

Abstract
Nephronophthisis (NPHP), Joubert (JBTS) and Meckel-Gruber (MKS) syndromes are autosomal-recessive ciliopathies presenting with cystic kidneys, retinal degeneration, and cerebellar/neural tube malformation. Whether defects in kidney, retinal, or neural disease primarily involve ciliary, Hedgehog, or cell polarity pathways remains unclear. Using high-confidence proteomics, we identified 850 interactors copurifying with nine NPHP/JBTS/MKS proteins, and discovered three connected modules: “NPHP1-4-8” functioning at the apical surface; “NPHP5-6” at centrosomes; and “MKS” linked to Hedgehog signaling. Assays for ciliogenesis and epithelial morphogenesis in 3D renal cultures link renal cystic disease to apical organization defects, whereas ciliary and Hedgehog pathway defects lead to retinal or neural deficits. Using 38 interactors as candidates, linkage and sequencing analysis of 250 patients identified ATXN10 and TCTN2 as new NPHP-JBTS genes and our Tctn2 mouse knockout shows neural tube and Hedgehog signaling defects. Our study further illustrates the power of linking proteomic networks and human genetics to uncover critical disease pathways.
Introduction
Ciliopathies are a heterogeneous group of diseases that present with a broad constellation of clinical phenotypes, including renal cysts, retinal degeneration, polydactyly, mental retardation, and obesity (reviewed by Hildebrandt et al., 2009a; Zaghloul and Katsanis, 2009). Studies of these diseases suggest that their pathogenesis relates to dysfunction of the microtubule-based primary cilium. It is hypothesized that the primary cilium is a sensory organelle, acting as a mechanosensor in the kidney, and organizing sensory receptors including rhodopsin in the retina. Cilia are also key components of the Hedgehog (Hh) signaling pathway (Corbit et al., 2005; Huangfu et al., 2003). The consistent finding of kidney, retinal, liver, limb and brain defects among the ciliopathies in turn suggests that cilia-dependent sensory and signaling functions are critical in the development, tissue organization and physiological function of multiple organ systems. However, whether all “ciliopathies” are simply caused by the absence of cilia themselves remains unclear. Specific ciliary signaling pathways, ciliary receptors, additional ciliary effectors, or even centrosomes, may be at the root of specific types of tissue or sensory failure, and these relationships have remained elusive.
Three linked ciliopathies called Nephronophthisis (NPHP), Joubert Syndrome (JBTS) and Meckel-Gruber Syndrome (MKS) are autosomal recessive disorders, initially described as distinct entities, but recently found to share phenotypic overlap, notably in cystic kidney disease. Nephronophthisis is the least severe of the group, characterized primarily by renal cysts, but sometimes involving retinal degeneration (called Senior-Loken syndrome), situs inversus, and mental retardation (Hildebrandt et al., 2009a). Joubert Syndrome involves both renal and non-renal manifestations of NPHP disease, but is distinguished by cerebellar vermis aplasia, a significant malformation of the cerebellum linked to ataxia (Parisi et al., 2007). As a perinatal lethal disease, Meckel-Gruber Syndrome is the most severe, characterized by occipital encephalocele, polydactyly, liver fibrosis, and severe renal cysts (Salonen and Paavola, 1998).
To date, mutations in sixteen genes have been linked to this group of disorders: NPHP1-9, AHI1/Jouberin, ARL13B, INPP5E, TMEM216, MKS1, MKS3/TMEM67, and MKS6/CC2D2A (Tallila et al., 2008; Hildebrandt et al., 2009a; Lee and Gleeson 2010; Supplemental References). Interestingly, different alleles of the same gene can result in the phenotypic spectrum of NPHP, JBTS and MKS. For example, NPHP1 mutations were reported in patients with both NPHP and JBTS (Parisi et al., 2007), whereas mutations in NPHP3, NPHP6/CEP290 and NPHP8/RPGRIP1L are linked to all forms of disease along the NPHP-JBTS-MKS spectrum (Baala et al., 2007) (Figure 1A). Because these three diseases can be caused by mutation of the same genes, we considered the hypothesis that these disorders may link to defects in a specific set of cellular mechanisms, and that the associated proteins may interact and function in common pathways. NPHP/JBTS/MKS proteins are rich in protein-protein interaction domains (Figure S1A), suggesting extensive protein-protein interactions. Studies using yeast two-hybrid and co-immunoprecipitation discovered interactions of NPHP1 with NPHP2, NPHP3, and NPHP4 (Mollet et al., 2002; Mollet et al., 2005; Olbrich et al., 2003; Otto et al., 2003). However, a systematic connection between NPHP, JBTS and MKS proteins has not been explored.
Figure 1. Mapping the NPHP-JBTS-MKS disease protein network using G-LAP-Flp strategy (see also Figures S1 and S2; Tables S1-3; Table S5; Supplemental Bioinformatics Tools).
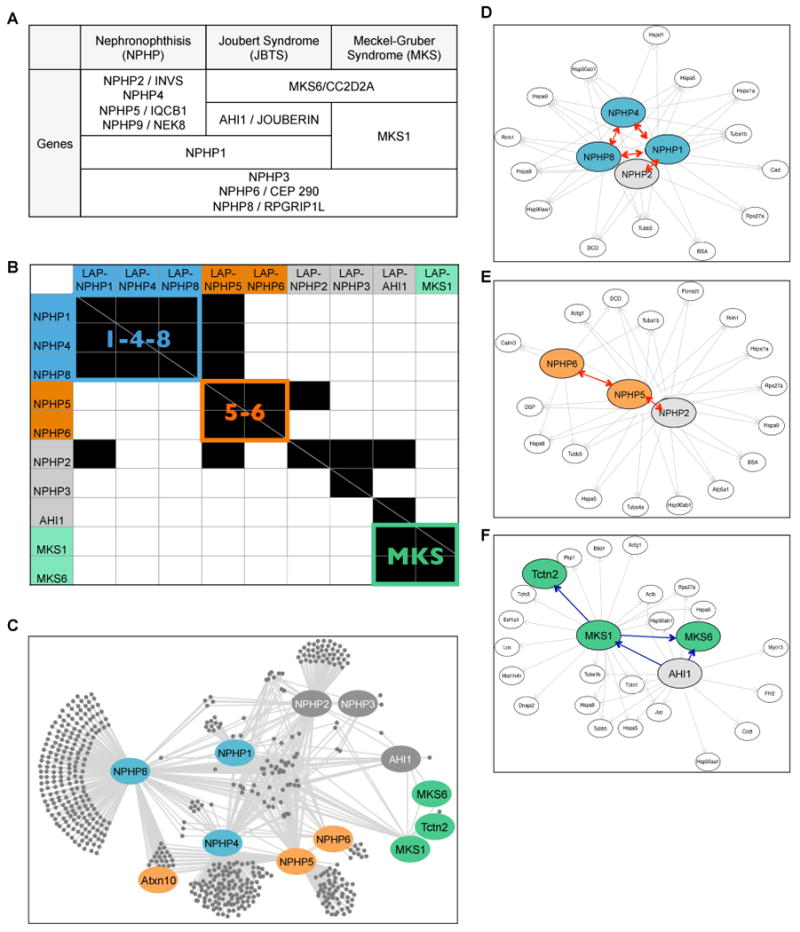
A. List of genes mutated in NPHP-JBTS-MKS ciliopathies.
B. Heat map summarizing MS/MS interactions among NPHP proteins discovered using G-LAP-Flp strategy. Horizontal axis = LAP-tagged “bait” proteins; vertical axis = interacting proteins. Identified interactions are shown in black. The NPHP1-4-8 (“1-4-8”), NPHP5-6 (“5-6”) and MKS modules are color coded in blue, orange and green.
C-F. The NPHP-JBTS-MKS interaction network generated from the R script and visualized using Cytoscape. The entire network is shown in C; individual subgraphs that illustrate the “1-4-8” (IMCD3), “5-6” (IMCD3), and “MKS” (NIH 3T3) modules are shown in D, E, and F. Ellipse = protein; single headed arrows = unreciprocated interactions (pointing to the hits); double headed arrows (red) = reciprocal interactions. Bait proteins and a subset of interactors are highlighted using the color scheme described in Figure 1B.
The proteins encoded by genes mutated in NPHP, JBTS and MKS are found either within the primary cilium, or at the basal body and several have homologs in Chlamydomonas (Hildebrandt et al., 2009a), suggesting that they participate in conserved ciliary machinery. However, some of these proteins are centrosomal even in the absence of cilia (Chang et al., 2006), suggesting that these machines may function at the centrosome, possibly to organize cell polarity or receptor trafficking, rather than simply being a structural component of the primary cilium.
To better understand the molecular mechanisms underlying NPHP-JBTS-MKS, we developed a high-confidence proteomic strategy using the G-LAP tandem-affinity method (Torres et al., 2009) to identify interactors associated with nine disease proteins. Our data show that NPHP-JBTS-MKS proteins form an interaction network that can be classified into three distinct modules: “NPHP1-4-8”, “NPHP5-6”, and “Mks”. Assays for ciliogenesis and epithelial morphogenesis using three-dimensional (3D) renal cultures suggest that NPHP1-4-8 links primarily to apical organization defects seen in nephronophthisis, whereas ciliary NPHP5-6 deficits appear central to retinal and potentially neural deficiency, with Mks module and Hedgehog signaling deficits linked to the neural tube defects. Excitingly, our network building strategy allowed us to propose candidates for new ciliopathy disease genes, leading to the identification of the first human mutations in the NPHP gene Ataxin10 (ATXN10) and JBTS gene Tectonic2 (TCTN2).
