman p81 filter papers followed by washing with 75 mm phosphoric acid. The 32P incorporation into CREBtide was determined by scintillation counting.
Statistical analysis.
Statistical analysis of the data were performed using one- or two-way ANOVA followed by post hoc tests.
Results
High levels of PDK1 expression in rat forebrain
To evaluate whether PDK1 may contribute to the antiapoptotic transduction pathways in the developing rat brain we determined its forebrain expression at P1, P7, and P21. At P1 and P7, survival of cortical neurons depends on extracellular survival signals including BDNF/TrkB and/or glutamate/NMDAR signaling (Alcántara et al., 1997; Olney, 2004). At P7, abundant PDK1 immunoreactivity was detected in the perikaria of neurons in the cortex and hippocampus (Fig. 1). Similar pattern of PDK1 distribution was observed at P21 (data not shown). Importantly, the active status of the cortical PDK1 was indicated by high basal levels of autophosphorylated PDK1 at the Ser-241 residue (pPDK1) with the ratio of pPDK/PDK1 remaining similar at all ages investigated (Fig. 2A,B). The presence of active PDK1 in the developing cortex was further confirmed by the observation of PDK1-phosphorylated RSK1/2 at the Ser-221/227 residue (Fig. 2A,B). Of note, the levels of pSer221/227-RSK1/2 gradually declined over the studied period of cortical development reaching 30% of P1 values at P21 (p < 0.05) (Fig. 2A,B). These results indicate that in cerebral cortex the relatively higher activity of PDK/RSK signaling coincides with the stronger cortical neuron dependence on extracellular survival signals at P1 and P7 compared with P21.
Figure 1.
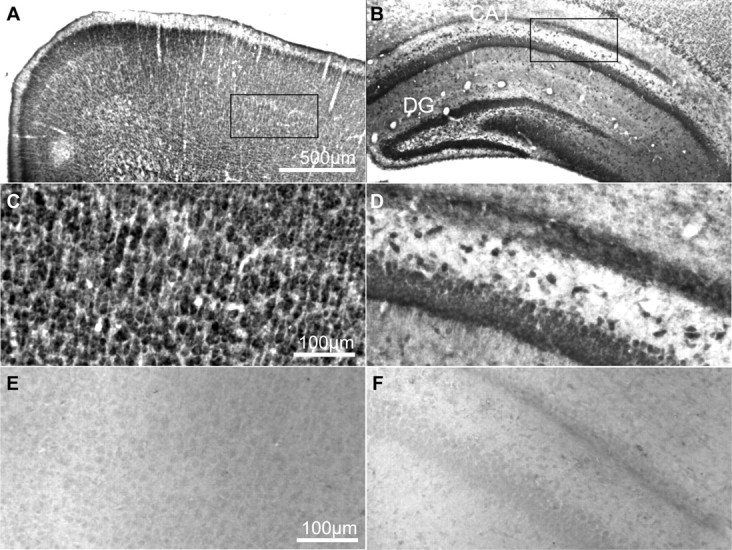
PDK1 expression in the rat brain. A–D, PDK1 immunoreactivity in coronal sections through forebrains of rat pups at P7. PDK1 was visualized using ABC/DAB immunohistochemistry. E, F, The specificity of PDK1 labeling was confirmed in control experiments, in which the primary antibody was replaced by the nonimmune rabbit IgG. Intense PDK1 staining was found in neuron-like cells in the neocortical layers II–VI (A, C) as well as the pyramidal cell layer of the Ammon's horn and granule cell layer of the dentate gyrus (B, D). Similar pattern of PDK1 immunoreactivity was observed at P21 (data not shown). CA, Cornu amonis; DG, dentate gyrus.
Figure 2.

Regulation of PDK1 during rat cortical development. A, B, At P1, P7, and P21, both total PDK1 and phospho-Ser-241 PDK1 were detected by Western blotting of lysates from rat cortex. Note that the pSer241-PDK1/total PDK1 ratio remained similar at all developmental stages examined. In contrast, the PDK1-mediated phosphorylation of RSK1/2 Ser221/227 decreased over the studied ages. Thus, PDK1-RSK1/2 signaling is active in developing forebrain during synaptogenesis when neurons are highly dependent on extracellular survival signals. At P1 and P7, electromobility of total or pSer241 PDK1 was lower than at P21, indicating possible hyperphosphorylation at non-Ser241 sites. In A, same amounts of protein lysates were analyzed in each lane. In B, data represent averages of three animals at each developmental stage ±SEM; the pSer241 PDK1 or pSer221/227 RSK1/2 levels were normalized against total PDK1 or RSK1/2, respectively. *p < 0.05.
BDNF activates the PDK1-RSK1/2 signaling in cultured rat cortical neurons
Next, we used cultured cortical neurons from newborn rats to determine whether BDNF regulates PDK1. Although BDNF did not affect the activity of the immunoprecipitated PDK1 activity in vitro (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material), the PDK1-mediated phosphorylation of RSK1/2 Ser221/227 residues increased in BDNF-stimulated neurons (Fig. 3A). The fourfold increase was observed as early as 10 min after 10 ng/ml BDNF exposure (Fig. 3A). The elevated levels of pSer221/227 persisted at least up to 6 h after initiation of BDNF treatment (Fig. 3A).
Figure 3.
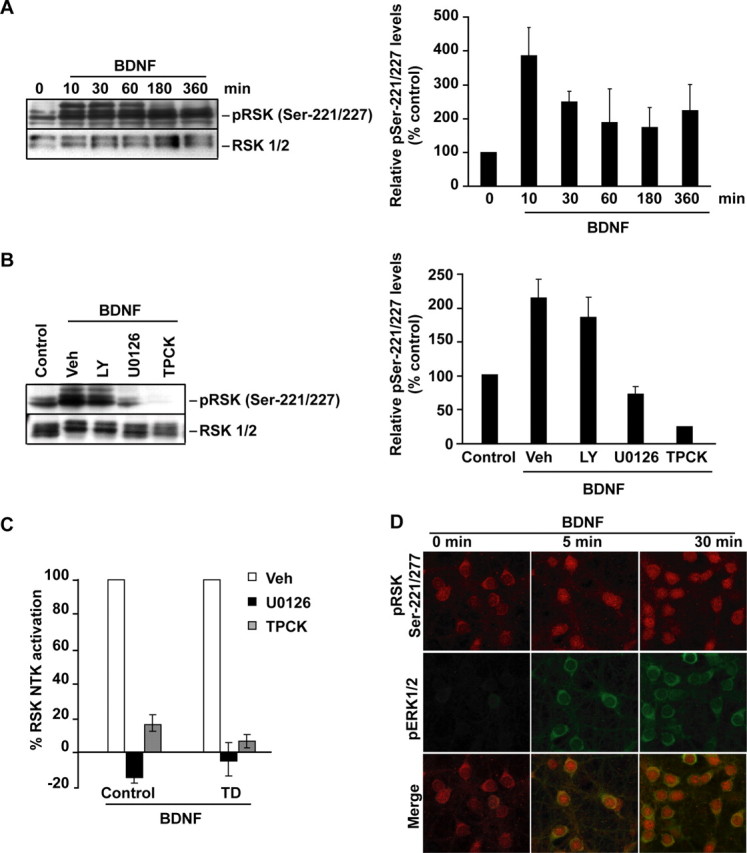
PDK1-mediated activation of RSK1/2 in BDNF-stimulated neurons. Neurons were stimulated with 10 ng/ml BDNF as indicated. In A, B, Western blot analysis of RSK1/2 phosphorylation at Ser221/227 is presented. Blots were reprobed to detect total RSK1/2. For quantifications, the ratios of pSer221/227 to total RSK1/2 were compared with unstimulated controls; averages of two independent experiments ±SD are presented. A, BDNF increased PDK1-mediated phosphorylation of RSK1/2. B, The BDNF-mediated increase of pSer221/227 required activities of the ERK1/2 pathway and PDK1. Although in neurons treated with either the vehicle (Veh; 0.2% DMSO) or the 30 μm PI3K inhibitor LY294002 (LY), a 1 h BDNF treatment elevated pSer221/227, the 50 μm MKK1/2 inhibitor U0126 or the 50 μm PDK1 pathway inhibitor TPCK blocked that effect. Experiments providing additional validation of the inhibitors are presented in supplemental Figure S1B, available at www.jneurosci.org as supplemental material. C, Without drug inhibitors (Veh), cortical neurons that were sham treated (control) or trophic deprived for 3 h and then stimulated with BDNF for 1 h showed increased RSK1/2 NTK activity as determined by immunocomplex kinase assay. The ERK1/2 or PDK1 pathway inhibition abolished RSK1/2 NTK activation by BDNF. Data represent two independent experiments ±SD. D, The coincidence of the MKK1/2-ERK1/2 and PDK1-RSK1/2 signaling in BDNF stimulated neurons. Representative confocal images are of neurons that were coimmunostained for pERK1/2 (pThr183-pTyr185, green) and pSer221/227 (red). BDNF stimulations were performed 1 h after placing neurons in serum-free media supplemented with MK801 to reduce the basal ERK1/2 signaling. Note nuclear localization of PDK1-phosphorylated RSK1/2 in BDNF-treated neurons. Most pERK appeared cytosolic. Similar patterns were observed in two independent experiments. Additional data illustrating the coincidence of the BDNF-mediated RSK1/2 activations at the NTK and CTK are presented in supplemental Figure S2, available at www.jneurosci.org as supplemental material.
