

Abstract
N-Terminal acetylation of α-synuclein (aS), a protein implicated in the etiology of Parkinson’s disease, is common in mammals. The impact of this modification on the protein’s structure and dynamics in free solution and on its membrane binding properties has been evaluated by high-resolution nuclear magnetic resonance and circular dichroism (CD) spectroscopy. While no tetrameric form of acetylated aS could be isolated, N-terminal acetylation resulted in chemical shift perturbations of the first 12 residues of the protein that progressively decreased with the distance from the N-terminus. The directions of the chemical shift changes and small changes in backbone 3JHH couplings are consistent with an increase in the α-helicity of the first six residues of aS, although a high degree of dynamic conformational disorder remains and the helical structure is sampled <20% of the time. Chemical shift and 3JHH data for the intact protein are virtually indistinguishable from those recorded for the corresponding N-terminally acetylated and nonacetylated 15-residue synthetic peptides. An increase in α-helicity at the N-terminus of aS is supported by CD data on the acetylated peptide and by weak medium-range nuclear Overhauser effect contacts indicative of α-helical character. The remainder of the protein has chemical shift values that are very close to random coil values and indistinguishable between the two forms of the protein. No significant differences in the fibrillation kinetics were observed between acetylated and nonacetylated aS. However, the lipid binding properties of aS are strongly impacted by acetylation and exhibit distinct behavior for the first 12 residues, indicative of an initiation role for the N-terminal residues in an “initiation–elongation” process of binding to the membrane.
The link between mutations in the gene encoding α-synuclein (aS) and familial early onset Parkinson’s disease1,2 has stimulated a very wide array of biophysical studies of this protein.3,4 In aqueous solution, this 140-residue protein adopts a dynamically disordered backbone conformation, but its 13Cα chemical shifts also have been interpreted as being indicative of a slight propensity to adopt α-helical torsion angles for several sections of its backbone.5,6 Electrostatic interaction between the, on average, 100 positively charged N-terminal residues and the negatively charged C-terminal tail results in a hydrodynamic radius (Rh = 28 Å)7 that is smaller than expected for a true random coil (∼37 Å) but larger than the value of ≈15 Å for a globular protein with the mass of aS.
A recent study suggests that when expressed in mammalian cells, aS can also form a stable helical tetramer,8 and the same conclusion was reached for a form of aS that included an N-terminal decapeptide extension, remaining after cleavage of its GST tag.9 In both cases, aS was purified without the heating step, usually employed in aS purification as an effective way to precipitate the vast majority of other, folded proteins. However, the conclusions of both studies remain the subject of much debate.10
The N-terminal 100-residue segment of aS has a high affinity for negatively charged lipids11,12 and adopts an α-helical conformation when bound to the surface of small unilamellar vesicles (SUVs) or detergent micelles.13−17 However, virtually all biophysical studies of aS conducted to date have focused on bacterially overexpressed protein, which lacks post-translational acetylation of the N-terminal residue, a modification commonly found in mammalian proteins.18 Indeed, extensive N-terminal acetylation of aS was detected when it was isolated from erythrocytes.8 Considering the critical role of the N-terminal aS residues in its interaction with phospholipids,17,19,20 it therefore is of interest to evaluate the impact of N-terminal acetylation. A very recent study reports that N-terminal acetylation is responsible for the formation of folded, α-helical aS tetramers.21
Selective chemical acetylation of bacterially expressed aS is challenging because of the many Lys amino groups in the protein. However, a recently developed recombinant expression system22 includes a plasmid for overexpression of the requisite acetylation enzyme NatB and permits bacterial expression of N-terminally acetylated and isotopically enriched aS, needed for NMR studies. Using the recently described “soft” purification protocol,21 we were unable to generate aS samples of sufficient purity for detailed biophysical studies. In an unsuccessful attempt to detect the putatitive tetramer, we also employed an aS construct extended with a C-terminal His tag, allowing for effective protein purification by using a nickel affinity column, thereby eliminating the heat denaturation step.
Here, we report primarily on the impact of N-terminal acetylation on the structural and lipid binding properties of wild-type aS (lacking the His tag), as viewed by NMR and CD spectroscopy. We evaluate the effect of the acetylation both on chemical shifts, which are extremely sensitive to even minute structural differences, and on 3JHN–Hα couplings that are accurate residue-specific reporters of the distribution of the backbone torsion angle ϕ. Our results show that in the absence of lipids the effect of acetylation is completely restricted to the 12 N-terminal residues. We therefore also evaluate the impact of acetylation on a 15-residue N-terminal peptide fragment of aS by both CD and NMR spectroscopy. Although the structural impact of acetylation remains strictly limited to transient formation of a very short α-helical segment at the very N-terminus of the protein, we find a strong increase in lipid binding affinity for acetylated aS.
Materials and Methods
Protein Expression and Purification
Expression and purification of nonacetylated wild-type (WT) aS were conducted largely as described previously.17 Briefly, bacteria were grown in M9 medium at 37 °C to an OD600 of 0.6, and the protein production was induced by addition of 1 mM IPTG. Cells were harvested by centrifugation 3 h later and immediately frozen at −80 °C. Lysis of cells and initial protein purification were achieved by several freeze–thaw cycles followed by heat precipitation (15 min at 85 °C) in 50 mM Tris (pH 7.4) and 500 mM NaCl. Streptomycin was added at a concentration of 10 mg/mL to partially precipitate DNA. The solution was then centrifuged at 50000g for 30 min, and the supernatant was diluted 10-fold with 50 mM Tris buffer (pH 7.4). Anion-exchange chromatography on a Q-Sepharose column was performed, and fractions containing aS were pooled together. As the final purification step, which principally served to remove residual DNA, size-exclusion chromatography was performed on a Superdex 75 HiLoad 16/60 prep grade column in 20 mM Tris (pH 7.4) and 50 mM NaCl. The protein was then dialyzed into water and lyophilized for storage.
Expression of Acetylated Protein
N-Terminal acetylation of WT aS was achieved by coexpression of a plasmid carrying the components of the NatB complex with a plasmid containing the wild-type aS gene, following the protocol described by Johnson et al.22 We observed that the restrictive conditions of M9 media have a strong effect on the acetylation reaction. Essentially complete acetylation (≥98%) was observed only when using protonated M9 medium, supplemented with 1 g/L protonated IsoGro (Sigma, St. Louis, MO). IsoGro is a protein hydrolysate and to some extent mimics LB medium. In protonated M9 supplemented with MEM vitamins, but lacking IsoGro, the extent of acetylation was only ∼85%. In fully deuterated M9, including 99% D2O solvent, supplemented with 1 g/L [2H,15N]IsoGro, the extent of acetylation was only ∼40%. Notably, upon expression of aS in fully deuterated M9 supplemented with MEM vitamins but in the total absence of IsoGro, no significant acetylation could be detected. Unless noted otherwise, all NMR experiments with acetylated aS were performed on 15N- and 13C-labeled aS samples, obtained using [15N,13C]IsoGro.