Results
Mapping of an NPHP-JBTS-MKS ciliopathy protein network
To better define physical interactions among the NPHP-JBTS-MKS disease proteins, we used the G-LAP-Flp purification strategy (Torres et al., 2009) to identify interacting proteins that copurify with the nine disease gene products known at the time this study was initiated. The G-LAP-Flp strategy is an optimized system for rapid generation of mammalian stable cell lines that facilitates high-confidence and high-throughput proteomic studies. We created clonal Flp-In cell lines stably expressing a LAP-tag (EGFP-TEV-S-peptide) fused to the amino-terminus of each individual disease protein (NPHP1, NPHP2/Inversin, NPHP3, NPHP4, NPHP5/IQCB1, NPHP6/CEP290, NPHP8/RPGRIP1L, AHI1/Jouberin, and MKS1) in NIH 3T3 fibroblasts, mouse kidney inner medullary collecting duct (IMCD3) cells, or human retinal pigment epithelial (RPE) cells. These ciliated cell models provide a physiologically relevant context for studying ciliary assembly, signaling and cystogenesis pathways. We then isolated complexes associated with each bait protein by tandem affinity purification and identified interacting proteins by mass spectrometry (MS) (Figure S1B).
To identify bona fide interactors, we optimized the LAP purification procedure to avoid the possibility of carryover, which would severely confound the analysis of a protein network (Supplemental Experimental Procedures). For each candidate interactor, we evaluated metric MS data including the total number of peptides, the sum of ion currents contributing to a specific mass spectrum (Sum TIC), and the percent sequence coverage. Data from fifteen affinity-purification protein mass-spectrometry (APMS) experiments were compiled into a single dataset. If any single spectrum from an identifiable peptide was found, the corresponding protein was included (Table S1; msdata.txt). Using the method developed by Scholtens and Gentleman (Scholtens, 2004; Scholtens et al., 2005), we performed a proteomic network analysis (Supplemental Experimental Procedures; nphp.networks.R), which we visualized using the Cytoscape software package (nphp.cys) (Shannon et al. 2003).
This method creates a ranked list of the most probable subgraphs, representing interactions (edges) between proteins (nodes) in a graphical form (Figure 1D-F; Figure S2). Here, the nodes represent the individual NPHP, JBTS, and MKS protein baits plus the identified hits. Hits identified in an APMS experiment are hypothesized to interact with the bait protein, and this connection is represented by an edge between the bait node and the node representing the hit. If a newly identified protein (node) shows a single interaction (edge) with a bait, it is of lower informational value compared to a node that shows multiple interactions with different baits; the presence of symmetric interactions, where two baits both identify the other efficiently in the APMS experiment, is of the highest value. Here, we began our analysis by first constructing a network based solely on the physical interactions seen in our APMS dataset, and later incorporated additional functional data to further validate the network.
When the physical interaction data are analyzed using this method, a striking disease protein network emerges. As highlighted in Figure 1B and C, we find that the most significant interactors for the NPHP-JBTS-MKS disease proteins are in fact other disease proteins from the same group. Our data confirm some previously reported interactions, including the NPHP1-NPHP2 and NPHP1-NPHP4 interactions (Mollet et al., 2002; Otto et al., 2003). However, our analysis is more comprehensive and provides a quantitative view of the most abundant interactors (Table S1). To validate our network, we tested several newly identified interactions by co-immunoprecipitation, and by in vitro binding to identify direct interactions. Our analysis reveals that NPHP-JBTS-MKS proteins do not form a single complex, but instead cluster into three biochemically distinct modules: (1) NPHP1, NPHP4, NPHP8 (“1-4-8”), (2) NPHP5, NPHP6 (“5-6”) and (3) MKS1, MKS6 (“MKS”).
Below we describe interactions within the specific modules.
NPHP1, NPHP4 and NPHP8 interact and localize to cell-cell contacts and the ciliary transition zone
NPHP1, NPHP4 and NPHP8 show strong mutual interactions via LAP-tagging. LAP-NPHP1 purifications contained endogenous NPHP4 and NPHP8 peptides in high abundance. Reciprocally, NPHP1 was identified in LAP-NPHP4 and LAP-NPHP8 purifications (Tables S1 and S2). Visualizing purified LAP-NPHP1 on silver-stained gels, revealed a substoichiometric band of NPHP8, and barely detectable NPHP4. LAP-NPHP4 purifications showed a band of NPHP8, NPHP1, and distinctive breakdown products of NPHP4. LAP-NPHP8 showed high efficiency interactions with NPHP1 and NPHP4 (Figure 2A). Quite possibly NPHP1-4-8 form multiple or processed complexes.
Figure 2. Validation of NPHP-JBTS-MKS interactions using co-immunoprecipitation and in vitro binding assays (see also Figure S3).
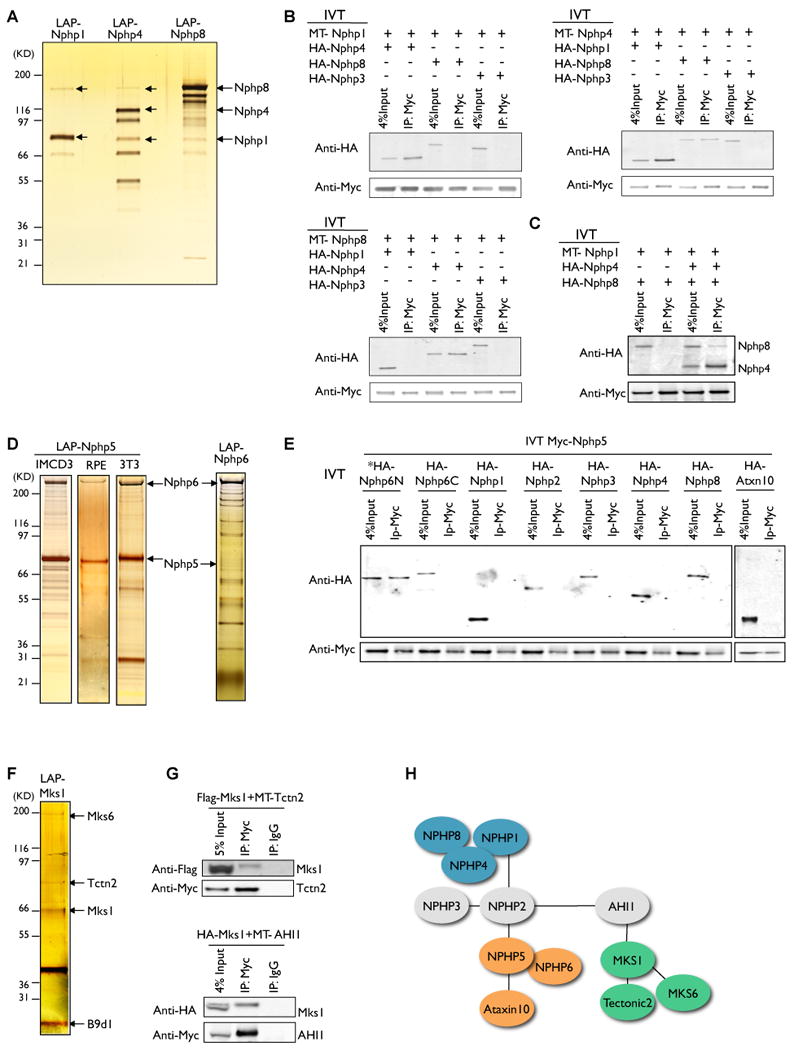
A. LAP-NPHP1, LAP-NPHP4 and LAP-NPHP8 were immunopurified from IMCD3 cells using anti-GFP antibody beads, eluted with TEV protease and recaptured on S-protein agarose. Eluates were separated on 4-12% SDS-polyacrylamide gradient gels and visualized by silver staining. NPHP1, NPHP4 and NPHP8 species are noted by arrows. B-C. NPHP4 bridges the interaction between NPHP1 and NPHP8 in vitro. Myc (MT)-tagged and HA-tagged NPHP1, NPHP4 and NPHP8 were in vitro translated using cell-free wheat germ extract. Each Myc-tagged protein was incubated with HA-tagged protein (s) and immunoprecipitated using anti-Myc beads. Eluates were separated by SDS-PAGE and immunoblotted with an anti-HA antibody. HA-NPHP3 was used as a negative control. D. LAP-NPHP5 complexes were immunopurified from IMCD3 (left), RPE (middle) and NIH 3T3 (right) cells, and LAP-NPHP6 complexes were immunopurified from IMCD3 cells as described in Figure 2A. Identified NPHP5 and NPHP6 species are noted by arrows. E. Interactions between NPHP5 and its associated proteins in vitro. Myc-tagged NPHP5 and HA-tagged interactors were in vitro translated and tested for direct binding using the procedure described above. NPHP5 binds directly to NPHP6 via its N-terminal domain (NPHP6N). F. LAP-MKS1 complexes were immunopurified from NIH 3T3 cells using the procedure described in Figure 2A. Identified Mks1, B9d1, Tctn2 and Mks6 species are noted by arrows. G. Validation of the interactions between Mks1 and copurified proteins Tctn2 and Ahi1. Myc-tagged Tctn2 or Ahi1 were coexpressed with Flag- or HA-tagged Mks1 in HEK293T cells and immunoprecipitated using anti-Myc beads or control IgG beads. Eluates were separated by SDS-PAGE and immunoblotted with an anti-Flag or anti-HA antibody. H. Cartoon summarizing the interactions among NPHP-JBTS-MKS proteins. Ellipse = protein, black line = interaction identified by MS/MS, touching ellipses = direct interactions validated by in vitro binding. NPHP1-4-8, NPHP5-6 and MKS modules are highlighted in blue, orange and green.
We used in vitro binding to test if these NPHP proteins interact directly. We assayed whether in vitro translated Myc-tagged NPHP1, NPHP4 and NPHP8 immunoprecipitate HA-tagged proteins produced by in vitro translation in wheat germ extracts. We find that NPHP4 directly binds both NPHP1 and NPHP8 in vitro, and can bridge the interaction between NPHP1 and NPHP8, whereas NPHP1 and NPHP8 do not appear to bind directly (Figure 2B-C, 2H).