The increase of pSer221/227 was abolished by the PDK1 signaling inhibitor, TPCK, and the ERK1/2 pathway inhibitor, U0126, but not the PI3K inhibitor, LY294002 (Fig. 3B). Although in some systems PDK1 may be upstream of the MKK1-ERK1/2 pathway (Corbit et al., 2000; Sato et al., 2004), in cortical neurons, TPCK did not affect BDNF-mediated activation of ERK1/2 but reduced the constitutive autophosphorylation of PDK1 at Ser241 (supplemental Fig. S1B, available at www.jneurosci.org as supplemental material). Therefore, the PDK1 and ERK pathway effects on pSer221/227 may arise by convergent signaling at the RSK level. Such a possibility is in good agreement with the proposed model of RSK1/2 activation by the initial ERK1/2-mediated phosphorylation that stimulates the CTK to generate a PDK1 binding site via autophosphorylation at the Ser380/386 residue (Hauge and Frödin, 2006). PDK1-mediated phosphorylation at the Ser221/227 would then directly activate NTK. Indeed, this activation scenario likely applies to neuronal RSK1/2 as indicated by the concomitant increases in CTK and NTK activities in response to BDNF (supplemental Fig. S2A–D, available at www.jneurosci.org as supplemental material), and the similar sensitivity of the BDNF-mediated NTK activation to PDK1 or ERK1/2 inhibition (Fig. 3C). Last, in BDNF-stimulated neurons, coincident increases of pSer221/227 and pERK1/2 were observed by immunofluorescence analysis (Fig. 3D).
To complement the pharmacological data supporting a role for PDK1 in BDNF-mediated signaling to RSK1/2, shRNA constructs were developed which in cortical neurons knocked down PDK1, ERK1 and ERK2 (Fig. 4A,B). At 72 h after electroporation into cortical neurons, neither shPDK1 nor shERK1 nor shERK2 nor shERK1/2 significantly affected the basal RSK1/2 NTK activity compared with the control shRNA plasmid targeting GFP (shGFP vs shPDK1s or shERK1s or shERK2s or shERKs, p > 0.05) (Fig. 4C). However, in neurons that were treated for 30 min with 10 ng/ml BDNF, the shPDK1 reduced RSK NTK activation by 41% (BDNF treatment, shGFP vs shPDK1, p < 0.05) (Fig. 4C). Similar inhibitory effect was observed with shERK1 (Fig. 4C). Conversely, knockdown of ERK2 removed BDNF activation of RSK NTK (BDNF treatment, shGFP vs shERK2, p < 0.001; shERK2 transfection, 0 vs10 ng/ml BDNF, p > 0.05) (Fig. 4C). These results indicate that BDNF activation of RSK1/2 NTK is reduced or disrupted by the knockdown of ERK1/PDK1 or ERK2, respectively. Because shERKs, but not shPDK1, reduced the BDNF activation of the ERK-regulated transcription factor Elk, these results further support the notion that, in neurons, PDK1 is required to activate RSK NTK in an ERK1/2-dependent manner (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). As just 10% of normal PDK1 activity is sufficient to regulate RSK1/2 (Lawlor et al., 2002), the weaker reduction of BDNF-mediated RSK1/2 activation with shPDK1 s compared with the drug inhibitor TPCK, likely reflects different levels of PDK1 inhibition using these two approaches (compare Fig. 3C and supplemental Fig. S1, available at www.jneurosci.org as supplemental material, with Fig. 4A,C). Indeed, although TPCK inhibition of PDK1 appeared almost complete (Fig. 3B,C; supplemental Fig. S1, available at www.jneurosci.org as supplemental material), shPDK1 reduced the PDK1 levels by no >80% (Fig. 4A). However, we cannot rule out that the greater potency of TPCK than shPDK1 to block RSK1/2 activation is attributable to TPCK off-target activities. Altogether, in BDNF-stimulated cortical neurons, PDK1 phosphorylates and activates RSK1/2 in an ERK1/2-dependent manner. Thus, PDK1 contributes to the BDNF-activated signaling pathway that may be important for the survival of developing neurons.
Figure 4.

Knockdown of PDK1 reduces ERK-dependent activation of RSK1/2. A, Freshly isolated cortical neurons were coelectroporated with two different shRNA constructs targeting PDK1 (shPDK1-1 or -2) together with an expression plasmid for FLAG-Bcl2 that was added to reduce electroporation toxicity (4 + 0.2 μg of each plasmid DNA/5 × 106 cells, respectively). The shRNA against GFP was used as a control (shGFP). After 72 h, Western blot analysis revealed that shPDK1-1 and shPDK1-2 reduced the levels of endogenous PDK1 by 70 or 80% of control, respectively. Numbers under the blot represent the ratios of endogenous PDK1 to Flag-Bcl2 levels. B, Freshly isolated cortical neurons were coelectroporated with pcDNA3-HA-ERK1, pcDNA3-HA-ERK2, and pcDNA3-FLAG-Bcl2 together with one of the following: shGFP, shERK1-1, shERK1-2, shERK2-1, and shERK2-2, as indicated (0.8 + 0.8 + 0.2 + 2.5 μg of each plasmid DNA/5 × 106 neurons, respectively). After 72 h, the levels of HA-ERK1/2 were analyzed by Western blotting with an anti-HA antibody. Numbers under the blot represent the ratios of the indicated HA-ERKs to Flag-Bcl2 levels. Reduced levels of HA-ERK1 or HA-ERK2 compared with Flag-Bcl2 indicate efficient knockdowns with shERK1 or shERK2, respectively. C, Cortical neurons were electroporated with the indicated shRNAs (3 μg plasmid DNA/10 × 106 cells). To reduce electroporation toxicity, the dominant negative p53 expression plasmid (p53-DD) was included in all samples (1 μg plasmid DNA/10 × 106 cells). After 72 h, neurons were stimulated with BDNF for 30 min and the NTK activity of the endogenous RSK1/2 was analyzed by immunocomplex kinase assay. Immunoprecipitates contained similar levels of RSK1/2 as determined by Western blotting (shown at the bottom of the graph). Depletion of PDK1 or ERK1 reduced the BDNF activation of RSK NTK. Depletion of ERK2 abolished that activation. Therefore, ERK1, ERK2, and PDK1 are required for full activation of neuronal RSK1/2 by BDNF. In A, B, similar results were obtained in two independent experiments. In C, averages of three independent experiments ±SEM are presented. *p < 0.05; **p < 0.01; ***p < 0.001; nonsignificant (NS), p > 0.05.
PDK1 mediates antiapoptotic effects of BDNF
To test PDK1 involvement in BDNF-mediated neuronal survival, we determined the effects of PDK1 knockdown on BDNF protection against TD-induced apoptosis. The shPDK1-transfected cortical neurons were trophic deprived by placement in serum-free medium that was supplemented with an NMDAR antagonist, MK801 (10 μm). The TD was performed in the presence or absence of 10 ng/ml BDNF. After 24 h, apoptosis was analyzed in the transfected neurons that were identified by the presence of β-gal immunostaining (Fig. 5A). In shGFP-transfected neurons, BDNF suppressed apoptosis from 75 to 44% (p < 0.001) (Fig. 5B). In PDK1-depleted neurons, the antiapoptotic effect of BDNF was reduced (82 or 63% apoptosis with 0 or 10 ng/ml BDNF, respectively, p < 0.05) (Fig. 5B). These data indicate the requirement of PDK1 for BDNF-mediated neuronal survival.