Expression and Purification of Acetylated His-Tagged aS
For the purposes of efficient purification while avoiding a heat denaturation step, we used a C-terminally His-tagged aS construct. The sequence of the protein matched the full sequence of wild-type aS, followed by one Gly and six His residues. The gene was synthesized and placed into the pGS21a plasmid, carrying ampicillin resistance, by GenScript.
Escherichia coli carrying aS-His and NatB plasmids were cultured in 1 L of fully protonated M9 medium supplemented with [15N]IsoGro. Protein production was induced at an OD600 of 0.6 via addition of 1 mM IPTG. Bacteria were collected by centrifugation 3 h after induction. The cell pellet was immediately resuspended in 20 mL of BOG buffer matching that described by Trexler and Rhoades21 [100 mM HEPES, 20 mM NaCl, 10% glycerol, and 0.1% BOG (pH 7.4)] with the addition of one full Complete protease inhibitor tablet (Roche). The cell suspension was lysed using a pressure cell, and the lysate then was cleared by centrifugation at 50000g for 30 min. The supernatant was loaded on a 5 mL Ni column. The column was washed with BOG buffer, including 20 mM imidazole. Target protein was then eluted with BOG buffer and 250 mM imidazole, yielding 10 mL of eluate. This solution was dialyzed overnight at 4 °C into a buffer better suited for NMR measurements [1× PBS, 5% glycerol, and 0.05% BOG (pH 7.4)]. On the basis of the UV absorption measurement, the final protein concentration was 150 μM and the total protein yield was 22 mg. NMR measurements were performed on the dialyzed protein after the addition of 7% D2O and a decrease in the pH to 6 by titration with a 0.1 M HCl stock solution. The time between lowering the pH and the start of the NMR experiments was ∼15 min.
Preparation of SUVs
Phospholipids were purchased from Avanti Polar Lipids (Alabaster, AL) as lyophilized powders of pure DOPC and a DOPE/DOPS/DOPC mixture with a 5:3:2 weight ratio (coagulation reagent I). A lipid mixture that consisted of approximately 15% DOPS, 25% DOPE, and 60% DOPC was also prepared by codissolving equal amounts of coagulation reagent I and DOPC in chloroform followed by solvent evaporation under a stream of N2 gas. SUVs were prepared as described previously17 in 20 mM sodium phosphate buffer [pH 6, 10% (w/v)].
NMR Spectroscopy
Diffusion measurements were performed on a 600 MHz Bruker spectrometer equipped with a triple-gradient room-temperature probe using the water-sLED experiment23 with observation optimized for the methyl region of the spectrum. The samples contained 100 μM protein and 5 mM dioxane (internal reference) in 20 mM sodium phosphate buffer (pH 6). Experiments were performed at 288 K using a diffusion delay of 300 ms and 1 ms x,y-gradient pulses with intensities set to 2, 8, 10, 12, 20, 25, 30, 35, 40, 44, 46, and 48% of the maximal value. An acquisition time of 200 ms was used, and 2048 scans with a 1.6 s interscan delay were collected for each gradient strength. Peak attenuations were measured in the methyl region of the spectrum, by scaling relative to the spectrum collected for the weakest gradient, using the dual-mode feature of Bruker XwinNMR, and hydrodynamic radii were extracted using the dioxane hydrodynamic radius (2.12 Å) as an internal reference.
For the backbone chemical shift assignments, three-dimensional (3D) TROSY-HNCO and TROSY-HNCACB spectra were recorded on 15N- and 13C-labeled WT aS (0.35 mM nonacetylated aS and 0.45 mM N-terminally acetylated aS) at 288 K, using a 600 MHz Bruker Avance II spectrometer running Topspin 2.1 and equipped with a z-axis gradient TCI cryogenic probe. All data were collected with two scans per free induction decay. The HNCO spectra comprised 70* × 200* × 575* complex points, for acquisition times of 70.7, 150, and 76.7 ms in the 13C, 15N, and directly detected 1H dimensions, respectively. An acquisition time in the 15N dimension much longer than the 1JNC′ refocusing INEPT delay, typically around 30 ms, was achieved by using the mixed-time (MT) evolution approach24 without requiring additional pulses. With an interscan delay of 0.8 s, the total measuring time for each HNCO spectrum was approximately 1.5 days.
The TROSY-HNCACB spectra were collected using the acquisition parameters listed above in the 15N and 1H dimensions. The 13C chemical shift was recorded using a 28 ms constant-time evolution (212 complex pairs of data points) to remove the 1JCC coupling and achieve an increased resolution. All the TROSY-HNCACB data were collected using 15% sparse sampling, with the sampling schedule in the two indirect dimensions generated randomly. The final spectra were reconstructed on a Linux computer containing dual six-core Xeon processors using 400 iterations of the recently described IST method25 at a threshold value of 0.98. The interscan delay was set to 1.4 s, and the total measuring time for each experiment was approximately 24 h. Standard data processing and most analyses were conducted using NMRPipe.26 Backbone assignments were traced using SPARKY.27
The 3JHN–Hα couplings were measured at 288 K from the cross-peak intensity modulation in a series of constant-time 1H–15N HMQC spectra,28 recorded at 900 MHz. In total, nine constant-time delays with durations of 50, 60, 75, 95, 140, 180, 210, 240, and 280 ms were used, which samples a J modulation curve with up to two zero crossing points for the majority of residues. Depending on the length of the JHH modulation time, the data matrix size ranges from 79* × 1024* in the 15N and 1H dimensions for the shortest constant-time duration to 579* × 1024* points for the longest constant-time duration, with a 103.2 ms 1H acquisition time in all the experiments. Cross-peaks were picked from the spectrum with the longest 15N acquisition time and therefore the highest resolution, unless it was near the zero crossing point and the peaks were too weak for a reliable determination of the peak positions, in which case the next best resolved spectrum was used. With the peak positions accurately determined, the autoFit.tcl routine in the NMRPipe software package26 was applied to extract the peak intensity in each spectrum, keeping the position fixed. Nonlinear least-squares fitting of the J-modulated peak intensities to eq 4 of ref (28) was performed to obtain the 3JHH couplings.
Two-dimensional (2D) TOCSY and NOESY spectra were recorded at 278 K on peptide samples containing 1.6 mM nonacetylated and 1.0 mM acetylated N-terminal 15-residue peptides, both amidated at V15, using a 900 MHz Bruker Avance III spectrometer with a cryogenic probe. For the NOESY spectra, a mixing time of 300 ms was used. In all the spectra, 512* × 2048* complex points in the indirect and direct dimensions were collected for acquisition times of 69.6 and 227 ms. The interscan delay was set to 1.5 s, with four and eight scans per free induction decay, yielding total measuring times of 2 and 4.5 h for the 2D TOCSY and NOESY spectra, respectively.