To explore functional interactions among NPHP1, NPHP4 and NPHP8, we investigated their subcellular localization. LAP-NPHP1, LAP-NPHP4, and LAP-NPHP8 each localize diffusely in the cytoplasm of IMCD3 cells seeded at low density. Strikingly, as cells approach confluence and develop into polarized epithelial monolayers, these NPHP proteins accumulate to cell-cell contacts, mostly basolateral of tight junctions (Figure 3A; Figure S4A-B). The NPHP1-4-8 proteins can also be found at a specified compartment that extends between the basal body to the base of the axoneme, shown by co-staining with the mother centriole marker ODF2, centriole distal appendage marker OFD1 and axonemal acetylated tubulin (Figure 3A-B). This NPHP1-4-8 compartment is reminiscent of the ciliary transition zone, believed to function as part of the ciliary sensory machinery in worms (Fliegauf et al., 2006; Winkelbauer et al., 2005). The localization of NPHP1-4-8 at transition zones can appear even in sparse cells, before the cell monolayer is fully organized into an epithelial sheet with complete adherens and apical junctions. This suggests that NPHP1-4-8 can be organized above the basal body independently from its organization at cell-cell junctions, and the basal body may be sufficient to organize the “1-4-8” compartment at the transition zone. NPHP1-4-8 each contain C2 domains, which might mediate interactions with phospholipids at cell-cell junctions or at the ciliary base.
Figure 3. Localization of NPHP “1-4-8”, NPHP “5-6” and NPHP2 to the ciliary transition zone, centrosome and the inversin compartment (see also Figure S4).
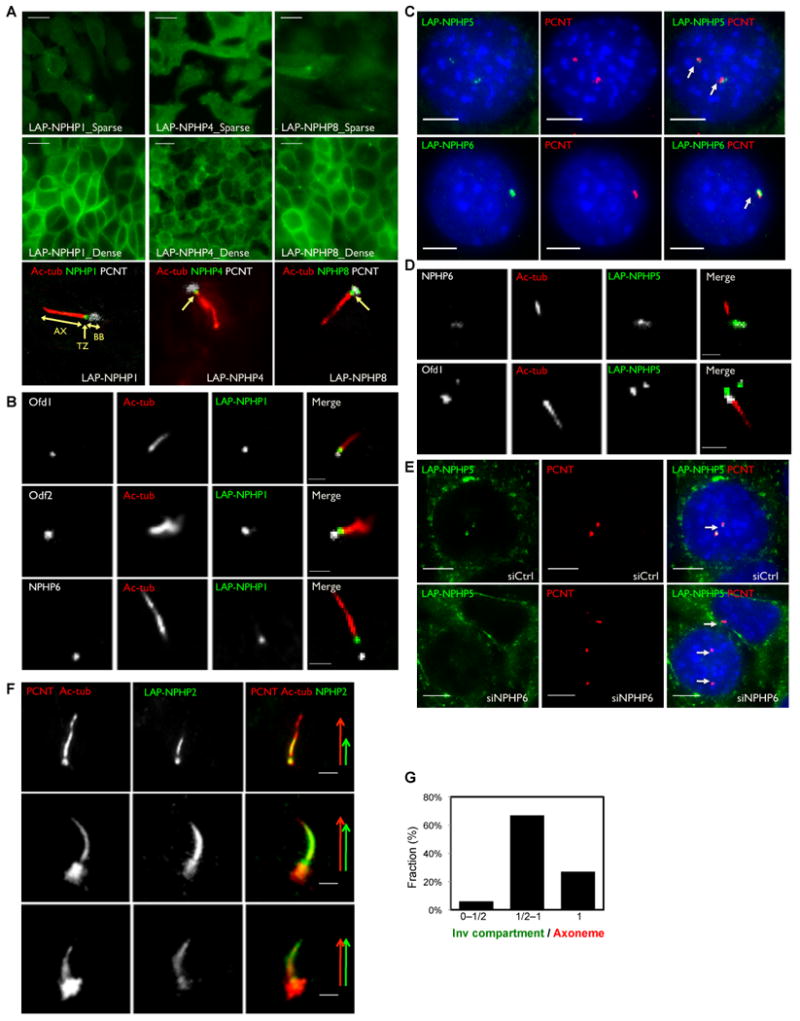
A. IMCD3 cells stably expressing LAP-NPHP1 (green), LAP-NPHP4 (green) or LAP-NPHP8 (green) were immunostained for pericentrin (PCNT, white) and acetylated α-tubulin (ac-tub, red). AX: Axoneme; TZ: Transition Zone; BB: Basal Body. B. IMCD3 cells stably expressing LAP-NPHP1 (green) were immunostained for acetylated α-tubulin (ac-tub, red) and Ofd1 (white), Odf2 (white), or NPHP6 (white) C-D. NPHP5 and NPHP6 co-localize to the centrosome. C. IMCD3 cells stably expressing LAP-NPHP5 (green) or LAP-NPHP6 (green) were immunostained for pericentrin (PCNT, red). D. IMCD3 cells stably expressing LAP-NPHP5 (green) were immunostained for acetylated α-tubulin (ac-tub, red) and NPHP6 (white) or Ofd1 (white). E. Centrosomal localization of LAP-NPHP5 is disrupted upon depletion of NPHP6. IMCD3 LAP-NPHP5 (green) cells were transfected with siRNA against NPHP6 or control, and then immunostained for pericentrin (PCNT, red). Nuclei were stained with Hoechst 33258 (blue). F. NPHP5 interacting protein NPHP2 localizes to the centrosome and to the cilium. IMCD3 LAP-NPHP2 cells (green) were immunostained for pericentrin (PCNT, red) and acetylated α-tubulin (ac-tub, red). Arrows exemplify variable NPHP2/Inversin compartment extensions along the axoneme. G. Percentage of cilia with a range of “Inversin compartment / Axoneme” ratios. Scale bars, 10um (A), 5um (C) and (E), 2um (B), (D) and (F).
Some previously reported interactors of NPHP1-4-8 were not seen in our purifications, including NPHP3, NPHP6, and a group of cortical regulators (Pyk2, p130Cas, PALS1, PATJ, Par6) (Benzing et al., 2001; Delous et al., 2009; Donaldson et al., 2000; Olbrich et al., 2003; Murga-Zamalloa et al., 2010). These absences likely reflect (1) differences in the efficiency of detecting interactions by APMS versus co-immunoprecipitation/immunoblot; (2) the difficulty of detecting membrane protein interactions in detergent lysates; or (3) more interestingly, rewired interactions in different tissues. Notably, we did validate that NPHP1 interacts with NPHP3 by coimmunoprecipitation (co-IP), showing that co-IP is more sensitive to show some interactions, compared to copurification (Figure S3D).
NPHP5 and NPHP6 form a complex and localize to the centrosome
Purifications of NPHP5 and NPHP6 consistently demonstrated strong binding between NPHP5 (MW∼ 68kD) and NPHP6/CEP290 (MW ∼ 290kD) in NIH 3T3, IMCD3 and human RPE cells (Figure 2D; Table S1-2). We validated this interaction using in vitro translated proteins, and confirmed that NPHP5 binds directly to NPHP6 via an N-terminal domain spanning amino acids 1-1207, consistent with a previously published study (Schafer et al., 2008). In mammalian cells, NPHP6 has been shown to localize to centrosomes (Chang et al., 2006). Consistently, LAP-NPHP5 and LAP-NPHP6 both co-localize with the centrosome marker pericentrin in IMCD3 cells (Figure 3C). LAP-NPHP5 also co-localizes with endogenous NPHP6, but not with the centriole distal appendage marker OFD1 (Figure 3D). We tested if either protein was required to recruit the other to the centrosome by siRNA depletion of NPHP5 or NPHP6. We found that NPHP5 failed to localize to the centrosome in the absence of NPHP6 (Figure 3E). In 53% of cells, LAP-NPHP5 is fully absent from pericentrin-positive centrosomes, compared to only 9% of cells transfected with a non-targeting siRNA. Conversely, depletion of NPHP5 had no effect on NPHP6 localization (not shown). We conclude that NPHP6 binds NPHP5 and recruits NPHP5 to the centrosome.
NPHP5 also interacts with other NPHP proteins. We observed co-purification of NPHP5 with NPHP1-4-8 only in NIH 3T3 cells, and with NPHP2 only in IMCD3 cells. These interactions were validated by co-IP in HEK293T cells, but likely require additional proteins to bridge the interactions, since NPHP5 does not bind directly to NPHP1, NPHP4, NPHP8 or NPHP2 in vitro (Figure 2E, 2H). In contrast to the basal body localization of NPHP5, NPHP2 was observed at the basal body, but also within the primary cilium (Figure 3F), A recent study suggested that NPHP2 localized primarily to the proximal segment of the axoneme (Shiba et al., 2010), which the authors termed the “inversin compartment”. Intriguingly, we observed NPHP2/Inversin compartment in a range of extensions along the axonemal structure, beginning proximally, but extending distally along the axoneme (Figure 3F-G; Figure S4C). The distinct localizations of NPHP1-4-8, NPHP5-6 and NPHP2 suggest a hierarchy for how these interactions are organized. Further, the connections between these modules vary notably among IMCD3, RPE and NIH 3T3 cells, suggesting that these modules may have distinct organization and functions in polarized epithelial cells not seen in fibroblasts.
NPHP5 additionally copurified with proteins previously linked to ciliogenesis. In all three cell lines, NPHP5 copurifies with Sec3; in IMCD3 cells, NPHP5 also copurified with Sec8 and Sec10 (Figure S2D). Sec3, Sec8 and Sec10 are components of the exocyst complex, a protein complex important in membrane trafficking and recently shown to be required for ciliogenesis (Zuo et al., 2009). NPHP5 at the basal body may help recruit the exocyst to a membrane compartment at the cilia base. In IMCD3 cells, we also observed co-purification of NPHP5 with Ataxin 10 (Figure S2D), a protein linked to Spinocerebellar Ataxia, a disease with distinctive deficiencies in cerebellar signaling and function (Matsuura et al., 2000; discussed below). We validated the NPHP5-Ataxin10 interaction by co-IP, but this interaction does not appear to be direct based on in vitro binding (Figure S3A; Figure 2E, 2H).