Figure 5.

Requirement of PDK1 for antiapoptotic activity of BDNF in trophic-deprived neurons. Neurons were cotransfected with shGFP or an equimolar mix of shPDK1-1 and shPDK1-2 (shPDK1) together with the β-galactosidase expression plasmid, pON260 (1 + 0.2 μg plasmid DNA/5 × 105 neurons, respectively). After 24 h, neurons were sham treated or trophic deprived in the presence of 0 or 10 ng/ml BDNF for the next 24 h. A, Representative photomicrographs illustrating morphology of the transfected neurons, which were identified by β-galactosidase immunostaining. Counterstaining with Hoechst 33258 revealed apoptotic alterations including condensation and fragmentation of the chromatin (arrows). Arrowheads point β-galactosidase-positive cells without signs of apoptosis. B, The knockdown of PDK1 reduced BDNF protection against TD-induced apoptosis. The individual shPDK1 plasmids affected BDNF survival similarly to their mix (data not shown). Averages of triplicate determinations from three independent experiments ±SEM are presented. *p < 0.05; ***p < 0.001.
To test whether increased PDK1 signaling is sufficient to support neuronal survival, we used the mutant form of PDK1 with the Ala-280 residue substituted by valine (PDK1A280V). This mutation, which has been identified in a genetic screen for suppressors of PI3K deficiency in Caenorhabditis elegans, greatly increases PDK1 activity through a mechanism that remains to be identified (Paradis et al., 1999; Wick et al., 2000). Also, although PDK1A280V has been shown to activate Akt, its effects on activity of RSK1/2 NTK have not been reported (Wick et al., 2000). Therefore, we coelectroporated cortical neurons with PDK1A280V and Flag-tagged wild-type RSK1. At 20 h after electroporation, PDK1A280V increased activity of Flag-RSK1 NTK by 2.5-fold of PDK1-empty vector controls (p < 0.001) (Fig. 6A). Conversely, in neurons receiving PDK1-empty vector control, 3 h exposure to 10 ng/ml BDNF resulted in relatively greater Flag-RSK1 activation reaching 8.4-fold of unstimulated control (p < 0.001) (Fig. 6A). These results indicate that PDK1A280V may at least partially bypass the requirement of the elevated ERK1/2 activity to engage and activate RSK. Of note, PDK1A280V has been reported to activate p70S6K despite the fact that wtPDK1/p70S6K interactions require previous p70S6K phosphorylation by mTor (Rintelen et al., 2001; Mora et al., 2004). Therefore, the mechanism of PDK1 activation by A280V mutation may involve constitutive PDK1 interactions with its substrates including Akt, p70S6K and RSKs.
Figure 6.
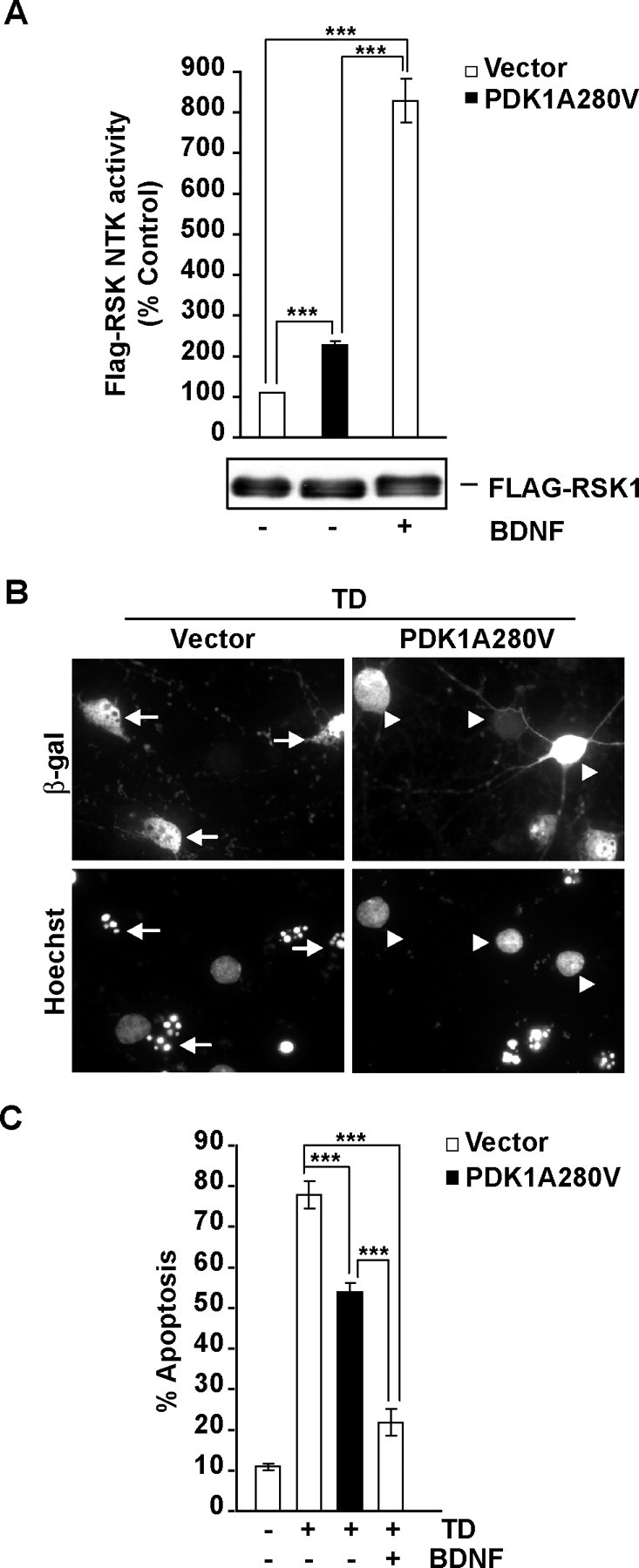
Neuroprotection against the TD-induced apoptosis by the activated mutant form of PDK1, PDK1A280V. A, Cortical neurons were coelectroporated with Flag-RSK1 and either pcDNA3 (vector) or the activated PDK1 mutant PDK1A280V as indicated (3 + 3 μg/10 × 106 cells). At 17 h after electroporation, cells were stimulated with 0 (−) or 10 (+) ng/ml BDNF for 3 h. At 20 h after electroporation, the effects of PDK1A280V or BDNF were analyzed by immunocomplex kinase assay with the immunoprecipitated Flag-RSK1. Although similar levels of Flag-RSK1 were detected in all immunoprecipitates (as indicated by the anti-flag Western blot shown under the graph), BDNF treatment or PDK1A280V increased Flag-RSK1 NTK activity. B, C, Neurons were cotransfected with expression plasmids for β-galactosidase (EF1αLacZ) or the activated PDK1A280V mutant (0.2 + 1 μg of plasmid DNA/5 × 105 neurons, respectively). The empty expression vector pcDNA3 was used as a control (Vector). After 24 h, neurons were sham treated (control) or trophic deprived with or without 10 ng/ml BDNF, as indicated. Apoptosis was evaluated after the next 24 h. B, Representative photomicrographs of the transfected neurons, which were detected by β-galactosidase immunoreactivity. Transfected neurons with normal chromatin morphology or its apoptotic rearrangements are pointed by arrowheads or arrows, respectively. C, Compared with BDNF, the overexpressed PDK1A280V offered partial protection against TD-induced apoptosis. In A and C, averages of three independent experiments ±SEM are presented. ***p < 0.001.
To study the survival effects of PDK1A280V, cortical neurons were transfected with the PDK1A280V expression plasmid or an empty vector control. At 24 h after transfection, neurons were trophic-deprived for 24 h. TD-induced apoptosis was reduced in neurons receiving PDK1A280V (78 or 54% apoptosis in empty vector or PDK1A280V-transfected cells; p < 0.001) (Fig. 6B,C). Conversely, in empty vector-receiving neurons, presence of 10 ng/ml BDNF in the TD media had stronger antiapoptotic effect than PDK1A280V overexpression (22 vs 54% apoptosis, respectively; p < 0.001) (Fig. 6C). Thus, in the absence of elevated ERK1/2 activity, PDK1A280V was sufficient to partially activate RSK and to reduce but not abolish TD-induced apoptosis. These data indicate that in trophic-deprived cortical neurons, endogenous PDK1 is required for antiapoptotic activity of BDNF whereas an overexpressed activated mutant form of PDK1 suffice to partially reduce apoptosis. Hence, PDK1 appears as an essential element of the survival signaling networks activated by BDNF.