TROSY-HSQC spectra were recorded at 1H frequencies of 500 and 800 MHz on Bruker spectrometers equipped with cryogenic probes. At 500 MHz, 200* × 560* complex points were collected for acquisition times of 165 (t1) and 80 ms (t2). At 800 MHz, 300* × 900* complex points were collected for acquisition times of 154 (t1) and 80 ms (t2).
CD Measurements
All CD measurements were taken at 20 °C, using a 1 mm path-length cuvette. Samples contained 20 mM sodium phosphate (pH 6), 150 mM NaCl buffer, and either 100 μM 15-residue peptides or 10 μM full-length protein.
Fibrillation Experiments
Experiments were performed using a Tecan Infinite 200 Pro multimode microplate reader. The sample volume was 200 μL per well. Sample conditions were as follows: 200 μM WT aS, either acetylated or nonacetylated, PBS buffer, pH 7.4. The plate was shaken at 88 rpm and 37 °C. A 1 mm diameter glass bead was placed into each sample well to accelerate fibrillation. Fibril formation was followed using the fluorescence of thioflavin T (ThT), which was added at a concentration of 10 μM to all samples. The instrument took fluorescence readings of all samples at regular intervals of 50 min for a total duration of 92 h. The excitation wavelength was set to 415 nm, and the ThT emission was measured at 480 nm. Four samples for acetylated and three for nonacetylated aS were measured.
Results and Discussion
The recently introduced bacterial expression system coexpresses the fission yeast NatB enzyme complex together with the protein of interest. It provides an effective method for obtaining N-terminal acetylation of proteins such as aS, which starts with a Met-Asp pair at its N-terminus.22 Using the regular bacterial strain BL21(DE3), we obtained yields of acetylated WT aS that were comparable to amounts normally harvested for nonacetylated protein (∼20 mg/L) when expressed in minimal media, required for incorporation of 13C and 15N stable isotopes. With no resonances visible in the 15N–1H HSQC NMR spectrum at the positions of the nonacetylated protein, levels of acetylation were established to be >98%. However, when an attempt was made to express perdeuterated aS, which can generate extremely well resolved 15N–1H correlation spectra for aS,29 the level of acetylation was at most 40% (for more details, see Materials and Methods). All results presented in this study therefore are for fully protonated material.
Effect of Acetylation on NMR Characteristics of aS
A comparison of the 15N–1H HSQC NMR spectra of N-terminally acetylated and nonacetylated WT aS reveals the impact of acetylation to be strictly limited to the first 12 residues of the protein, and both line widths and resonance positions are essentially indistinguishable for the remainder of the protein, despite the very high precision at which these values were recorded (Figure 1). Backbone assignments (Tables S1 and S2 of the Supporting Information) were obtained from nonuniformly sampled HNCACB 3D spectra. Evaluation of the actual chemical shift values in terms of secondary structure propensity, using the latest sets of sequence, pH, and temperature correction factors,30 shows root-mean-square differences (rmsds) from random coil values that are even considerably smaller (Figure S2 of the Supporting Information) than previously reported for both aS and other intrinsically unstructured proteins.31 The newer chemical shift evaluation method of Camilloni et al.31 identifies secondary structure by using all available backbone chemical shifts as input parameters. With this program, δ2D, the population of α-helix for aS in free aqueous solution is estimated to be even lower than initially deduced5 from older, less advanced approaches (Figure 2). This result is consistent with the random coil characteristics of its CD spectrum recorded in the absence of lipids (Figure 3A). However, δ2D indicates a small but distinct increase in the population of α-helix near the N-terminus for the acetylated form of the protein (Figure 2). Although the effect of N-terminal acetylation on the chemical shifts of residues 6–12 rapidly decreases with the distance from the N-terminus (Figure 1), it is rather remarkable that the effect of this small covalent modification in a disordered protein propagates as far as residue 12, an effect explained by the cooperative formation of a transient short α-helix of the N-terminal residues.
Figure 1.
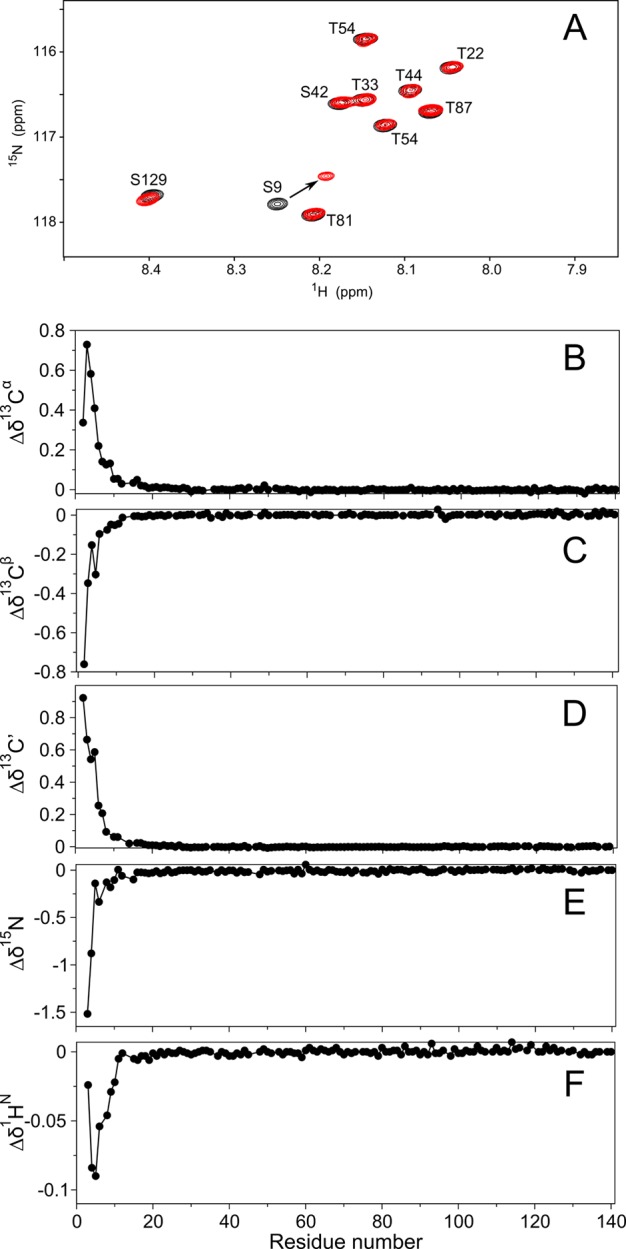
Impact of N-terminal acetylation on the NMR spectrum of aS. (A) Overlay of a small expanded region of the 800 MHz HSQC spectra of nonacetylated (black) and acetylated (red) aS. The pronounced change in the peak position for Ser9 is marked with an arrow. For the overlay of full spectra, see Figure S1 of the Supporting Information. Differences in chemical shifts in parts per million between acetylated and nonacetylated aS for different backbone atoms as a function of residue number: (B) 13Cα, (C) 13Cβ, (D) 13C′, (E) 15N, and (F) 1HN. Pairwise rmsds calculated over residues 13–140 are 0.008 ppm (13Cα), 0.007 ppm (13Cβ), 0.005 ppm (13C′), 0.019 ppm (15N), and 0.002 ppm (1HN).