Mks1 binds to proteins important for neural tube closure including Mks6 and Tectonic
Extending from our core interaction network in Figure 1C, we identified a third module consisting of Mks1 and its interacting proteins (Figure 1F). Mks1 localizes to the base of the cilium in vertebrates and nematodes (Bialas et al., 2009) and MKS1 is mutated in type 1 Meckel-Gruber Syndrome. Recent studies show that Mks1 loss of function mouse mutant kerouac (krc) exhibits neural tube patterning defects, polydactyly, exencephaly, and biliary malformations. These are similar to the known defects in human MKS, believed to be linked to disruption of Hh signaling (Weatherbee et al., 2009). To further characterize this pathway, we purified proteins associated with Mks1 from Hh-responsive NIH 3T3 cells. Mks1 copurified with all three members of the Tectonic family of proteins (Tectonic 1-3) (Tables S1 and S2). Tectonic is a family of three potentially secreted or transmembrane proteins. Tectonic1 has been implicated in Hh-mediated patterning of the neural tube in mouse (Reiter and Skarnes, 2006), and our data suggest that Tectonic2 (Tctn2) is also important for Hh signaling (presented below). Other Mks1 interacting proteins include Mks6/CC2D2A (MW∼ 188kD) and B9d1 (MW∼ 23kD), both of which can be visualized on the silver stained gel (Figure 2F). Mutations of MKS6 have been identified in MKS and JBTS patients, and are associated with reduced ciliogenesis and neural tube defects in these patients (Mougou-Zerelli et al., 2009; Tallila et al., 2008). B9d1, along with B9d2 and Mks1, are the three known mammalian proteins containing a B9 domain. C. elegans B9 proteins form a complex that localizes to the ciliary base (Williams et al., 2008). C. elegans B9 proteins function redundantly with nephrocystins to regulate sensory cilia morphology and behavior, while individual mammalian B9 protein appears to function more independently. Like Mks1, mouse B9d1 is important for Hh signal transduction (B. Chih and A. Peterson, personal communication). Therefore, Mks1 and its interactors may function as key regulators of the Hh signaling cascade to regulate proper patterning of the neural tube. Mks1, Mks6, and Tectonic1, also bind to the Joubert Syndrome protein Ahi1/Jouberin, which in turn copurifies with NPHP2 (Figure 1F, 2G; Figure S2B and Table S1), suggesting Ahi1 as a potential bridging molecule.
Functional Requirements for Ciliation, 3D Spheroid Formation, and Hh signaling Show Distinct Activities for the NPHP “1-4-8”, NPHP “5-6” and MKS modules
NPHP, JBTS and MKS are hypothesized to be diseases of ciliary dysfunction. We therefore tested whether NPHP-JBTS-MKS proteins are required for ciliogenesis. In IMCD3 cells, we found 61% of siRNA control treated cells formed primary cilia, as detected by staining for acetylated α-tubulin and pericentrin (Figure 4A-B). As a positive control, we depleted Ift88, an intraflagellar transport component previously shown to be required for cilia formation (Pazour et al., 2000). As expected, depletion of Ift88 caused a dramatic decrease in ciliation (Figure 4A-B). We then depleted Nphp5, Nphp6, Nphp2, Nphp3, Nphp1, Nphp4, Nphp8, Ahi1 or Mks1 mRNAs by ∼ 60-95% using siRNAs (Figure S5B). Depletion of centrosomal proteins Nphp5 and Nphp6, along with Mks1 led to ciliogenesis defects. In contrast, normal numbers of cilia were observed in cells depleted of Nphp1, Nphp4, Nphp8, Nphp2, Nphp3, or Ahi1 (Figure 4B; Figure S5A). Our data suggest that Nphp5, Nphp6 and Mks1 are critical for ciliogenesis, whereas the other NPHP-JBTS-MKS proteins might not be strictly required for ciliogenesis under standard cell culture conditions, or without much more efficient knockdown. These proteins may instead be important for establishing tissue architecture, regulating ciliary signaling, or only affect ciliogenesis in vivo.
Figure 4. Functional requirements for ciliation and 3D spheroid formation show distinct activities for the NPHP “1-4-8”, NPHP “5-6”, and MKS modules (see also Figures S4 and S5; Tables S4, S6 and S7).
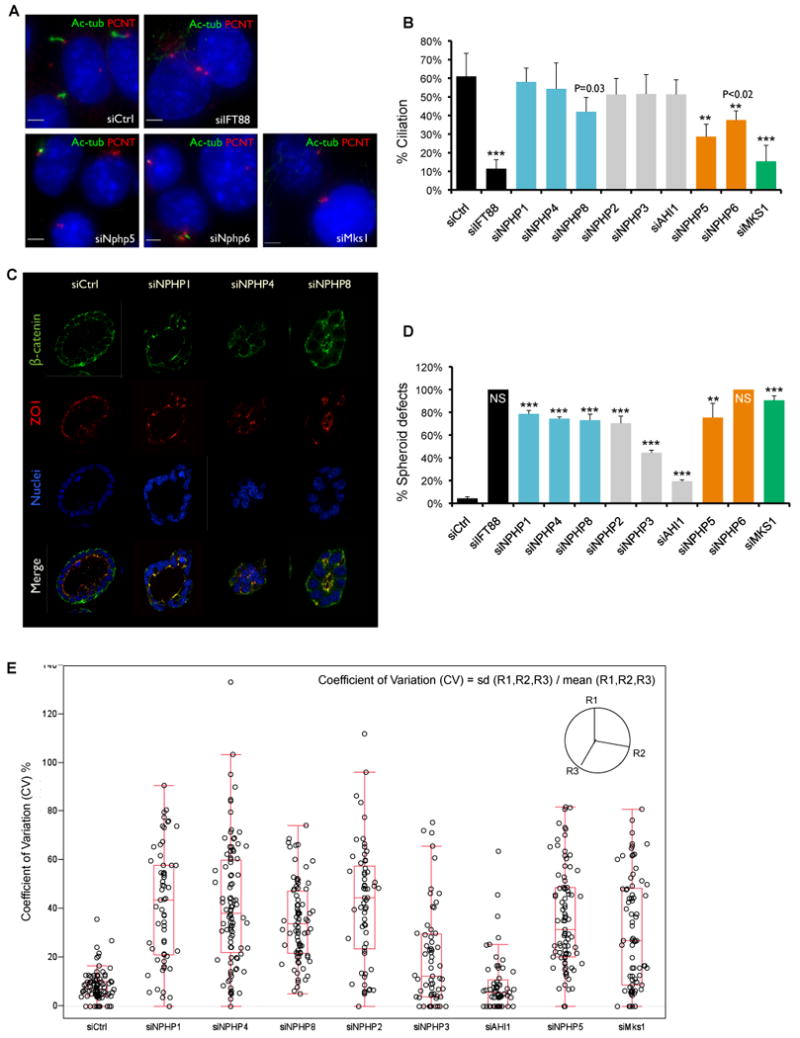
A. Depletion of NPHP5, NPHP6, and MKS1 causes ciliation defects. IMCD3 cells were transfected with siRNAs against individual disease genes, IFT88, or control. Cells were fixed 72 hrs post-transfection and stained for acetylated α-tubulin (green), pericentrin (red), and DNA dye Hoechst 33528 (blue). Scale bar, 5um.
B. Cilia were scored based on positive, adjacent staining of both pericentrin and acetylated α-tubulin. Percentage of nuclei with cilia was plotted (500-700 cells counted). Error bars represent standard error. *** p <0.002; ** p <0.02 (student's t test).
C. Depletion of NPHP1, NPHP4 or NPHP8 cause spheroid defects in 3D kidney culture. IMCD3 cells were transferred to 3D collagen/Matrigel culture 24 hour post-transfection. Spheroids were fixed 72 hours later and immunostained for β-catenin (green) and ZO1 (red). Nuclei were stained with Hoechst 33528 (blue).
D. Percentage of spheroids with defects. 400 – 700 spheroids counted and error bars represent standard error. NS, “No spheroids formed”, shown as 100% defective. *** p <0.001; ** p <0.01 (student's t test).
E. The sphericity of a spheroid was defined using the three radii (R) measurements, which were sampled on each spheroid at 100 degree intervals. The coefficient of variation (CV) was calculated using the formula: CV= standard deviation (R1,R2,R3) / mean (R1,R2,R3). Raw CV data from each knockdown are plotted along with the outlier box plot. Lower quartile = 25th percentile; upper quartile = 75th percentile; top line = upper quartile + 1.5 × interquartile range; bottom line = lower quartile - 1.5 × interquartile range; middle line = 50th percentile; data points outside the lines are outliers.