The RSK1/2 as a target for antiapoptotic activity of PDK1
In a TD model similar to ours including withdrawal of serum and the NMDAR activity, BDNF-mediated survival of cerebellar granule neurons required the PDK1 target RSK1/2 (Bonni et al., 1999). Therefore, we tested whether RSK1/2 is required for PDK1-dependent survival of trophic-deprived cortical neurons. The shRSK1 and shRSK2 expression plasmids were developed and shown to efficiently knock down overexpressed flag-tagged RSK1 and RSK2, respectively (Fig. 7A). Next, neurons were transfected with shGFP or shRSK1-1 or shRSK2-1 and, at 48 h after transfection, trophic deprived for the next 24 h. The TD treatment was performed with or without 10 ng/ml BDNF. In neurons receiving shGFP, BDNF reduced TD-induced apoptosis (75 or 50% apoptosis with 0 or 10 ng/ml BDNF, respectively, p < 0.001) (Fig. 7B). In contrast, shRSK1-1 abolished BDNF protection (62 or 60% with 0 or 10 ng/ml BDNF, respectively, p > 0.05) (Fig. 7B). Similar removal of BDNF neuroprotection was observed with shRSK2-1, as well as with shRSK1-2, and shRSK2-2 (Fig. 7B) (data not shown). Therefore, in trophic-deprived cortical neurons, both RSK1 and RSK2 are required for the antiapoptotic activity of BDNF indicating their possible involvement in PDK1-dependent survival.
Figure 7.
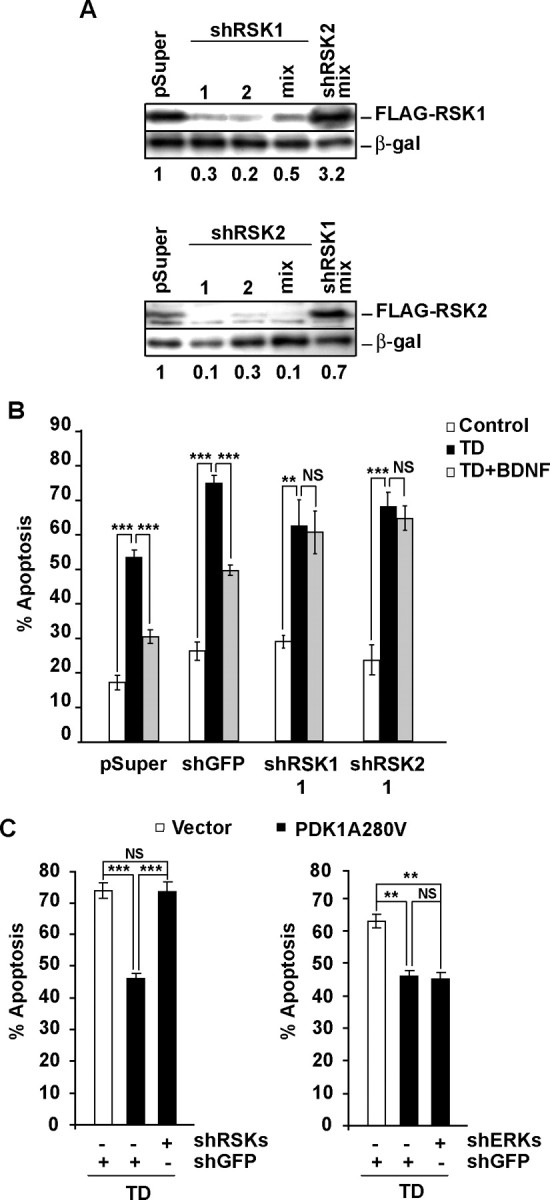
RSK1/2 is required for PDK1-mediated neuroprotection. A, Freshly isolated cortical neurons were coelectroporated with indicated shRNA constructs, together with expression plasmids for β-galactosidase (EF1αLacZ) and either FLAG-RSK1 or FLAG-RSK2 as indicated (2.5 + 0.2 + 0.3 μg of each plasmid DNA/5 × 106 neurons, respectively). If mixed, shRNA plasmids were combined eqimolarly and applied at 2.5 μg of total plasmid DNA/5 × 106 neurons. The controls included the shRNA vector pSuper and the shRSK2 or shRSK1 mixes for shRSK1 or shRSK2, respectively. After 72 h, the levels of FLAG-RSK1 or FLAG-RSK2 but not β-galactosidase decreased in response to isoform-specific shRNAs indicating efficient and selective knockdown. B, Neurons were cotransfected with the β-galactosidase plasmid (pON260) and the indicated shRNAs (0.2 + 0.5 μg of plasmid DNA/0.5 × 106 neurons, respectively). After 48 h, neurons were sham- or TD-treated with or without 10 ng/ml BDNF for additional 24 h. In neurons receiving control plasmids (pSuper or shGFP), BDNF reduced the apoptotic responses to TD. The protection against TD was removed by knocking down either RSK1 or RSK2 indicating that each of these RSK isoforms was required for BDNF to suppress apoptosis. C, Neurons were cotransfected with pON260, the indicated shRNAs, and the activated PDK1A280V mutant (0.2 + 0.2 + 0.8 μg of plasmid DNA/0.5 × 106 neurons, respectively). After 24 h, neurons were trophic deprived for the next 24 h. PDK1A280V reduced TD-induced apoptosis in neurons receiving shGFP, whereas it failed to protect the recipients of the equimolar mix of shRSK1-1, shRSK1-2, shRSK2-1, and shRSK2-2 (shRSKs). In contrast, the equimolar mix of shERK1-1, shERK1-2, shERK2-1, and shERK2-2 (shERKs) did not affect the PDK1A280V-mediated neuroprotection. Results shown in A were replicated in two independent experiments. In B, C, averages of triplicate determinations from three independent experiments ±SEM are shown. **p < 0.01; ***p < 0.001; nonsignificant (NS), p > 0.05.
To further test that possibility, we examined the effects of shRSK1/2 on the antiapoptotic activity of PDK1A280V. In neurons receiving shGFP, PDK1A280V reduced TD-induced apoptosis to 46% compared with 74% in empty vector-transfected cells (Vector, p < 0.001) (Fig. 7C). Knockdown of RSK1/2 with an equimolar mix of all four shRNAs against these kinases blocked PDK1A280V neuroprotection against the TD increasing apoptosis back to 74% (A280V plus shGFP vs A280V plus shRSK, p < 0.001) (Fig. 7C). In contrast, antiapoptotic effects of PDK1A280V were not affected by knockdown of ERK1/2 (Fig. 7C). Thus, in trophic-deprived cultured cortical neurons, PDK1-mediated survival requires RSK1/2. Also, the antiapoptotic interactions between PDK1 and RSK1/2 appear downstream of ERK1/2.
To further verify the prosurvival liaisons between PDK1 and RSK1/2, we tested whether PDK1 may suppress apoptosis by regulating the active overexpressed RSK1. Our first approach was to determine PDK1 contribution to antiapoptotic effects of the combined overexpression of the wild-type RSK1 (wtRSK1) with the low BDNF concentration of 2 ng/ml. Overexpression of either wtRSK1 or wtRSK2 or both together was insufficient to protect against TD (data not shown). Likewise, at 2 ng/ml, BDNF exerted suboptimal neuroprotection compared with 10 ng/ml (data not shown). However, 2 ng/ml BDNF activated PDK1-RSK1/2 signaling (Fig. 8A). If combined with wt RSK1 or RSK2 or both, 2 ng/ml BDNF reduced TD-induced apoptosis (40 or 60% with wtRSK1 or empty vector, respectively, p < 0.001) (Fig. 8B; supplemental Fig. S4, available at www.jneurosci.org as supplemental material). After PDK1 knockdown, the protective effects of 2 ng/ml BDNF plus wtRSK1 were abolished (62 or 63% apoptosis with wtRSK1 or its empty vector control, respectively, p > 0.05) (Fig. 8B). Therefore, in trophic-deprived neurons that were treated with 2 ng/ml BDNF, PDK1 was required for the antiapoptotic activation of the overexpressed wtRSK1.
Figure 8.