Figure 2.
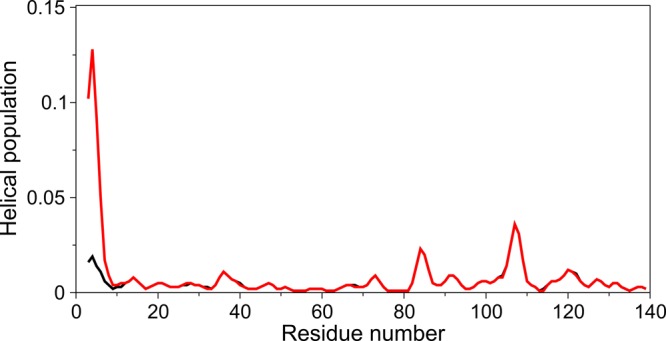
Population of α-helix for nonacetylated (black) and acetylated (red) WT aS as derived from the measured 13Cα, 13Cβ, 13C′, 15N, and 1HN chemical shifts using δ2D.31
Figure 3.
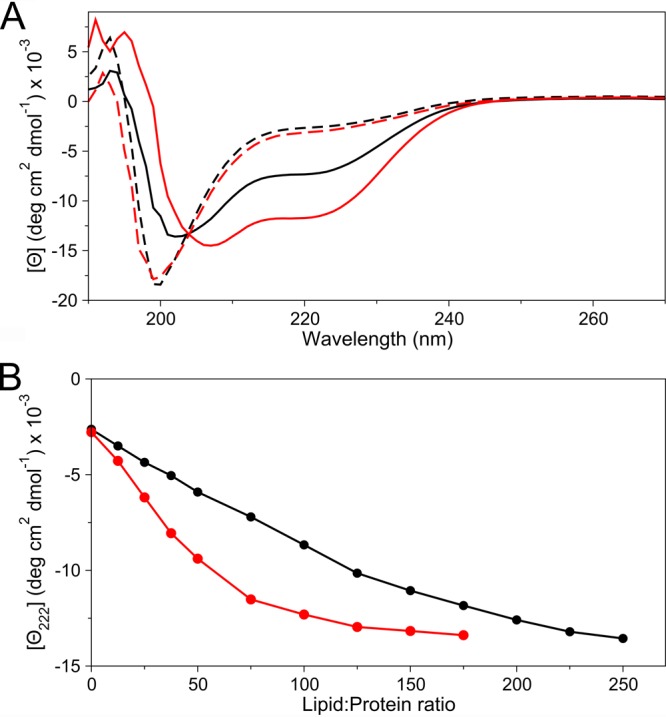
CD data for the interaction of nonacetylated (black) and acetylated (red) WT aS with lipid vesicles. (A) Solid lines represent CD spectra obtained at a 75:1 lipid:protein molar ratio. Dashed lines show CD spectra in the absence of lipids. (B) Graph showing the change in CD signature at 222 nm, reflecting the amount of α-helical structure, as a function of lipid:protein molar ratio. Buffer conditions were 20 mM phosphate (pH 6) and 150 mM NaCl. Measurements were performed on samples containing 10 μM protein. The lipid consisted of 30% DOPS, 50% DOPE, and 20% DOPC.
Remarkably, there is no discernible impact of the acetylation on the chemical shifts of the acidic C-terminal tail of the protein, despite the decrease in positive charge and increase in helical population at the N-terminus. An interaction between the net positively charged N-terminus and the acidic C-terminal region of the protein was previously established on the basis of paramagnetic relaxation experiments and was postulated to be primarily electrostatic in nature. The presence of such a long-range interaction is also reflected in a hydrodynamic volume that is ∼2.3 times smaller than expected for a true random coil of 140 residues.7,32 The absence of any detectable chemical shift perturbation for residues 13–140 upon losing the positive N-terminal charge indicates that the acetylation has no discernible effect on the time-averaged backbone angles of these residues, highlighting the absence of specific interactions between the C-terminal region and the N-terminus. However, very weak, nonspecific interactions between the N-terminal and C-terminal regions are reflected in a slight decrease in the global electrostatic compaction of the protein, as indicated by a small ∼1.5% decrease in the rate of translational diffusion upon acetylation of WT aS (Figure 4). The 1.5% decrease in translational diffusion corresponds to a 1.5% (0.5 Å) increase in the hydrodynamic radius and requires only extremely small (≪1°) changes in the time-averaged backbone torsion angle distributions. Therefore, the absence of any significant difference in backbone torsion angles between the acetylated and nonacetylated forms of the protein, deduced from the indistinguishable chemical shifts, is not inconsistent with the slight difference in the hydrodynamic radii of the two forms of the protein.
Figure 4.
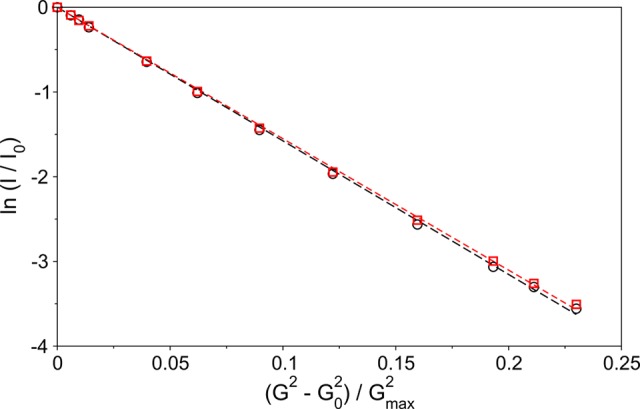
Linearized (logarithmic) PFG diffusion plots for nonacetylated (black) and acetylated (red) WT aS. Proton peak intensities were observed in the methyl region. Dashed lines represent linear fits to the symbols of the matching color.