To investigate the roles of NPHP-JBTS-MKS proteins in tissue architecture, we tested the effect of depleting these proteins in a system that reflects the cell biology of the kidney collecting duct and thus reports on defects seen in cystic kidney diseases. Spheroid growth in three-dimensional (3D) culture allows epithelial cells to organize into polarized, ductal structures that resemble their in vivo architecture. The spheroid systems are unique models of epithelial cell polarity and signaling (Supplemental References). Notably, IMCD3 cells are derived from collecting ducts at the cortical-medullary border, thought to be the key target cells in nephronophthisis. Using this model system, we transfected IMCD3 cells with siRNAs for Ift88, Nphp1, Nphp4, Nphp8, Nphp5, Nphp6, Nphp2, Nphp3, Ahi1, or Mks1, and then plated the transfected cells in Matrigel to induce 3D spheroid growth. After 3 days, control siRNA-treated cells formed spheroid structures with a clear lumen, apical cilia, defined tight junctions, and clear basolateral structures. Cells depleted of Ift88 or Nphp6 developed few or grossly affected spheroids, appearing as clumps of cells with few cilia evident, suggesting that ciliary genes are strongly important in spheroid formation. Consistently, depletion of Nphp5, or Mks1, genes required for ciliation, likewise caused spheroids to form with severe defects and few cilia evident. In striking contrast, cells depleted of Nphp1, Nphp4, Nphp8 and Nphp2 developed spheroids with irregular lumens, reduced sphericity, fewer ZO1 positive tight junctions, and perturbed localization of β-catenin (Figure 4C-E). However, no gross abnormality of ciliogenesis was evident in these spheroids (not shown). The apparent partition in functional requirements between ciliation and 3D spheroid formation suggests a distinct activity for the Nphp “1-4-8” module in organizing apical junctions versus the Nphp “5-6” and Mks1 modules in organizing cilia. We observed very modest disorganization of spheroids in cells depleted of Ahi1 or Nphp3 (Figure 4D-E), suggesting these NPHP proteins may participate in mechanisms distinct from ciliation or apical organization.
Studies in zebrafish further supported the importance of NPHP “1-4-8”, “5-6” and MKS modules in apical organization and ciliary function. Knockdown of nphp2, nphp5 and nphp6 leads to body curvature defects (Otto et al, 2003; Schafer et al, 2008;). We have also found that knockdown of additional NPHP, JBTS and MKS proteins similarly results in body axis alterations (Figure S5D). Kupffer's vesicle (KV), the ciliated organ implicated in zebrafish left–right patterning (Essner et al., 2005), is also disrupted in these mutants (Figure S5C). The KV arises from dorsal forerunner cells (DFCs). DFCs migrate attached to the overlying surface epithelium and rearrange into rosette-like epithelial structures, and then coalesce into a single rosette that differentiates into the KV with a ciliated lumen at its apical center. In addition to cilia integrity, polarity cues and apical organization are also crucial for KV morphogenesis and function (Oteiza et al., 2008), consistent with our observation that NPHP1/4/8, NPHP5 and MKS1 morphants all show KV defects.
Disruption of Hh signaling is thought to partly account for the neural tube and limb phenotypes seen in MKS patients (Weatherbee et al., 2009). MKS1 is critical for ciliogenesis, spheroid formation, and Hh signaling. However, there is no clear evidence demonstrating that NPHP5/6 are directly involved in Hh signaling. To investigate the functions of NPHP5/6 in Hh signaling, we used the standard S12 Gli-luciferase Hh signaling assay and observed that siRNA knockdowns of Nphp5 and Nphp6 had no effects on Hh signal transduction (Table S4). Surprisingly, ciliogenesis was also not perturbed in these cells (Table S4), suggesting that NPHP5/6 may function differently in osteoblasts (S12 cells) versus in polarized epithelial cells (IMCD3). In contrast to the requirement of Mks1 in Hh signal transduction, the specific roles of NPHP5 and NPHP6 in Hh signaling remain to be clarified.
Identification of Ataxin10 and Tectonic2 as new NPHP-JBTS disease proteins
Our analysis of the NPHP-JBTS-MKS network reveals extensive physical interactions among known disease proteins as well as with proteins not currently implicated in cilia-associated disorders. We reasoned that the physical interactions between specific disease proteins lead to the phenotypic overlap of these diseases and therefore hypothesized that some interacting proteins from our analysis may represent unrecognized disease loci. Remarkably, we found multiple recently reported disease proteins identified independently from our interaction network. These proteins include CC2D2A/MKS6 (Mougou-Zerelli et al., 2009; Tallila et al., 2008), which interacts with AHI1 and MKS1, and Nek8/NPHP9, which interacts with NPHP2 (Figure 1F; Figure S2E; Figure S3C).
With the potential to use proteomic network analysis as an unbiased means to discover new disease genes, we submitted 38 candidate genes to total genome linkage analysis to look for extensive homozygosity within candidate chromosomal intervals. This method is based on SNP analysis to predict candidate intervals that are linked to causal mutations. Genes within these intervals can then be sequenced to establish the presence of homozygous mutation. In small families lacking pedigree information, the number of candidate intervals can be large, thus make sequencing of candidate genes impractical. Recently, a systematic approach with complete exome sequencing provided one solution to identifying specific disease alleles (Hildebrandt et al., 2009b). Here, we imagined that using our high-confidence proteomic hits would provide an enriched set to discover disease genes in NPHP/JBST/MKS patients. Using this method, we were able to identify two new genes linked to NPHP/JBTS disease: Ataxin10 (ATXN10) and Tectonic2 (TCTN2).
First, we noted a genomic region of extensive homozygosity of 11.6 Mb on chromosome 22 with a high nonparametric linkage score for 3 affected siblings in a consanguineous family (A1197) from Turkey (Figure S6A). After genome analysis with 1M Affymetrix SNP chip, a homozygous ATXN10 mutation (IVS8-3T>G) was identified in all 3 affected siblings. All affected children died at age 2 years from kidney failure and renal biopsies, consistent with nephronophthisis (Figure 5A). One of the siblings additionally suffered from seizures and had evidence of cerebral atrophy by imaging. This mutation was absent from 90 healthy Caucasian control samples and 86 ethnically matched control individuals. We identified the protein encoded by ATXN10, Ataxin10, as an NPHP5 interacting protein.
Figure 5. Identification of ATXN10 and TCTN2 as new NPHP and JBTS disease genes (see also Figure S6).
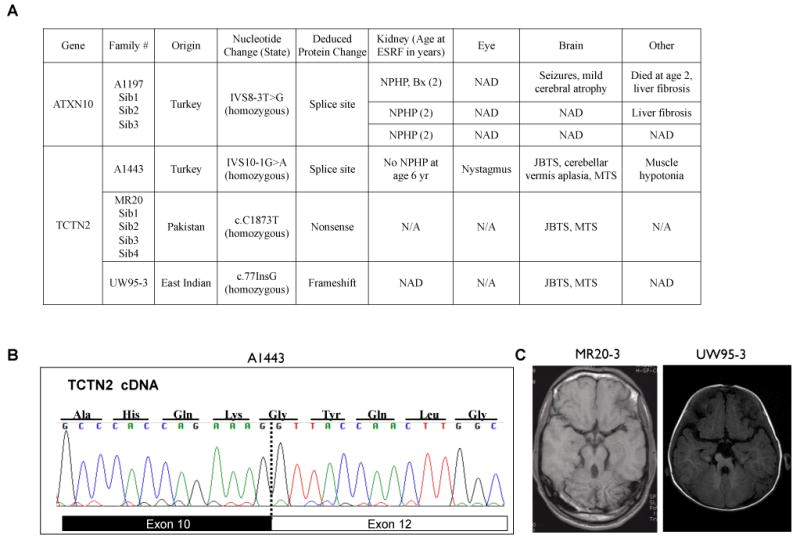
A. Genotype and phenotype of patients with mutations in ATXN10 and TCTN2. Bx, Biopsy compatible with NPHP; MTS, “molar tooth sign”; NAD, nothing abnormal detected; N/A, clinical data not available.
B. RT-PCR was performed in Joubert syndrome patient A1443 using cDNA primers to exons 7 and 14 of TCTN2. Sequencing revealed an in-frame skipping of exon 11.
C. MRI images (T1) of Joubert syndrome patients MR20-3 and UW95-3 showing the molar tooth sign (MTS).
Similarly, we observed a region of extensive homozygosity in a particular locus near the gene TCTN2 in 6 patients with consanguineous background (Figure S6B). After sequencing all exons of TCTN2, a mutation was found in one family (A1443) from Turkey at IVS10-1G>A, affecting the obligatory splice acceptor site, which resulted in skipping of exon 11 (Figure 5A-B). The mutation was also found in a heterozygous state in the parents of a 6 -year old female who was homozygous for the mutation. She had been previously diagnosed with Joubert syndrome due to cerebellar vermis aplasia and hypotonia, and had no evidence for renal disease. Again, this mutation was absent from the same control sets of Caucasian and ethnically matched healthy individuals.
With the evidence linking TCTN2 to Joubert Syndrome, we screened additional patients and identified another two Joubert families with frameshift or nonsense mutations in TCTN2. Patient UW95-3 had gross motor and communication delays and increased tone and reflexes, as well as bilateral talipes equinovarus deformity (clubfeet). At 15 months of age, he had no evidence of cardiovascular, renal or liver disease by ultrasound and laboratory testing (Figure 5A). Brain MRI revealed the molar tooth sign (Figure 5C), consistent with Joubert Syndrome. A homozygous mutation was identified in TCTN2 exon 1 (c.77InsG; p.D26GfsX51) that results in frame shift and a premature stop codon (Figure S6C). Family MR20 has four affected siblings with consanguineous Pakistani background. All four patients developed childhood-onset Joubert Syndrome with extremely poor learning abilities. Brain MRI revealed the molar tooth sign (Figure 5C). A homozygous nonsense mutation in TCTN2 exon 16 (c.C1873T; p.Q625X) was identified in all 4 affected, but not in unaffected siblings (Figure 5A; Figure S6D).
Thus, from a list of 38 candidates curated by our proteomic network analysis and a modest number of patients tested, two new human disease genes were identified.