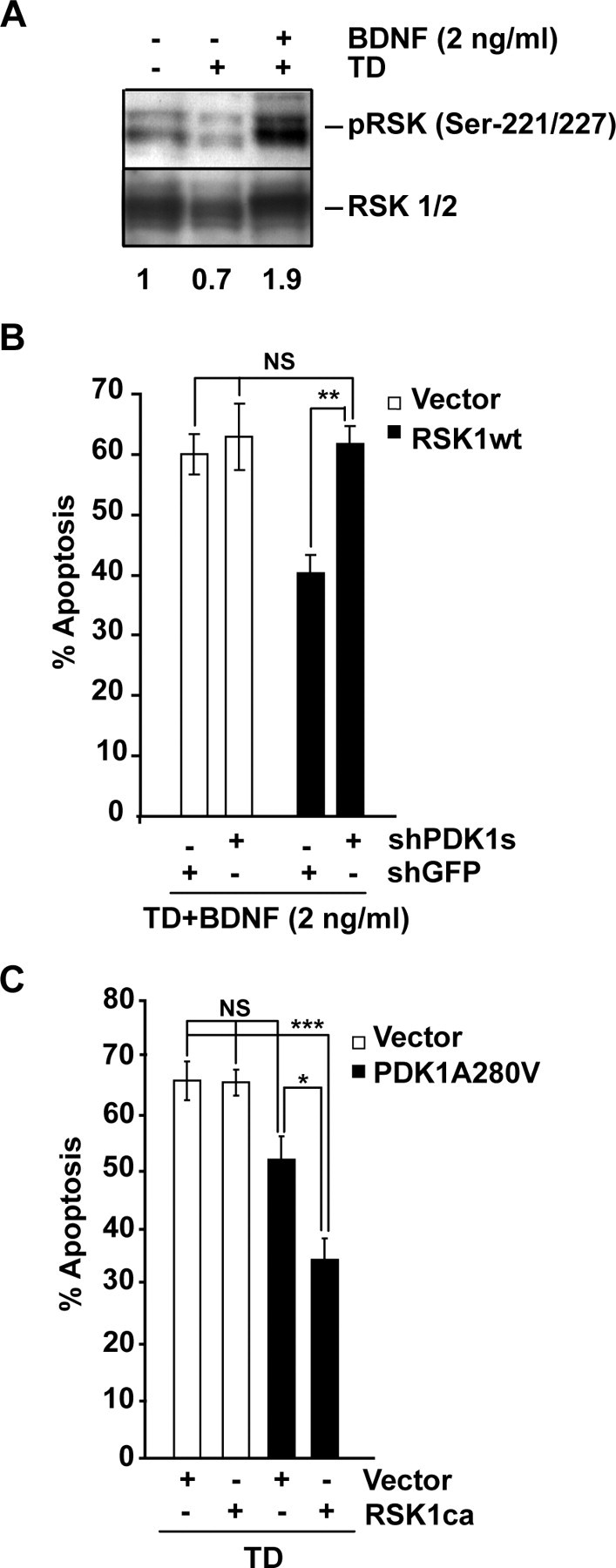
Requirement of PDK1 for the antiapoptotic activity of the overexpressed RSK1. A, After 3 h of TD, cortical neurons were stimulated with 0 or 2 ng/ml BDNF for 1 h. At that low concentration, BDNF increased pSer-221/227 levels suggesting RSK1/2 activation. Numbers under the blots indicate the relative levels of pSer-221/227 after normalization against total RSK1/2 (fold of control). B, Neurons were cotransfected with expression plasmids for β-galactosidase (pON260) or the wild-type RSK1 (wtRSK1) together with shGFP or shPDK1 as indicated (0.2 + 0.4 + 0.4 μg of plasmid DNA/0.5 × 106 neurons, respectively). An empty expression vector (pcDNA3, Vector) was used as a control for wtRSK1. After 24 h, neurons were trophic deprived for the next 24 h in the presence of 2 ng/ml BDNF. Combining this low concentration of the neurotrophin together with wtRSK1 protected against the TD- induced apoptosis. The protection was PDK1-dependent. C, Neurons were cotransfected with expression plasmids for β-galactosidase (pON260), the caRSK1 and the activated PDK1A280V mutant as indicated (0.2 + 0.4 + 0.4 μg of plasmid DNA/0.5 × 106 neurons, respectively). Empty expression vector (pcDNA3, Vector) was used as a control. At 24 h after transfection, cells were TD-treated for the next 24 h. At the low plasmid dosage used for these experiments, the PDK1A280V was unable to suppress the TD-induced apoptosis (p > 0.05). However, coexpression of caRSK1, and PDK1A280V resulted in neuroprotection. Results presented in A were replicated in two independent experiments. In B, C, data represent averages of triplicate determinations from three independent experiments ±SEM. *p < 0.05; **p < 0.01; ***p < 0.001; nonsignificant (NS), p > 0.05.
The second approach to probe the antiapoptotic effects of PDK1/RSK signaling was to test whether PDK1A280V neuroprotection would increase if combined with an active RSK1 mutant (caRSK1). Despite the ability to constitutively bind PDK1 in an ERK1/2-independent manner (Silverman et al., 2004), caRSK1 alone was unable to support survival of TD-treated cortical neurons (66 or 66% apoptosis with vector or caRSK1, respectively, p > 0.05) (Fig. 8C). As in these experiments, lower amount of PDK1A280V plasmid DNA was applied (0.4 vs 1 or 0.8 μg plasmid DNA/5 × 105 cells in Fig. 6A,B or Fig. 7C, respectively), PDK1A280V did not produce significant neuroprotection against the TD (67 or 52% apoptosis with vector control or PDK1A280V, respectively, p > 0.05) (Fig. 8C). However, if combined with caRSK1, PDK1A280V suppressed TD-induced apoptosis (35 or 52% apoptosis with A280V plus caRSK1 or A280V plus Vector, respectively, p < 0.05) (Fig. 8C). Altogether, these data support the notion that PDK1-mediated neuronal survival requires RSK1/2.
PDK1 contributions to antiapoptotic signaling by the ERK1/2 pathway
Since in BDNF-stimulated cortical neurons both PDK1 and ERK1/2 regulate RSK1/2 (Figs. 3–4), we determined whether neuroprotective activities of PDK1 and the ERK1/2 pathway converge. Similarly to shPDK1 or shRSK1/2, also the ERK1 or ERK2 knockdown abolished BDNF protection against TD-induced apoptosis (Fig. 9A). Conversely, the constitutively active MKK1 (caMKK1) that selectively activates ERK1/2 (Mansour et al., 1994), suppressed TD-induced apoptosis (14 or 69 or 73% with caMKK1 or empty vector or wtMKK1, respectively, p < 0.001) (Fig. 9B). Hence, consistent with previously published reports, the ERK1/2 signaling pathway is both required and sufficient for BDNF protection against the TD consisting of serum withdrawal combined with reduced/blocked NMDAR activity (Bonni et al., 1999; Szatmari et al., 2007).
Figure 9.

Role of PDK1 in antiapoptotic activity of the ERK1/2 pathway. A, Neurons were transfected with equimolar mixes of shERK1-1 plus shERK1-2 (shERK1) or shERK2-1 plus shERK2-2 (shERK2). Plasmid dosing and treatments were as described for Figure 5. BDNF-mediated suppression of TD-induced apoptosis was abolished by knockdown of either ERK. B, Neurons were cotransfected with expression plasmids for β-galactosidase (pON260) and either wild-type (wt), dominant negative (dn), or constitutively active (ca) mutant forms of MKK1 (0.2 + 1 μg of plasmid DNA/5 × 105 neurons, respectively). Empty expression vector (pcDNA3, Vector) was used as a control. After 24 h, neurons were sham treated (control) or trophic deprived for the next 24 h. The caMKK1 reduced the apoptotic response to TD. C, Neurons were cotransfected with pON260, MKK1ca and the indicated shRNAs (0.2 + 0.8 + 0.2 μg of plasmid DNA/5 × 105 neurons, respectively). The shERK1/2 or shRSK1/2 consisted of equimolar mixes of shERK1-1 plus shERK1-2 plus shERK2-1 plus shERK2-2 or shRSK1-1 plus shRSK1-2 plus shRSK2-1 plus shRSK2-2, respectively. The empty expression vector (pcDNA3, vector) or shGFP were used as controls for MKK1ca or shRNAs, respectively. Cells were treated as in B. Either shERK1/2 or shRSK1/2 or shPDK1 reduced the caMKK1-mediated neuroprotection against TD, indicating a critical role of PDK1-RSK1/2 interactions for the survival signaling by the MKK1-ERK1/2 pathway. D, Neurons were cotransfected with pON260, caMKK1, and the activated PDK1A280V mutant (0.2 + 0.2 + 0.4 μg of plasmid DNA/0.5 × 106 neurons, respectively). The pcDNA3.1 was used as a vector control (Vector). Cells were treated as in B. At the plasmid dosage applied in these experiments, neither caMKK1 nor PDK1A280V alone were sufficient to protect against TD. However, their coexpression suppressed the TD-induced apoptosis indicating the survival synergy of the PDK1 and MKK1-ERK1/2 pathways. In A–D, averages of triplicate determinations from three independent experiments ±SEM are shown. **p < 0.01; ***p < 0.001; nonsignificant (NS). E, Our data supports a model that BDNF- or MKK1ca-mediated neuroprotection against the TD-induced apoptosis employs PDK1-dependent activation of RSK. Also, the survival requirement for the PDK1-RSK signaling suggests that RSK-NTK substrates are critical for the antiapoptotic neuroprotection.