Next to chemical shifts, 3JHN–Hα couplings are particularly reliable quantitative reporters of the time-averaged distribution of the backbone torsion angles, ϕ.33 For folded proteins with known structures, the quantitative relation between these torsion angles and 3JHN–Hα couplings is defined by the empirical Karplus equation. Recent work has demonstrated that this equation is remarkably robust and typically is limited by the accuracy of the atomic coordinates that define the ϕ angles and by small out-of-peptide-plane deviations of the amide protons.34 When atomic coordinates were used that had been refined by the use of residual dipolar couplings, the level of agreement between 3JHN–Hα couplings and backbone torsion angles decreased to within the 0.35 Hz uncertainty of the measurement, indicating that no correction for amino acid type (excluding Gly) or H-bonding is needed.34 We therefore measured a nearly complete set of 3JHN–Hα couplings in both acetylated and nonacetylated aS. With the exception of small decreases in 3JHN–Hα for the first few residues in the protein upon acetylation (Table S4 of the Supporting Information), the values measured for the two forms of the protein are essentially indistinguishable (rmsd of 0.05 Hz).
It is interesting to note, however, that the different residue types show distinct clustering of their 3JHN–Hα values (Table S4 of the Supporting Information), with the smallest values found for Ala residues (⟨3JHN–Hα⟩ = 5.64 ± 0.26 Hz, excluding C-terminal A140) and the largest values found for β-branched residues such as Val (⟨3JHN–Hα⟩ = 7.60 ± 0.29 Hz), reflecting the natural tendencies of these amino acids to populate more helical or extended regions of Ramachandran space in intrinsically unstructured peptides and proteins. The first six residues of acetylated aS show decreases relative to these random coil values that fall outside the standard deviation, again indicative of α-helix. Residues following G7 do not show such an effect.
Effect of Acetylation on an N-Terminal 15-Residue Peptide Fragment
As the impact of acetylation appears to be restricted to the first few residues of aS, we also studied the synthetic 15-residue N-terminal fragment of the protein in the absence and presence of N-terminal acetylation. Because of their much smaller number of residues, these peptides exhibit far less 1H resonance overlap than the full-length protein and make it possible to study their structure by the conventional 2D NOESY method. With the exception of residues 13–15, proximate to the C-terminus of the peptide, the chemical shifts of the two peptides match those seen in the corresponding full-length proteins very closely, lending validity to the study of these peptides. The nonacetylated peptide shows essentially random coil characteristics. Its 2D NOESY spectrum lacks medium-range (i, i + n; 2 ≤ n ≤ 5) NOEs at the lowest contour level. By contrast, weak dαβ(i,i+3), dαN(i,i+3), and dαN(i,i+4) NOEs point to α-helical character for the first six residues of the acetylated peptide (Figure S3 of the Supporting Information). However, upon quantitative comparison of the intraresidue dαN(i,i) and sequential dαN(i–1,i) NOE intensities, these ratios remain considerably below unity. As pointed out previously,35,36 the dαN(i,i)/dαN(i–1,i) NOE intensity ratio is very sensitive to the value of the ψ angle of residue i – 1 and ranges from ∼6 for α-helix to ∼0.25 for β-sheet. Ratios observed in the absence of acetylation are very close to values of ∼0.35, typical of fully disordered proteins. A decrease of ∼50% in the intensity of the sequential dαN(i–1,i) NOEs for residues V3 and F4 increases the NOE intensity ratio to >0.5 (Figure 5), consistent with a shift to an increased population of helical backbone angles compared to random coil. However, the fact that dαN(i,i)/dαN(i–1,i) ratios remain much below unity clearly indicates that the population of α-helical conformations remains low, fully consistent with the analysis of chemical shifts, described above.
Figure 5.
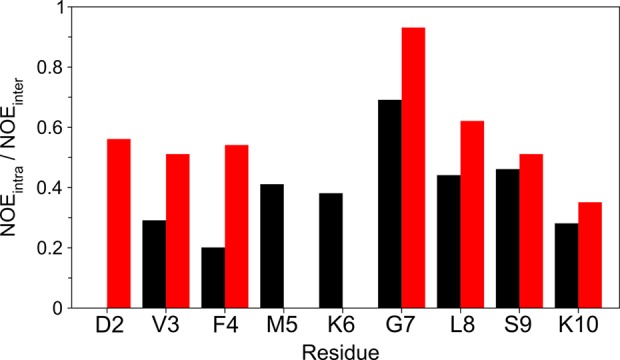
Ratios of intraresidue to sequential Hα–HN NOE intensities, dαN(i,i)/dαN(i–1,i), for the first 10 residues of the nonacetylated (black) and acetylated (red) 15-residue N-terminal peptides. The ratio for residue G7 was divided by 2, and the ratio for L8 was multiplied by 2 to roughly account for the presence of two Hα protons on G7. Ratios for the full-length, nonacetylated protein can be found in ref (36).
Only a very slight increase in α-helicity is observed in the CD spectrum of the full aS protein upon acetylation (Figure 3A), because the CD signal remains dominated by the random coil character of the ∼95% of the chain that is not impacted by the N-terminal acetylation. However, upon comparison of the CD spectra of the N-terminal 15-residue peptides, an increase in α-helical character is clearly observed upon acetylation (Figure S4 of the Supporting Information). Consistent with the NMR results, the degree of α-helicity remains low, however.
Estimated Increase in the Helicity of the First Five Residues
N-Terminal acetylation is known to stabilize α-helices.37,38 For the first few residues of N-terminally acetylated aS in the absence of lipids and detergents, comparison of 3JHN–Hα couplings as well as 13Cα and 13Cβ chemical shifts with those of the nonacetylated protein shows a small increase in the propensity for α-helical structure. Acetylation results in an ∼0.5 ppm increase in the 13Cα chemical shifts for the first five residues and progressively smaller increases for the following few residues. Assuming an ∼3 ppm secondary chemical shift for an ideal α-helix, this indicates an ∼17% increase in the α-helical population for the first five residues.
Upon acetylation, 3JHN–Hα decreases by ∼0.5 Hz for residues 3–5, whereas the value for K6 (6.1 Hz) falls about two standard deviations or 0.5 Hz below the values observed for Lys residues in the remainder of the chain for both forms of the protein (Table S4 of the Supporting Information). With an average 3JHN–Hα of 4 Hz expected for an ideal α-helix and a 3JHN–Hα of ≈7 Hz for random coil, the approximate increase in the level of α-helical conformation is also estimated to be ∼17%.
CD measurements show a markedly stronger helical signature for the acetylated N-terminal 15-residue peptide than for the nonacetylated peptide (Figure S4 of the Supporting Information). If this increase is attributed solely to a change in helical content, then the average increase in helicity is ∼6% (according to JFit). Considering that on the basis of our NMR chemical shift perturbation this change can be attributed to the first five residues, the 6% increase in average helicity corresponds to an ∼18% increase in helicity for these five residues.
Thus, the three independent indicators of helical propensity all indicate an increase in helicity for the first five residues of ∼17%, a number that is slightly larger than the smoothed 11% increase reported by the δ2D program of Camilloni et al.,31 which takes into account all backbone secondary chemical shifts.