Tctn2 regulates Hh signaling and ciliogenesis
Tctn2 was identified as an interactor of Mks1, itself shown to regulate Hh-dependent neural tube patterning in vivo (Weatherbee et al., 2009). The human TCTN2 mutations we identified are associated with neural developmental defects. Based on this observation, we hypothesized that Tctn2 could be a regulator of Hh signaling. To test this hypothesis, we generated Tctn2 null mice. On a mixed 129/Bl6 background Tctn2 mutants have fully penetrant neural tube closure defects and exencephaly is apparent at E13.5 (Figure 6A). On a Bl6 background Tctn2-/- embryos exhibit microphthalmia, cleft palate, and polydactyly (Figure 6B-C), consistent with altered Hedgehog signaling. Tctn2 mutants also have ventricular septal defects (Figure 6D) and can display right-sided stomach (Figure 6E), phenotypes characteristic of ciliary defects. To determine if Tctn2 is required for cilia function or formation, we examined primary cilia in mouse embryonic fibroblasts (MEFs) and neural tubes. Tctn2 was required for ciliogenesis in isolated cells and in vivo, consistent with TCTN2 being a ciliopathy gene (Figure 6F-G).
Figure 6. Tctn2 is required for ciliogenesis and Hh signaling transduction (see also Figure S7).
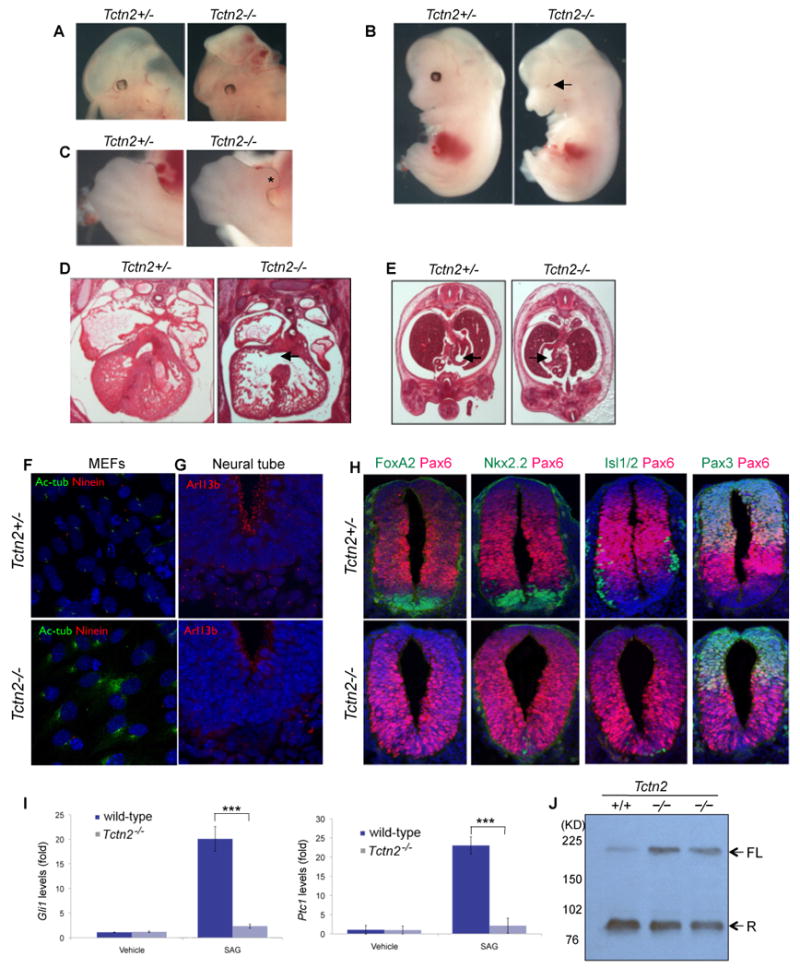
A. E13.5 Tctn2-/- embryos on a mixed 129/Bl6 background have fully penetrant cranial exencephaly. On a Bl6 background, E13.5 Tctn2-/- embryos display (B.) microphthalmia (arrow) and (C.) single hindlimb preaxial polydactyly, either bilaterally or unilaterally (asterisk). D. Hematoxylin and eosin staining of E14.5 Tctn2-/- embryos reveals ventricular septal defects (black arrow) and (E.) laterality defects as evidenced by randomized stomach situs. F. Mouse embryonic fibroblasts (MEFs) derived from E12.5 Tctn2+/- embryos are ciliated, whereas Tctn2-/- embryo-derived MEFs rarely generate cilia. Acetylated tubulin (green) marks cilia, Ninein (red) marks basal bodies and centrosomes, DAPI (blue) marks nuclei. G. Immunofluorescent detection of Arl13b (red) in E9.5 transverse neural tube sections indicates that Tctn2-/- embryos display few and abnormal cilia. H. Tctn2 is required for patterning of the ventral neural tube. Immunofluorescence of E9.5 transverse sections between the heart and hind limb stained for Pax6 in red and, in green, FoxA2, Nkx2.2, Islet1/2 or Pax3. I. Levels of the Hh transcriptional targets Gli1 and Ptc1 were assessed by qPCR in MEFs derived from E12.5 Tctn2+/+ and Tctn2-/- littermate embryos stimulated with DMSO (vehicle) or Smoothened agonist (SAG) for 18 hours. Tctn2+/+ MEFs upregulate Gli1 and Ptc1 20-25 fold following SAG addition, whereas Tctn2-/- MEFs are unresponsive. Experiments were performed three times in triplicate and values normalized to β-actin and presented as relative levels +/- SEM. *** p < 0.001 (student's t test). J. Lysates from E13.5 Tctn2+/+ and Tctn2-/- littermate embryos immunoblotted for Gli3. In wild-type embryos, the majority of Gli3 is processed into a truncated repressor form (R). Tctn2 mutants have increased levels of unprocessed full-length (FL) Gli3.
High level Hh signaling is required for formation of the floor plate (Sasaki and Hogan, 1994). Tctn2 mutants lack a morphologically distinct floor plate, and examination of FoxA2 expression in Tctn2-/- embryos revealed that the floor plate was not specified. Similarly, Pax6, which is repressed by Hh signaling, was ventrally expanded in the absence of Tctn2. A severe reduction in Nkx2.2-expressing V3 interneuron progenitors and Islet1/2-expressing motor neurons further suggested defects in Hh-dependent patterning in the absence of Tctn2 (Figure 6 H).
To directly determine if Tctn2 is important for Hh transduction, we assayed Ptc1 and Gli1, general transcriptional targets of Hh signaling, in wild -type and Tctn2-/- MEFs. Following pathway activation by addition of a Smoothened agonist (SAG) both Ptc1 and Gli1 are induced ∼20-fold in wild-type MEFs, while Tctn2-/- MEFs display negligible responsiveness (Figure 6I). Tctn2-/- embryos also exhibited increased amounts of full length, unprocessed (Gli3-190) Gli3 protein (Figure 6J), indicating that Tctn2 is important for Gli3 processing and function. Thus, Tctn2-/- neural tubes have defects characteristic of altered Hh signal transduction, and Tctn2-/- cells fail to respond to Hh agonists, suggesting that cerebellar defects in affected individuals with TCTN2 mutations may reflect defects in ciliogenesis linked to reduced Hh signaling.
Discussion
The NPHP-JBTS-MKS Interactome: A Multi-Module Network Linking Ciliopathies
Using a high-confidence proteomic strategy, we have discovered and begun a systematic mapping of an NPHP-JBTS-MKS interaction network. In contrast to our earlier studies on the Bardet-Biedl syndrome (BBS; Nachury et al., 2007), where seven highly conserved BBS proteins formed a single, monodisperse complex, the NPHP-JBTS-MKS proteins do not form a single complex. Instead, this large group of disease proteins can be clustered into three biochemically and functionally distinct modules, where proteins within the first two modules show notable colocalization (Figure 7). Our studies support that genetic loss-of-function within each individual module drives a unique mechanism contributing to the specific histopathologic features of these disorders. The first module consists of NPHP1, NPHP4 and NPHP8, localized to cell-cell contacts and to the ciliary transition zone. They are not strongly required for ciliation, as assayed in our in vitro models. However, when kidney epithelial cells are deprived of these proteins, they form disorganized spheroids in 3D culture characterized by irregular lumens, loss of tight junctions, and perturbed localization of β-catenin. NPHP1 and NPHP4 have been reported to interact with polarity proteins PALS1, PATJ and Par6 and depletion of NPHP1 or NPHP4 in MDCK cells results in delayed tight junction formation (Delous et al., 2009). These observations together with our data support the hypothesis that NPHP1-4-8 module organizes specialized structures at the apical surface of polarized cells and thus may participate in pathways important for epithelial morphogenesis and the establishment of tissue architecture. Given the effects of NPHP1-4-8 deficiency in pediatric renal disease, it will be important to examine whether this module is affected in tissues that are frequently impaired in patients with cystic kidney disease, including liver and pancreas.
Figure 7. A Model for the NPHP-Joubert-Meckel-Gruber Network.

Three interacting modules link centrosomal proteins to apical organization and to a Hedgehog regulatory network.
Unlike the 1-4-8 module, the centrosomal module proteins NPHP5 and NPHP6 are indispensable for ciliation in IMCD3 cells; furthermore, cells fail to develop normal spheroids when depleted of NPHP5 or NPHP6, underscoring the role of centrosome/cilia integrity in tissue organization. MKS1 and its interacting proteins are grouped in the third module, characterized by their functional connection to neural tube development and Hh signal transduction.
Several of the NPHP proteins appear to bridge the three major modules, notably NPHP2 and AHI1. NPHP2, -3, and -9 interact in the Inversin compartment, but the role of this new structure remains mysterious. Our preliminary data suggest NPHP3 is particularly important for localizing specific G-protein coupled receptors (GPCRs) to the cilia (L. Sang and P. Jackson, unpublished), which provides a first clue linking NPHP proteins to specific ciliary GPCR signaling pathways.