To determine whether the antiapoptotic signaling by the ERK1/2 pathway involves PDK1 and RSK1/2, neurons were cotransfected with caMKK1 and shGFP or shPDK1 or shRSK1/2. In addition, shERK1/2 was used as a positive control as it was expected to block the caMKK1-mediated neuroprotection. At 24 h after transfection, neurons were sham or trophic deprived. In neurons receiving shGFP, caMKK1 abolished apoptotic response to TD (18 or 26% apoptosis after sham or TD treatment, respectively, p > 0.05) (Fig. 9C). The knock down of ERK1/2 blocked the neuroprotective activity of caMKK1 (16 or 67% apoptosis after sham or TD treatment, respectively, p < 0.001) (Fig. 9C). Similar trends were observed in neurons receiving shRSKs or shPDK1 (Fig. 9C). Thus, in trophic-deprived cortical neurons, RSK1/2 and PDK1 are indispensable for the antiapoptotic effects of the ERK1/2 pathway.
Finally, we tested whether overexpressed caMKK1 and PDK1A280V can synergize suppressing the TD-induced apoptosis. For these experiments, we cotransfected caMKK1 and PDK1A280V at subprotective plasmid DNA dosage of 0.2 and 0.4 μg/5 × 105 neurons, respectively. After 24 h, neurons were sham or TD-treated for next 24 h. Cells that received either caMKK1 or PDK1A280V were not protected against TD-induced apoptosis (66 or 63 or 52% apoptosis in vector- or MKK1ca- or PDK1A280V-transfected neurons, respectively, p > 0.05) (Fig. 9D). However, when combined, caMKK1 plus PDK1A280V reduced apoptosis to 33% (vector vs combined plasmids, p < 0.001) (Fig. 9D). These results further support the critical role of PDK1 for the survival signaling via the ERK1/2 pathway. Altogether, our data indicate that in trophic deprived-neurons, the antiapoptotic effects of BDNF or the caMKK1 are mediated by the ERK1/2-dependent signaling between PDK1 and RSK1/2 (Fig. 9E).
Discussion
In this study, we have provided the first direct evidence for the PDK1 involvement in neurotrophin-mediated neuronal survival. We also have demonstrated that RSK1/2 is among major antiapoptotic targets of PDK1 in neurotrophin-protected neurons. Finally, we have shown that the neurotrophin-activated ERK1/2 is a critical regulator for the survival signaling by the PDK1-RSK1/2 pathway.
Requirement of PDK1 for neurotrophin-mediated neuronal survival
Our data indicate that PDK1 is indispensable for BDNF-mediated survival of rat cortical neurons. First, active PDK1 was expressed during the active cortical synaptogenesis period when neuronal survival is strongly dependent on extracellular signals including neurotrophins (Alcántara et al., 1997; Olney, 2004). Second, phosphorylation of the PDK1 substrate RSK1/2 Ser221/227 was high at these developmental stages while increasing in BDNF-stimulated cultured cortical neurons. Third, PDK1 knockdown removed the antiapoptotic effects of BDNF in TD-exposed cortical neurons. Last, overexpression of the active mutant form of PDK1 was sufficient to reduce the TD-induced apoptosis. These results identify PDK1 as an important regulator of neuronal survival in the developing rat forebrain.
In developing neurons, the survival role of PDK1 has been proposed based on the phenotypes of PDK1-deficient mice including lack of dorsal root ganglia (DRG) and various degrees of reduction in forebrain size (Lawlor et al., 2002; Collins et al., 2003; McManus et al., 2004; Collins et al., 2005). Also, it is well established that survival of developing DRG and forebrain neurons requires extracellular signals including neurotrophins (Silos-Santiago et al., 1995; Alcántara et al., 1997). The findings from the PDK1-deficient animals together with the results of our study indicate the critical involvement of PDK1 in neurotrophin-mediated suppression of developmental apoptosis in the nervous system.
Neuronal survival signaling by the PDK1-RSK1/2 pathway
We showed that PDK1-RSK1/2 signaling was active in the developing rat brain during synaptogenesis/developmental apoptosis while appearing relatively reduced as these processes subsided. Conversely, in BDNF-stimulated cultured cortical neurons, RSK1/2 became activated in a PDK1-dependent manner. RSK1/2 was necessary for BDNF or active PDK1-mediated protection against TD-induced apoptosis. Also, protection by overexpressed RSK1 that was activated by BDNF required PDK1. Last, on coexpression, active mutant forms of PDK1 and RSK1 synergized suppressing the TD-induced apoptosis. Therefore, our data identify RSK1/2 as a relevant survival target for PDK1 signaling.
Although the essential role of PDK1 in activating RSK1/2 was discovered nearly a decade ago (Jensen et al., 1999), the physiological significance of this regulatory interaction has not been directly addressed before this study. Indirect approaches suggested that PDK1/RSK2 signaling may be important for NGF-stimulated neurite outgrowth in PC12 cells or Notch-induced thymocyte proliferation (Silverman et al., 2004; Kelly et al., 2007). Thus, in addition to providing the first direct demonstration of functional importance for the PDK1-mediated regulation of RSK1/2, we also identify neuronal survival as a previously unrecognized response to BDNF-activated signaling by the PDK1-RSK1/2 pathway.
In addition to RSK1/2, PDK1 regulates other members of the AGC kinase family that may be involved in neurotrophin-mediated neuronal survival (Mora et al., 2004). Of those, the PI3K-dependent Akt has been implicated as a major survival transducer in neurons, and, also as an important target for the PDK1 survival signaling in non-neuronal systems (Datta et al., 1999; Flynn et al., 2000; Kaplan and Miller, 2000; Cho et al., 2001; Sato et al., 2002; Liang et al., 2006; Hayashi et al., 2007). Although PDK1 is the major PI3K-dependent kinase that phosphorylates Akt Thr-308, other activities targeting this site were reported (Yano et al., 1998). Thus, at least in neurotrophin-stimulated neurons, the relative contribution of PDK1 to the antiapoptotic activation of Akt is yet to be examined. Obviously, in the case of the BDNF protection against TD, it is likely that both PDK1-RSK1/2 and PDK1-Akt pathways are required to efficiently block apoptosis, for instance, by cooperative inactivation of the proapoptotic protein Bad (Datta et al., 1997; Bonni et al., 1999). Last, at least in some systems, RSK1/2 has been suggested to mediate the PDK1 effects on Akt (Pan et al., 2004). Future experiments will determine whether in TD-exposed cortical neurons, the BDNF-mediated survival involves PDK1-dependent Akt activation, and if so, whether the PDK1-Akt represents a parallel survival pathway to the PDK1-RSK one, and/or, whether they both belong to a single interconnected survival signaling network.
The survival signaling alliance of PDK1 and ERK1/2
We showed that in trophic-deprived cortical neurons, the antiapoptotic effect of BDNF required PDK1, RSK1/2 and ERK1/2. In addition, selective activation of the ERK1/2 pathway with caMKK1 suppressed the TD-induced apoptosis in a PDK1-, and RSK1/2-dependent manner, whereas synergistic neuroprotection was observed after coexpressing activated forms of PDK1 and MKK1. Finally, we demonstrated corequirement of PDK1 and ERK1/2 for activation of neuronal RSK1/2, and the essential role of the latter in PDK1-mediated survival signaling. Altogether, these results support a notion that the ERK1/2 and PDK1 are both required to jointly activate the antiapoptotic signaling by RSK1/2.