Impact of Acetylation on Lipid Binding
Interactions between aS and a variety of different types of lipid vesicles have previously been studied by optical spectroscopic methods, in particular CD,11,13 as well as by both continuous wave and pulsed EPR14,15,39−42 and solution NMR5,29 spectroscopies. All of these techniques indicate a transition from a disordered to an α-helical state upon lipid binding, with some debate remaining about whether the first ∼100 residues form a single contiguous helix or whether aS adopts a flexibly tethered two-helix structure, as was seen in the presence of small SDS micelles.16,43 The various studies unanimously agreed on the important role of negative membrane charge in the interaction, with the highest affinity for lipids with phosphatidylglycerol and phosphatidylserine headgroups and a very low affinity for neutral, zwitterionic phosphatidylcholine lipids, an observation solidified by fluorescence correlation spectroscopy.12
Even when using very dilute suspensions of negatively charged small unilamellar vesicles (SUVs), corresponding to a lipid:protein molar ratio as low as unity, significant attenuation of NMR resonance intensity of the N-terminal residues of aS was observed.17 Remarkably, the attenuated N-terminal HSQC cross-peaks are not shifted relative to those seen in lipid-free samples and show an only very small degree of line broadening, indicating a slow exchange process between the free, disordered state and the lipid-bound state. The absence of attenuation seen for the C-terminal residues indicates that these residues retain their dynamically disordered, random coil character even when the corresponding N-terminal amides have converted to an NMR-invisible “dark state”, induced by lipid binding.17,29
Here, we evaluated the impact of N-terminal acetylation of aS on lipid binding by both CD and NMR spectroscopy. CD measurements were taken under conditions of 20 mM sodium phosphate buffer (pH 6) and 150 mM NaCl at 20 °C, using SUVs composed of 30% DOPS, 50% DOPE, and 20% DOPC. This commercially available lipid mixture is convenient for generating highly stable and reproducible SUVs and includes the headgroups of some of the most abundant phospholipids found in synaptic vesicles.44 At an intermediate lipid:protein molar ratio of 75, the CD spectra show a substantially higher α-helical content for acetylated aS (Figure 3A). A full titration curve, probing the effect of lipid binding on helicity over a wide range of lipid:protein stoichiometries, indicates that acetylated aS has a roughly 2-fold higher affinity for the negatively charged lipid vesicles than does nonacetylated protein (Figure 3B).
NMR experiments were performed under the same buffer conditions but at 15 °C, with lipid vesicles composed of 15% DOPS, 25% DOPE, and 60% DOPC. Vesicles with lower charges were used because this decreases their affinity for aS and thereby weakens the effect of crowding on their surface when aS binds to them. These lower-affinity vesicles are less suitable for CD measurements because of interference from SUV scattering at high lipid concentrations but are well suited for NMR measurements. As one can see in Figure 6, the general shape of the attenuation profile observed for the nonacetylated protein shows the same stepwise attenuation profile as previously reported by Bodner et al. for the vesicles carrying a higher charge density.17 Remarkably, however, the acetylated aS shows a distinct additional attenuation for the first 12 residues, followed by the transition to the characteristic flat profile for residues 25–90 (Figure 6). The same average attenuation of residues 25–90 is achieved for acetylated aS at 2-fold lower lipid concentration, compared to that of the nonacetylated protein. This observation is consistent with the ∼2-fold higher lipid affinity of acetylated aS, indicated by the CD measurements.
Figure 6.
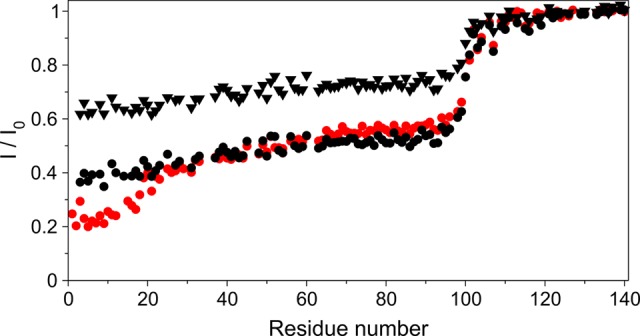
Ratios of WT aS TROSY-HSQC peak heights in the presence (I) and absence (Io) of lipids. Data from the following three samples are presented: acetylated aS at a lipid:protein (L:P) ratio of 22 (red circles), nonacetylated aS at an L:P ratio of 22 (black triangles), and nonacetylated aS at an L:P ratio of 44 (black circles). Experiments were performed on 200 μM protein samples in 20 mM phosphate (pH 6) and 150 mM NaCl buffer at 15 °C on an 800 MHz spectrometer. The lipid consisted of 15% DOPS, 25% DOPE, and 60% DOPC.
Effect of Acetylation on aS Fibrillation
The impact of N-terminal acetylation on the fibril forming propensity of aS, in the absence of lipids, was investigated by measurement of the fibrillation kinetics of the two forms of the protein, using a Tecan Infinite 200 Pro multimode microplate reader to monitor ThT fluorescence. Readings were taken automatically every 50 min for 92 h under conditions of continuous agitation at 37 °C, using four and three separate samples for the acetylated and nonacetylated aS, respectively. The amount of data available so far remained limited by the amounts of available protein and showed some spread in both the lag time and growth rate of the fibrils (Figure S5 of the Supporting Information). However, these data point to the absence of any significant impact of N-terminal acetylation on either the lag phase or the fibril elongation rate. Indeed, under our conditions, both acetylated and nonacetylated aS showed an ∼20 h lag phase and an ∼22 h fibril elongation phase. The absence of any effect on fibrillation could not be anticipated considering that mutations quite close to the N-terminus of the nonacetylated protein, such as V16P, have been reported to inhibit aS fibrillation substantially.45 On the other hand, this absence of any effect appears to be consistent with the notion that the structural changes induced by acetylation are very remote from the hydrophobic NAC region of aS (residues 61–95), the main region implicated in aS fibrillation.46
Concluding Remarks
The vast majority of the very wide array of biophysical measurements reported to date on the structural characterization of aS as well as its interaction with lipids have been taken on bacterially expressed protein that lacks the N-terminal acetylation, a post-translational modification that is present to a very high degree in mammalian cells. An initial report on mammalian-expressed aS suggested the protein exists as an α-helical tetramer,8 a finding subsequently attributed to its N-terminal acetylation21 but disputed by a number of other research groups.10
All of the work described above involving full-length protein was performed on aS that underwent a heating step during purification. We tried to replicate the procedure followed by Trexler and Rhoades21 but failed to isolate the putative tetrameric form of the protein, possibly because of difficulties in separating it from a host of other proteins. Although the isolated material showed considerable α-helical character as judged by CD, it was heterogeneous in mass and showed precipitation after the sample had been heated to 90 °C, characteristic of irreversible denaturation. To work around these difficulties with aS purification under nondenaturing conditions, we resorted to adding a C-terminal His tag to aS. Using this new construct, we were able to perform gentle purification and obtain highly pure N-terminally acetylated protein (see Materials and Methods for details). The resulting protein showed the same dynamic disorder that was seen for nonacetylated or acetylated monomeric protein, despite the absence of a heat denaturation step and very mild conditions used during purification. When using carefully matched buffer conditions, the only differences between the NMR spectra of the gently purified acetylated His-tagged aS and acetylated WT aS, lacking the His tag but purified using heat denaturation, are localized to the C-terminal residues, presumably reflecting the effect of a weak electrostatic interaction between the mostly positively charged His tag (at pH 6.0) and the mostly negatively charged C-terminal residues (Figure 7). It is conceivable that the C-terminal His tag prevents the formation of the putative tetramer. However, this scenario seems unlikely because in the proposed structure of the tetramer the C-terminal region of aS remains disordered.9
Figure 7.