Collectively, the NPHP-JBTS-MKS interactome has provided new biochemical evidence that these disorders are highly connected, and has suggested specific underlying mechanisms leading to disease progression. Our 3D culture system appears to effectively mirror requirements for genes that suppress cystic kidney disease, including those that function to organize apical structures in the cell, notably NPHP1-4-8. For other genes such as NPHP5, NPHP6, and Mks1, broader defects in centrosome/cilia integrity and ciliary signaling may lead to tissue failure not only in the kidney, but also in a variety of other organs, including the neural tube and eye. Therefore, to our understanding, not all the “ciliopathies” are simply caused by the absence of cilia per se. They rather represent a manifestation of defects in multiple interlinked cellular mechanisms. More intriguingly, whereas mild renal cysts may arise simply as a result of tissue organization defect, lack of centrosome/cilia integrity would generally predict more severe phenotypes, notably retinal degeneration; ciliary disruption with altered Hh signaling appears linked to cerebellar malformation and neural tube defects.
The NPHP-JBTS-MKS Network is Distinct from BBsome and IFT Complexes
Bardet-Biedl syndrome (BBS) shares a number of common phenotypes with NPHP, JBTS and MKS, including kidney cysts, retinal degeneration, polydactyly, and mental retardation. There is a single documented BBS family with mutations in each of MKS1/BBS13 and NPHP6/BBS14, suggesting at a minimum a genetic interaction between BBS and MKS (reviewed by Zaghloul and Katsanis, 2009). However, we did not observe notable physical interactions between the NPHP-JBTS-MKS network and components of BBSome, other BBS proteins, or over 14 BBS1 interacting proteins (C. Westlake. and P. Jackson, unpublished). A reasonable hypothesis is that the shared phenotypes are likely due to broader disruption of some key regulatory pathways, such as the Hh signaling, Wnt signaling, cell polarity, or centrosome control of the cytoskeleton.
We also evaluated whether the NPHP-JBTS-MKS network proteins link to intraflagellar transport (IFT). The IFT process is essential for the formation of cilia, and defects in IFT may lead to cystic kidney disease and retinal degeneration (Davenport et al., 2007; Rosenbaum and Witman, 2002). Interestingly, NPHP-JBTS-MKS network also does not overlap with components of IFT-A or IFT-B complex proteins (S. Mukhopadhyay and P. Jackson, unpublished), with the notable exception of an interaction between NPHP5 and IFT122 (Table S1). A recent publication suggests that Chlamydomonas NPHP6/CEP290 is a transition zone protein important for tethering flagellar membrane to the transition zone, and loss of NPHP6/CEP290 results in an imbalance of IFT complexes in the flagellum (Craige et al., 2010). This is consistent with our model that NPHP proteins participate in a mechanism anchoring the transition zone to the centrosome and cell cortex. It will be intriguing to determine if any of the mammalian transition zone proteins (such as NPHP1, 4 and 8) or centrosomal proteins NPHP5/6 functionally link to IFT.
High-Confidence Proteomic Analysis Accelerates Discovery of New Disease Genes
The identification of an NPHP-JBTS-MKS network not only linked human genetics with underlying cellular mechanisms, it also accelerated the discovery of new NPHP-JBTS disease genes. The causative genes are still unknown in approximately 70% of patients with NPHP (Hildebrandt et al., 2009a). Employing protein interaction data to predict new candidate genes involved in human genetic disorders has been explored (Lim et al., 2006; Goh et al., 2007; Supplemental References). However lack of orthogonal information remains a major challenge in discovering new disease genes (Sowa et al., 2009). Our proteomic strategy has proved to be a highly effective approach, confirming two new disease loci among 38 candidate genes suggested by our network.
A pentanucleotide expansion of ATXN10 results in Spinocerebellar ataxia type 10, a neurodegenerative disease involving cerebellar dysfunction leading to ataxia and seizures (Matsuura et al., 2000). Additionally, Atxn10 has a proposed role in cerebellar neuron survival and neuritogenesis through an interaction with G-protein β2 subunit (Waragai et al., 2006). Depletion of Atxn10 in 3D kidney culture leads to modest defects in both spheroid organization and ciliogenesis (Figure S6E-F). We hypothesize that these defects may directly contribute to the kidney phenotypes observed in the NPHP patients. Further investigation will help clarify the various functions of Atxn10 in Nephronophthisis and in Spinocerebellar ataxia.
Tctn2 is a member of the Tectonic family proteins (Reiter and Skarnes, 2006), and similar to Tctn1, is important for Hh signal transduction. Intriguingly, whereas Tctn2 is indispensible for ciliogenesis in the neural tube, knockdown of Tctn2 in kidney IMCD3 cells only causes modest ciliation and spheroid defects (Figure S6E-F). Such distinct requirements may reflect differences in expression profiles as well as the complexity of the organs involved. Indeed, Tctn2 is highly expressed in embryonic brain tissues but below the limit of detection in the kidney (Figure S6G). The tissue distribution pattern of Tctn2 and the differences in requirements for ciliogenesis are consistent with the TCTN2 patient phenotypes, which show cerebellar vermis aplasia, but no renal disease.
Included in our NPHP-JBTS-MKS network are numerous new interacting proteins. Many of those proteins are functioning in fundamental cellular processes, such as cytoskeletal organization, intracellular transport, and enzymatic reactions (Table S3). Therefore, the NPHP-JBTS-MKS network has provided a road map not only for discovering new ciliopathy genes, but also for unveiling novel disease pathways. Moreover, since cell polarity and ciliary signaling defects have been implicated in a number of other diseases, notably cancer, the NPHP-JBTS-MKS network may also help advance our understanding of cancer and suggest potential therapeutic targets.
Experimental Procedures
Proteomic Network Analysis
Data from fifteen affinity-purification protein mass-spectrometry experiments were compiled into a single data set. If any single spectrum from an identifiable peptide was found, the corresponding protein was included. Of the resulting 850 unique hits, twelve proteins under current investigation were excluded from the dataset; all protein identifiers were mapped to Ensemble Gene IDs, and to common gene names. This dataset is available as the tab-delimited file ‘msdata.txt’ in the supplemental material. Further processing reduced this dataset to one comprising the results of the 3T3 and IMCD3 experiments only, and removed proteins that are either annotated to the ‘Keratin filament’ term in the GO Cellular Component Ontology or that include ‘keratin’ in the gene name. The complete peptide and scoring information is available in Table S5. An adjacency matrix was compiled for each cell line experiments, and from these matrices we derived two interaction networks. Each network's maximal bait-hit complete subgraphs were found, and merged using sensitivity and specificity parameters of 0.7 and 0.75, respectively (Scholtens et al., 2005). These resulting estimates of protein complex composition were plotted from within R, and the entire networks were exported for display in Cytoscape (Shannon et al. 2003). Enrichment of each network and its inferred sub-complexes for GO terms was assessed using Fisher's exact test, with the universe of genes defined as the set of mouse Ensemble Gene IDs having GO term annotations. Although this test does not explicitly account for correlation of GO term enrichment due to the ontologies' graph structures, it serves to provide a general view of the processes, functions and locations of the networks and their subgraphs. All dataset manipulations and subsequent analyses were performed using R version 2.11.1. Analysis of subgraphs used the package ‘apComplex’ version 2.14.0 (Scholtens 2004), while gene set enrichment analysis for GO terms used ‘topGO’ version 1.16.2. The analysis script and session information are included in the file ‘nphp.networks.R’ in the supplemental material.