Of note, in several cell line systems including immortalized hippocampal cells, ERK1/2 activation has been shown to depend on PDK1 (Corbit et al., 2000, Sato et al., 2004). Thus, integration of the survival signaling by PDK1 and ERK1/2 pathways could also occur upstream of ERK1/2. However, in BDNF-stimulated primary cortical neurons, PDK1 inhibition did not affect ERK1/2 activation (supplemental Figs. S1B and S3, available at www.jneurosci.org as supplemental material). Likewise, the neuroprotection by active PDK1 was present in neurons depleted of ERK1/2 (Fig. 7C). These results further support the notion that the PDK1 contributes to the prosurvival activity of the ERK1/2 pathway downstream of ERK1/2.
Although PDK1 and ERK1/2 cooperation to protect from apoptosis has not been reported before this study, ERK1/2-RSK2 signaling has been proposed to mediate antiapoptotic effects of BDNF in TD-exposed cultured rat cerebellar granule neurons (Bonni et al., 1999). Also, in that system, the proposed antiapoptotic targets of RSK2 include the NTK substrates CREB and BAD. As PDK1 is required for activation of RSK NTK (Jensen et al., 1999), it is reasonable to speculate that, PDK1 contributes to neurotrophin-mediated survival of various neuronal populations that depend on RSK1/2 activity including cortical and cerebellar granule neurons.
The role of RSK1/2 NTK as a coincidence detector of PDK1- and ERK1/2-mediated survival signals is likely, although not tested yet. An alternative possibility is that phosphorylation of non-overlapping substrates underlies the survival alliance between PDK1 and ERK1/2 pathways. Thus, ERK1/2 and PDK1 but not RSK1/2 were required for BDNF neuroprotection against DNA damage (G. Kharebava and M. Hetman, unpublished observation). Indeed there are several PDK1-regulated kinases including Akt, p70 ribosomal S6 kinase or protein kinase C that were implicated in cell survival and that could synergize with ERK1/2 to suppress neuronal apoptosis (Mora et al., 2004).
Regulation of neuronal PDK1
Our results indicate that in rat forebrain, PDK1 is highly active during the time of developmental synaptogenesis when apoptotic avoidance is regulated by extracellular survival signals. Also, in cultured cortical neurons, BDNF increased PDK1 phosphorylation of RSK1/2 without changing PDK1 activity in an in vitro assay. Last, PDK1 activation of RSK1/2 required ERK but not PI3K. These results are consistent with the notion that PDK1 activity targeting RSK is PI3K-independent and is recruited to RSK by ERK (Jensen et al., 1999). However, the survival signals that drive PDK1-RSK1/2 signaling in the developing brain remain to be identified. Likewise, the role for ERK in PDK1 recruitment to RSK has yet to be shown in vivo.
Interestingly, in the cortex of P1 and P7 rats, the PDK1 had lower electromobility than in P21 rats indicating possible posttranslational modification(s) that coincide with increased activity of PDK1 toward RSK1/2 (Fig. 2A). Therefore, a novel regulatory mechanism may operate on PDK1 in a developmental stage-dependent manner that stimulates its interactions with RSK1/2. Such mechanism(s) may engage PDK activation by phosphorylations at several tyrosine residues as recently reported in non-neuronal systems (Yang et al., 2008). Last, as a CTK-independent activation of RSK NTK has been recently reported, it is possible that at P1 and P7 PDK1 activates RSK1/2 by a CTK-independent mechanism (Cohen et al., 2007). Our future experiments will address the developmental mechanisms of PDK1 regulation in the brain.
In summary, we identified PDK1 as a novel regulator of neuronal survival. Also, we uncovered previously unrecognized interactions between neuronal PDK1, ERK1/2 and RSK1/2 that are required for the antiapoptotic activity of BDNF. Our data support a possibility that the master AGC kinase regulator PDK1 provides one of the key links ensuring processing and integration of extracellular survival signals that act on the developing nervous system.
Footnotes
This work was supported by National Institutes of Health Grants NS047341-01 and RR015576-06, The Kentucky Spinal Cord and Head Injury Research Trust (Grant 3-5), The Commonwealth of Kentucky Challenge for Excellence, and Norton Healthcare. We thank Dr. Scott R. Whittemore, Dr. Theo Hagg, and Scott C. Smith for critical reading of this manuscript. Drs. Jeffrey Maller, John Blenis, Feng Liu, Moshe Oren, Andrew Sharrocks, and Jacek Jaworski provided reagents used in this work.
References
- Adams JP, Sweatt JD. Molecular psychology: roles for the ERK MAP kinase cascade in memory. Annu Rev Pharmacol Toxicol. 2002;42:135–163. doi: 10.1146/annurev.pharmtox.42.082701.145401. [DOI] [PubMed] [Google Scholar]
- Alcántara S, Frisén J, del Río JA, Soriano E, Barbacid M, Silos-Santiago I. TrkB signaling is required for postnatal survival of CNS neurons and protects hippocampal and motor neurons from axotomy-induced cell death. J Neurosci. 1997;17:3623–3633. doi: 10.1523/JNEUROSCI.17-10-03623.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayascas JR, Wullschleger S, Sakamoto K, García-Martínez JM, Clacher C, Komander D, van Aalten DM, Boini KM, Lang F, Lipina C, Logie L, Sutherland C, Chudek JA, van Diepen JA, Voshol PJ, Lucocq JM, Alessi DR. Mutation of PDK1 PH domain inhibits PKB/Akt leading to small size and insulin-resistance. Mol Cell Biol. 2008;28:3258–3272. doi: 10.1128/MCB.02032-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- Buss RR, Sun W, Oppenheim RW. Adaptive roles of programmed cell death during nervous system development. Annu Rev Neurosci. 2006;29:1–35. doi: 10.1146/annurev.neuro.29.051605.112800. [DOI] [PubMed] [Google Scholar]
- Cherrington JM, Mocarski ES. Human cytomegalovirus ie1 transactivates the alpha promoter-enhancer via an 18-base-pair repeat element. J Virol. 1989;63:1435–1440. doi: 10.1128/jvi.63.3.1435-1440.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho KS, Lee JH, Kim S, Kim D, Koh H, Lee J, Kim C, Kim J, Chung J. Drosophila phosphoinositide-dependent kinase-1 regulates apoptosis and growth via the phosphoinositide 3-kinase-dependent signaling pathway. Proc Natl Acad Sci U S A. 2001;98:6144–6149. doi: 10.1073/pnas.101596998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MS, Hadjivassiliou H, Taunton J. A clickable inhibitor reveals context-dependent autoactivation of p90 RSK. Nat Chem Biol. 2007;3:156–160. doi: 10.1038/nchembio859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins BJ, Deak M, Arthur JS, Armit LJ, Alessi DR. In vivo role of the PIF-binding docking site of PDK1 defined by knock-in mutation. EMBO J. 2003;22:4202–4211. doi: 10.1093/emboj/cdg407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins BJ, Deak M, Murray-Tait V, Storey KG, Alessi DR. In vivo role of the phosphate groove of PDK1 defined by knockin mutation. J Cell Sci. 2005;118:5023–5034. doi: 10.1242/jcs.02617. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Soh JW, Yoshida K, Eves EM, Weinstein IB, Rosner MR. Different protein kinase C isoforms determine growth factor specificity in neuronal cells. Mol Cell Biol. 2000;20:5392–5403. doi: 10.1128/mcb.20.15.5392-5403.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- del Peso L, González-García M, Page C, Herrera R, Nuñez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- Dimitri CA, Dowdle W, MacKeigan JP, Blenis J, Murphy LO. Spatially separate docking sites on ERK2 regulate distinct signaling events in vivo. Curr Biol. 2005;15:1319–1324. doi: 10.1016/j.cub.2005.06.037. [DOI] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Flynn P, Wongdagger M, Zavar M, Dean NM, Stokoe D. Inhibition of PDK-1 activity causes a reduction in cell proliferation and survival. Curr Biol. 2000;10:1439–1442. doi: 10.1016/s0960-9822(00)00801-0. [DOI] [PubMed] [Google Scholar]