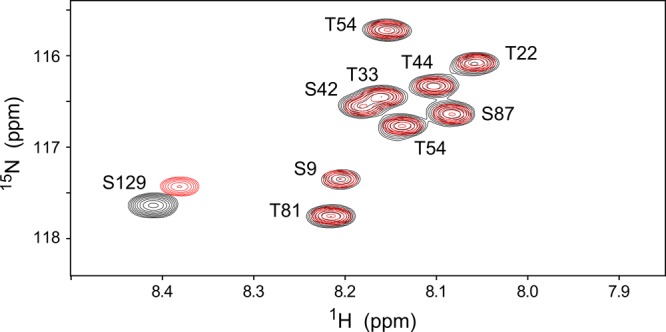
Overlay of small expanded regions of the TROSY-HSQC spectra of N-terminally acetylated, C-terminally His-tagged aS after soft purification (red) and of acetylated WT aS that had undergone heating during purification (black), recorded under matching buffer conditions [1× PBS, 5% glycerol, 0.05% BOG, and 7% D2O (pH 6)]. Spectra were acquired on a 500 MHz spectrometer equipped with a cryoprobe. The signature position of the S9 peak indicates that both proteins are indeed N-terminally acetylated. The only peaks that show small chemical shift differences between the two samples are in the acidic C-terminal region (e.g., S129) and result from the direct effect of the His tag. The full spectrum is shown in Figure S6 of the Supporting Information.
In the absence of lipids, the N-terminal acetylation of aS induces a modest ∼17% population of α-helical conformation for the first six residues. However, the exchange between α-helix and random coil takes place on a time scale faster than ∼1 μs, as judged by the absence of significant line broadening for these N-terminal residues. In the absence of lipids, the chemical shifts of only the very N-terminal residues of aS are visibly impacted by the acetylation and no significant perturbations can be seen in the NMR spectrum beyond residue 12. Considering that the NMR resonances are exquisitely sensitive to the distribution of backbone torsion angles sampled by the molecule, this strongly suggests the absence of any specific interactions of other aS residues with this transient small N-terminal helical segment. At first sight, the very small, ∼1.5% increase in the hydrodynamic radius of aS upon acetylation might appear to contradict the absence of long-range interactions. However, it appears that the 20% lower hydrodynamic radius of aS compared to a total random chain of the same length simply reflects a small electrostatic compaction caused by the opposite net charge between the 40 C-terminal residues and the 60 N-terminal residues. This charge difference, and thereby the compaction, is reduced by the removal of one elementary positive charge upon acetylation of the N-terminal amino group.
The distinct increase in lipid affinity seen for N-terminally acetylated aS, which extends well beyond the 20 N-terminal residues as judged by both CD and NMR (Figures 3 and 6), is perhaps surprising. In fact, one might have expected that the removal of the N-terminal positive charge associated with acetylation of the amino group would slightly decrease the affinity of aS for negatively charged lipid surfaces. Instead, we observed that the minor change in the protein’s chemical structure upon N-terminal acetylation, and its associated transient ∼17% α-helical character of the first few residues in free solution, strongly impacts the lipid binding properties of the entire protein. This observation points to a special and essential role for the N-terminus of the protein in initiating membrane binding, prior to elongation of the bound section of the protein beyond its dozen immediate N-terminal residues. This “initiation–elongation” model of binding is consistent with the stepwise decrease in NMR resonance attenuation seen in the presence of small quantities of SUVs when moving farther from the N-terminus17 and explains the strong increase in the level of lipid binding upon acetylation. Indeed, with the helical structure of aS in the lipid-bound state being well established, preformation of helical structure at the N-terminus in aqueous solution will significantly increase the on rate for binding of aS to lipid, while not significantly affecting the off rate. This kinetic change therefore results in an increase in lipid binding affinity. The distinct steps in the attenuation profile as a function of distance from the N-terminus (Figure 6) point to significant kinetic barriers between aS free in solution and the cases where either just the first dozen residues are lipid-bound or the entire N-terminal 100-residue region of the protein interacts with the membrane. A special role in initiating lipid binding has already been ascribed to the N-terminal residues.20,47 Our current data for acetylated aS amplify this role considerably and suggest that N-terminal acetylation of the protein is critical for the study of its interaction with lipid vesicles.
Acknowledgments
We thank Dr. Daniel Mulvihill (University of Kent, Kent, U.K.) for the N-acetylation B complex construct, Dr. Sven Hyberts for software modules used to allow processing of NUS 3D NMR spectra, Dr. Jennifer Lee [National Heart, Lung and Blood Institute (NHLBI), National Institutes of Health] for use of her multimode microplate reader, Dr. Zhiping Jiang for help with setting up fibrillation experiments, and Drs. Rodney L. Levine and Jue Chen (NHLBI) and Jennifer Lee for helpful discussions and suggestions.
Glossary
Abbreviations
- aS
α-synuclein
- BOG
octyl β-d-glucopyranoside
- NAC
non-amyloid component
- CD
circular dichroism
- DOPC
1,2-dioleoyl-sn-glycero-3-phosphocholine
- DOPE
dioleoylphosphatidylethanolamine
- DOPS
1,2-dioleoyl-sn-glycero-3-phospho-l-serine
- HSQC
heteronuclear single-quantum correlation
- PFG
pulsed field gradient
- ThT
thioflavin T
- TROSY
transverse relaxation-optimized spectroscopy
- NOE
nuclear Overhauser effect
- rmsd
root-mean-square difference
- WT
wild type, lacking the C-terminal His tag.