Supplementary Material
Acknowledgments
The authors acknowledge expert advice and contributions from Chris Westlake, Guowei Fang, Ben Chih, Andy Peterson, Cecile Chalouni, John S. Beck, Darryl Y. Nishimura, Charles C. Searby, Martin Griebel, John Neveu, Bogdan Budnik, Renee Robinson, Alex Loktev, Jorge Torres, Saikat Mukhopadhyay, Dirk Siepe and Kevin Wright. We acknowledge the following support: JJM, NIH Medical Scientist Training Program Grant GM07365-33; RHG, the Netherlands Organisation for Scientific Research VIDI grant 016.066.354 and the EU FP7 “Syscilia” project 241955; LMB, the Cardiovascular Center Interdisciplinary Research Fellowship, University of Iowa; JFO, NIH grant DK071108; DCS, NIH Grant CA112369; J.B.V., Canadian Institutes of Health Research grant MOP-102758; DAD, KL2RR025015 and R01NS064077; J.F.R., NIH Grant R01-AR054396, the March of Dimes, the Burroughs Wellcome Fund, the Packard Foundation, and the Sandler Family Supporting Foundation; VCS, NIH Grants R01-EY11298, R01-EY017168, the Roy J. Carver Charitable Trust, Carver Endowment for Molecular Ophthalmology, and Research to Prevent Blindness. VCS is an HHMI investigator. FH is an HHMI Investigator, a Doris Duke Distinguished Clinical Scientist, and a Frederick G. L. Huetwell Professor. FH acknowledges support from the NIH grants (DK1068306, DK1069274, and DK090917).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baala L, Audollent S, Martinovic J, Ozilou C, Babron MC, Sivanandamoorthy S, Saunier S, Salomon R, Gonzales M, Rattenberry E, et al. Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome. Am J Hum Genet. 2007;81:170–179. doi: 10.1086/519494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benzing T, Gerke P, Hopker K, Hildebrandt F, Kim E, Walz G. Nephrocystin interacts with Pyk2, p130(Cas), and tensin and triggers phosphorylation of Pyk2. Proc Natl Acad Sci U S A. 2001;98:9784–9789. doi: 10.1073/pnas.171269898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bialas NJ, Inglis PN, Li C, Robinson JF, Parker JD, Healey MP, Davis EE, Inglis CD, Toivonen T, Cottell DC, et al. Functional interactions between the ciliopathy-associated Meckel syndrome 1 (MKS1) protein and two novel MKS1-related (MKSR) proteins. J Cell Sci. 2009;122:611–624. doi: 10.1242/jcs.028621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang B, Khanna H, Hawes N, Jimeno D, He S, Lillo C, Parapuram SK, Cheng H, Scott A, Hurd RE, et al. In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd16 mouse. Hum Mol Genet. 2006;15:1847–1857. doi: 10.1093/hmg/ddl107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437:1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- Craige B, Tsao CC, Diener DR, Hou Y, Lechtreck KF, Rosenbaum JL, Witman GB. CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J Cell Biol. 2010;190:927–940. doi: 10.1083/jcb.201006105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport JR, Watts AJ, Roper VC, Croyle MJ, van Groen T, Wyss JM, Nagy TR, Kesterson RA, Yoder BK. Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr Biol. 2007;17:1586–1594. doi: 10.1016/j.cub.2007.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delous M, Hellman NE, Gaude HM, Silbermann F, Le Bivic A, Salomon R, Antignac C, Saunier S. Nephrocystin-1 and nephrocystin-4 are required for epithelial morphogenesis and associate with PALS1/PATJ and Par6. Hum Mol Genet. 2009;18:4711–4723. doi: 10.1093/hmg/ddp434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson JC, Dempsey PJ, Reddy S, Bouton AH, Coffey RJ, Hanks SK. Crk-associated substrate p130(Cas) interacts with nephrocystin and both proteins localize to cell-cell contacts of polarized epithelial cells. Exp Cell Res. 2000;256:168–178. doi: 10.1006/excr.2000.4822. [DOI] [PubMed] [Google Scholar]
- Essner JJ, Amack JD, Nyholm MK, Harris EB, Yost HJ. Kupffer's vesicle is a ciliated organ of asymmetry in the zebrafish embryo that initiates left-right development of the brain, heart and gut. Development. 2005;132:1247–1260. doi: 10.1242/dev.01663. [DOI] [PubMed] [Google Scholar]
- Fliegauf M, Horvath J, von Schnakenburg C, Olbrich H, Muller D, Thumfart J, Schermer B, Pazour GJ, Neumann HP, Zentgraf H, et al. Nephrocystin specifically localizes to the transition zone of renal and respiratory cilia and photoreceptor connecting cilia. J Am Soc Nephrol. 2006;17:2424–2433. doi: 10.1681/ASN.2005121351. [DOI] [PubMed] [Google Scholar]
- Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabasi AL. The human disease network. Proc Natl Acad Sci U S A. 2007;104:8685–8690. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol. 2009a;20:23–35. doi: 10.1681/ASN.2008050456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt F, Heeringa SF, Ruschendorf F, Attanasio M, Nurnberg G, Becker C, Seelow D, Huebner N, Chernin G, Vlangos CN, et al. A systematic approach to mapping recessive disease genes in individuals from outbred populations. PLoS Genet. 2009b;5:e1000353. doi: 10.1371/journal.pgen.1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426:83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- Lee JH, Gleeson JG. The role of primary cilia in neuronal function. Neurobiol Dis. 2010;38:167–172. doi: 10.1016/j.nbd.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Hao T, Shaw C, Patel AJ, Szabo G, Rual JF, Fisk CJ, Li N, Smolyar A, Hill DE, et al. A protein-protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell. 2006;125:801–814. doi: 10.1016/j.cell.2006.03.032. [DOI] [PubMed] [Google Scholar]
- Matsuura T, Yamagata T, Burgess DL, Rasmussen A, Grewal RP, Watase K, Khajavi M, McCall AE, Davis CF, Zu L, et al. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nature genetics. 2000;26:191–194. doi: 10.1038/79911. [DOI] [PubMed] [Google Scholar]
- Mollet G, Silbermann F, Delous M, Salomon R, Antignac C, Saunier S. Characterization of the nephrocystin/nephrocystin-4 complex and subcellular localization of nephrocystin-4 to primary cilia and centrosomes. Hum Mol Genet. 2005;14:645–656. doi: 10.1093/hmg/ddi061. [DOI] [PubMed] [Google Scholar]
- Mollet Gr, Salomon Rm, Gribouval O, Silbermann F, Bacq D, Landthaler G, Milford D, Nayir A, Rizzoni G, Antignac C, et al. The gene mutated in juvenile nephronophthisis type 4 encodes a novel protein that interacts with nephrocystin. Nature genetics. 2002;32:300–305. doi: 10.1038/ng996. [DOI] [PubMed] [Google Scholar]
- Mougou-Zerelli S, Thomas S, Szenker E, Audollent S, Elkhartoufi N, Babarit C, Romano S, Salomon R, Amiel J, Esculpavit C, et al. CC2D2A mutations in Meckel and Joubert syndromes indicate a genotype-phenotype correlation. Hum Mutat. 2009;30:1574–1582. doi: 10.1002/humu.21116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murga-Zamalloa CA, Desai NJ, Hildebrandt F, Khanna H. Interaction of ciliary disease protein retinitis pigmentosa GTPase regulator with nephronophthisis-associated proteins in mammalian retinas. Mol Vis. 2010;16:1373–1381. [PMC free article] [PubMed] [Google Scholar]
- Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peranen J, Merdes A, Slusarski DC, Scheller RH, Bazan JF, Sheffield VC, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129:1201–1213. doi: 10.1016/j.cell.2007.03.053. [DOI] [PubMed] [Google Scholar]
- Olbrich H, Fliegauf M, Hoefele J, Kispert A, Otto E, Volz A, Wolf MT, Sasmaz G, Trauer U, Reinhardt R, et al. Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapetoretinal degeneration and hepatic fibrosis. Nature genetics. 2003;34:455–459. doi: 10.1038/ng1216. [DOI] [PubMed] [Google Scholar]
- Otto EA, Schermer B, Obara T, O'Toole JF, Hiller KS, Mueller AM, Ruf RG, Hoefele J, Beekmann F, Landau D, et al. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat Genet. 2003;34:413–420. doi: 10.1038/ng1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oteiza P, Koppen M, Concha ML, Heisenberg CP. Origin and shaping of the laterality organ in zebrafish. Development. 2008;135:2807–2813. doi: 10.1242/dev.022228. [DOI] [PubMed] [Google Scholar]
- Parisi MA, Doherty D, Chance PF, Glass IA. Joubert syndrome (and related disorders) (OMIM 213300) Eur J Hum Genet. 2007;15:511–521. doi: 10.1038/sj.ejhg.5201648. [DOI] [PubMed] [Google Scholar]
- Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, Cole DG. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol. 2000;151:709–718. doi: 10.1083/jcb.151.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter JF, Skarnes WC. Tectonic, a novel regulator of the Hedgehog pathway required for both activation and inhibition. Genes Dev. 2006;20:22–27. doi: 10.1101/gad.1363606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum JL, Witman GB. Intraflagellar transport. Nature reviews. 2002;3:813–825. doi: 10.1038/nrm952. [DOI] [PubMed] [Google Scholar]
- Salonen R, Paavola P. Meckel syndrome. J Med Genet. 1998;35:497–501. doi: 10.1136/jmg.35.6.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki H, Hogan BL. HNF-3 beta as a regulator of floor plate development. Cell. 1994;76:103–115. doi: 10.1016/0092-8674(94)90176-7. [DOI] [PubMed] [Google Scholar]
- Schafer T, Putz M, Lienkamp S, Ganner A, Bergbreiter A, Ramachandran H, Gieloff V, Gerner M, Mattonet C, Czarnecki PG, et al. Genetic and physical interaction between the NPHP5 and NPHP6 gene products. Hum Mol Genet. 2008;17:3655–3662. doi: 10.1093/hmg/ddn260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholtens D. apComplex: Estimate protein complex membership using AP-MS protein data. R package version 2.14.0 2004 [Google Scholar]
- Scholtens D, Vidal M, Gentleman R. Local modeling of global interactome networks. Bioinformatics. 2005;21:3548–3557. doi: 10.1093/bioinformatics/bti567. [DOI] [PubMed] [Google Scholar]
- Shiba D, Manning DK, Koga H, Beier DR, Yokoyama T. Inv acts as a molecular anchor for Nphp3 and Nek8 in the proximal segment of primary cilia. Cytoskeleton (Hoboken) 2010;67:112–119. doi: 10.1002/cm.20428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallila J, Jakkula E, Peltonen L, Salonen R, Kestila M. Identification of CC2D2A as a Meckel syndrome gene adds an important piece to the ciliopathy puzzle. Am J Hum Genet. 2008;82:1361–1367. doi: 10.1016/j.ajhg.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres JZ, Miller JJ, Jackson PK. High-throughput generation of tagged stable cell lines for proteomic analysis. Proteomics. 2009;9:2888–2891. doi: 10.1002/pmic.200800873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waragai M, Nagamitsu S, Xu W, Li YJ, Lin X, Ashizawa T. Ataxin 10 induces neuritogenesis via interaction with G-protein beta2 subunit. J Neurosci Res. 2006;83:1170–1178. doi: 10.1002/jnr.20807. [DOI] [PubMed] [Google Scholar]
- Weatherbee SD, Niswander LA, Anderson KV. A mouse model for Meckel syndrome reveals Mks1 is required for ciliogenesis and Hedgehog signaling. Hum Mol Genet. 2009;18:4565–4575. doi: 10.1093/hmg/ddp422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CL, Winkelbauer ME, Schafer JC, Michaud EJ, Yoder BK. Functional redundancy of the B9 proteins and nephrocystins in Caenorhabditis elegans ciliogenesis. Mol Biol Cell. 2008;19:2154–2168. doi: 10.1091/mbc.E07-10-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkelbauer ME, Schafer JC, Haycraft CJ, Swoboda P, Yoder BK. The C. elegans homologs of nephrocystin-1 and nephrocystin-4 are cilia transition zone proteins involved in chemosensory perception. J Cell Sci. 2005;118:5575–5587. doi: 10.1242/jcs.02665. [DOI] [PubMed] [Google Scholar]
- Zaghloul NA, Katsanis N. Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J Clin Invest. 2009;119:428–437. doi: 10.1172/JCI37041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo X, Guo W, Lipschutz JH. The exocyst protein Sec10 is necessary for primary ciliogenesis and cystogenesis in vitro. Mol Biol Cell. 2009;20:2522–2529. doi: 10.1091/mbc.E08-07-0772. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.