- Habas A, Kharebava G, Szatmari E, Hetman M. NMDA neuroprotection against a phosphatidylinositol-3 kinase inhibitor, LY294002 by NR2B-mediated suppression of glycogen synthase kinase-3beta-induced apoptosis. J Neurochem. 2006;96:335–348. doi: 10.1111/j.1471-4159.2005.03543.x. [DOI] [PubMed] [Google Scholar]
- Hauge C, Frödin M. RSK and MSK in MAP kinase signalling. J Cell Sci. 2006;119:3021–3023. doi: 10.1242/jcs.02950. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Mo JH, Gong X, Rossetto C, Jang A, Beck L, Elliott GI, Kufareva I, Abagyan R, Broide DH, Lee J, Raz E. 3-Hydroxyanthranilic acid inhibits PDK1 activation and suppresses experimental asthma by inducing T cell apoptosis. Proc Natl Acad Sci U S A. 2007;104:18619–18624. doi: 10.1073/pnas.0709261104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetman M, Cavanaugh JE, Kimelman D, Xia Z. Role of glycogen synthase kinase-3beta in neuronal apoptosis induced by trophic withdrawal. J Neurosci. 2000;20:2567–2574. doi: 10.1523/JNEUROSCI.20-07-02567.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetman M, Hsuan SL, Habas A, Higgins MJ, Xia Z. ERK1/2 antagonizes glycogen synthase kinase-3beta -induced apoptosis in cortical neurons. J Biol Chem. 2002;277:49577–49584. doi: 10.1074/jbc.M111227200. [DOI] [PubMed] [Google Scholar]
- Jensen CJ, Buch MB, Krag TO, Hemmings BA, Gammeltoft S, Frödin M. 90-kDa ribosomal S6 kinase is phosphorylated and activated by 3-phosphoinositide-dependent protein kinase-1. J Biol Chem. 1999;274:27168–27176. doi: 10.1074/jbc.274.38.27168. [DOI] [PubMed] [Google Scholar]
- Kalita K, Kharebava G, Zheng JJ, Hetman M. Role of megakaryoblastic acute leukemia-1 in ERK1/2-dependent stimulation of serum response factor-driven transcription by BDNF or increased synaptic activity. J Neurosci. 2006;26:10020–10032. doi: 10.1523/JNEUROSCI.2644-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10:381–391. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- Kelly AP, Finlay DK, Hinton HJ, Clarke RG, Fiorini E, Radtke F, Cantrell DA. Notch-induced T cell development requires phosphoinositide-dependent kinase 1. EMBO J. 2007;26:3441–3450. doi: 10.1038/sj.emboj.7601761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor MA, Mora A, Ashby PR, Williams MR, Murray-Tait V, Malone L, Prescott AR, Lucocq JM, Alessi DR. Essential role of PDK1 in regulating cell size and development in mice. EMBO J. 2002;21:3728–3738. doi: 10.1093/emboj/cdf387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang K, Lu Y, Li X, Zeng X, Glazer RI, Mills GB, Fan Z. Differential roles of phosphoinositide-dependent protein kinase-1 and akt1 expression and phosphorylation in breast cancer cell resistance to Paclitaxel, Doxorubicin, and gemcitabine. Mol Pharmacol. 2006;70:1045–1052. doi: 10.1124/mol.106.023333. [DOI] [PubMed] [Google Scholar]
- Mansour SJ, Matten WT, Hermann AS, Candia JM, Rong S, Fukasawa K, Vande Woude GF, Ahn NG. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science. 1994;265:966–970. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- McManus EJ, Collins BJ, Ashby PR, Prescott AR, Murray-Tait V, Armit LJ, Arthur JS, Alessi DR. The in vivo role of PtdIns(3,4,5)P3 binding to PDK1 PH domain defined by knockin mutation. EMBO J. 2004;23:2071–2082. doi: 10.1038/sj.emboj.7600218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora A, Komander D, van Aalten DM, Alessi DR. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol. 2004;15:161–170. doi: 10.1016/j.semcdb.2003.12.022. [DOI] [PubMed] [Google Scholar]
- Olney JW. Fetal alcohol syndrome at the cellular level. Addict Biol. 2004;9:137–149. doi: 10.1080/13556210410001717006. discussion 151. [DOI] [PubMed] [Google Scholar]
- Pan ZZ, Devaux Y, Ray P. Ribosomal S6 kinase as a mediator of keratinocyte growth factor-induced activation of Akt in epithelial cells. Mol Biol Cell. 2004;15:3106–3113. doi: 10.1091/mbc.E04-01-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis S, Ailion M, Toker A, Thomas JH, Ruvkun G. A PDK1 homolog is necessary and sufficient to transduce AGE-1 PI3 kinase signals that regulate diapause in Caenorhabditis elegans. Genes Dev. 1999;13:1438–1452. doi: 10.1101/gad.13.11.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rintelen F, Stocker H, Thomas G, Hafen E. PDK1 regulates growth through Akt and S6K in Drosophila. Proc Natl Acad Sci U S A. 2001;98:15020–15025. doi: 10.1073/pnas.011318098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Fujita N, Tsuruo T. Interference with PDK1-Akt survival signaling pathway by UCN-01 (7-hydroxystaurosporine) Oncogene. 2002;21:1727–1738. doi: 10.1038/sj.onc.1205225. [DOI] [PubMed] [Google Scholar]
- Sato S, Fujita N, Tsuruo T. Involvement of 3-phosphoinositide-dependent protein kinase-1 in the MEK/MAPK signal transduction pathway. J Biol Chem. 2004;279:33759–33767. doi: 10.1074/jbc.M402055200. [DOI] [PubMed] [Google Scholar]
- Shaulian E, Zauberman A, Ginsberg D, Oren M. Identification of a minimal transforming domain of p53: negative dominance through abrogation of sequence-specific DNA binding. Mol Cell Biol. 1992;12:5581–5592. doi: 10.1128/mcb.12.12.5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silos-Santiago I, Greenlund LJ, Johnson EM, Jr, Snider WD. Molecular genetics of neuronal survival. Curr Opin Neurobiol. 1995;5:42–49. doi: 10.1016/0959-4388(95)80085-9. [DOI] [PubMed] [Google Scholar]
- Silverman E, Frödin M, Gammeltoft S, Maller JL. Activation of p90 Rsk1 is sufficient for differentiation of PC12 cells. Mol Cell Biol. 2004;24:10573–10583. doi: 10.1128/MCB.24.24.10573-10583.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatmari E, Kalita KB, Kharebava G, Hetman M. Role of kinase suppressor of Ras-1 in neuronal survival signaling by extracellular signal-regulated kinase 1/2. J Neurosci. 2007;27:11389–11400. doi: 10.1523/JNEUROSCI.3473-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vantaggiato C, Formentini I, Bondanza A, Bonini C, Naldini L, Brambilla R. ERK1 and ERK2 mitogen-activated protein kinases affect Ras-dependent cell signaling differentially. J Biol. 2006;5:14. doi: 10.1186/jbiol38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickers ER, Kasza A, Kurnaz IA, Seifert A, Zeef LA, O'donnell A, Hayes A, Sharrocks AD. Ternary complex factor-serum response factor complex-regulated gene activity is required for cellular proliferation and inhibition of apoptotic cell death. Mol Cell Biol. 2004;24:10340–10351. doi: 10.1128/MCB.24.23.10340-10351.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick MJ, Dong LQ, Riojas RA, Ramos FJ, Liu F. Mechanism of phosphorylation of protein kinase B/Akt by a constitutively active 3-phosphoinositide-dependent protein kinase-1. J Biol Chem. 2000;275:40400–40406. doi: 10.1074/jbc.M003937200. [DOI] [PubMed] [Google Scholar]
- Yang KJ, Shin S, Piao L, Shin E, Li Y, Park KA, Byun HS, Won M, Hong J, Kweon GR, Hur GM, Seok JH, Chun T, Brazil DP, Hemmings BA, Park J. Regulation of 3-phosphoinositide-dependent protein kinase-1 (PDK1) by Src involves tyrosine phosphorylation of PDK1 and Src homology 2 domain binding. J Biol Chem. 2008;283:1480–1491. doi: 10.1074/jbc.M706361200. [DOI] [PubMed] [Google Scholar]
- Yang P, Baker KA, Hagg T. A disintegrin and metalloprotease 21 (ADAM21) is associated with neurogenesis and axonal growth in developing and adult rodent CNS. J Comp Neurol. 2005;490:163–179. doi: 10.1002/cne.20659. [DOI] [PubMed] [Google Scholar]
- Yano S, Tokumitsu H, Soderling TR. Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway. Nature. 1998;396:584–587. doi: 10.1038/25147. [DOI] [PubMed] [Google Scholar]
- Yun MS, Kim SE, Jeon SH, Lee JS, Choi KY. Both ERK and Wnt/beta-catenin pathways are involved in Wnt3a-induced proliferation. J Cell Sci. 2005;118:313–322. doi: 10.1242/jcs.01601. [DOI] [PubMed] [Google Scholar]