Supporting Information Available
Overlay of 800 MHz HSQC spectra of nonacetylated and acetylated aS; plots of the differences between chemical shifts measured for nonacetylated aS and random coil values predicted on the basis of the work of Kjaergaard et al. for five nuclei; comparison of small expanded regions of the 900 MHz NOESY spectra of the nonacetylated and acetylated 15-residue N-terminal fragment of aS; CD spectra of nonacetylated and acetylated 15-residue peptides; changes in thioflavin T fluorescence as a function of time in the course of fibrillation experiments for nonacetylated and acetylated aS; overlay of 500 MHz HSQC spectra of acetylated wild-type aS and acetylated His-tagged aS, purified under soft conditions; tables of 13Cα, 13Cβ, 13C′, 15N, and 1HN chemical shift assignments measured for acetylated and nonacetylated aS at 15 °C and pH 6; a table of chemical shifts predicted on the basis of the work of Kjaergaard et al. for WT aS at 15 °C and pH 6; and a table of 3JHN–Hα couplings for the first 15 residues of acetylated and nonacetylated aS and the average 3JHN–Hα values for the residue types involved. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This work was funded by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (NIH), and the Intramural AIDS-Targeted Antiviral Program of the Office of the Director, NIH.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Cookson M. R. (2005) The biochemistry of Parkinson’s disease. Annu. Rev. Biochem. 74, 29–52. [DOI] [PubMed] [Google Scholar]
- Simon-Sanchez J.; Schulte C.; Bras J. M.; Sharma M.; Gibbs J. R.; Berg D.; Paisan-Ruiz C.; Lichtner P.; Scholz S. W.; Hernandez D. G.; Kruger R.; Federoff M.; Klein C.; Goate A.; Perlmutter J.; Bonin M.; Nalls M. A.; Illig T.; Gieger C.; Houlden H.; Steffens M.; Okun M. S.; Racette B. A.; Cookson M. R.; Foote K. D.; Fernandez H. H.; Traynor B. J.; Schreiber S.; Arepalli S.; Zonozi R.; Gwinn K.; van der Brug M.; Lopez G.; Chanock S. J.; Schatzkin A.; Park Y.; Hollenbeck A.; Gao J. J.; Huang X. M.; Wood N. W.; Lorenz D.; Deuschl G.; Chen H. L.; Riess O.; Hardy J. A.; Singleton A. B.; Gasser T. (2009) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 41, 1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breydo L.; Wu J. W.; Uversky V. N. (2012) α-Synuclein misfolding and Parkinson’s disease. Biochim. Biophys. Acta 1822, 261–285. [DOI] [PubMed] [Google Scholar]
- Dikiy I.; Eliezer D. (2012) Folding and misfolding of α-synuclein on membranes. Biochim. Biophys. Acta 1818, 1013–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliezer D.; Kutluay E.; Bussell R.; Browne G. (2001) Conformational properties of α-synuclein in its free and lipid-associated states. J. Mol. Biol. 307, 1061–1073. [DOI] [PubMed] [Google Scholar]
- Marsh J. A.; Singh V. K.; Jia Z. C.; Forman-Kay J. D. (2006) Sensitivity of secondary structure propensities to sequence differences between α- and γ-synuclein: Implications for fibrillation. Protein Sci. 15, 2795–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paleologou K. E.; Schmid A. W.; Rospigliosi C. C.; Kim H. Y.; Lamberto G. R.; Fredenburg R. A.; Lansbury P. T.; Fernandez C. O.; Eliezer D.; Zweckstetter M.; Lashuel H. A. (2008) Phosphorylation at Ser-129 but not the phosphomimics S129E/D inhibits the fibrillation of α-synuclein. J. Biol. Chem. 283, 16895–16905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels T.; Choi J. G.; Selkoe D. J. (2011) α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477, 107–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Perovic I.; Chittuluru J.; Kaganovich A.; Nguyen L. T. T.; Liao J.; Auclair J. R.; Johnson D.; Landeru A.; Simorellis A. K.; Ju S.; Cookson M. R.; Asturias F. J.; Agar J. N.; Webb B. N.; Kang C.; Ringe D.; Petsko G. A.; Pochapsky T. C.; Hoang Q. Q. (2011) A soluble α-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. U.S.A. 108, 17797–17802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauvet B.; Kamdem M. M.; Fares M. B.; Desobry C.; Michael S.; Ardah M. T.; Tsika E.; Coune P.; Prudent M.; Lion N.; Eliezer D.; Moore D. J.; Schneider B.; Aebischer P.; El-Agnaf O. M.; Masliah E.; Lashuel H. A. (2012) α-Synuclein in the central nervous system and from erytrhocytes, mammalian cells and E. coli exists predominantly as disordered monomer. J. Biol. Chem. DOI: 10.1074/jbc.M111.318949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo E. J.; McLaurin J.; Yip C. M.; St George-Hyslop P.; Fraser P. E. (2000) α-Synuclein membrane interactions and lipid specificity. J. Biol. Chem. 275, 34328–34334. [DOI] [PubMed] [Google Scholar]
- Rhoades E.; Ramlall T. F.; Webb W. W.; Eliezer D. (2006) Quantification of α-synuclein binding to lipid vesicles using fluorescence correlation spectroscopy. Biophys. J. 90, 4692–4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson W. S.; Jonas A.; Clayton D. F.; George J. M. (1998) Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 273, 9443–9449. [DOI] [PubMed] [Google Scholar]
- Jao C. C.; Hegde B. G.; Chen J.; Haworth I. S.; Langen R. (2008) Structure of membrane-bound α-synuclein from site-directed spin labeling and computational refinement. Proc. Natl. Acad. Sci. U.S.A. 105, 19666–19771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgieva E. R.; Ramlall T. F.; Borbat P. P.; Freed J. H.; Eliezer D. (2008) Membrane-bound α-synuclein forms an extended helix: Long-distance pulsed ESR measurements using vesicles, bicelles, and rodlike micelles. J. Am. Chem. Soc. 130, 12856–12857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmer T. S.; Bax A.; Cole N. B.; Nussbaum R. L. (2005) Structure and dynamics of micelle-bound human α-synuclein. J. Biol. Chem. 280, 9595–9603. [DOI] [PubMed] [Google Scholar]
- Bodner C. R.; Dobson C. M.; Bax A. (2009) Multiple Tight Phospholipid-Binding Modes of α-Synuclein Revealed by Solution NMR Spectroscopy. J. Mol. Biol. 390, 775–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J. P.; Walker D. E.; Goldstein J. M.; de Laat R.; Banducci K.; Caccavello R. J.; Barbour R.; Huang J. P.; Kling K.; Lee M.; Diep L.; Keim P. S.; Shen X. F.; Chataway T.; Schlossmacher M. G.; Seubert P.; Schenk D.; Sinha S.; Gai W. P.; Chilcote T. J. (2006) Phosphorylation of Ser-129 is the dominant pathological modification of α-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 281, 29739–29752. [DOI] [PubMed] [Google Scholar]
- Bisaglia M.; Schievano E.; Caporale A.; Peggion E.; Mammi S. (2006) The 11-mer repeats of human α-synuclein in vesicle interactions and lipid composition discrimination: A cooperative role. Biopolymers 84, 310–316. [DOI] [PubMed] [Google Scholar]
- Bartels T.; Ahlstrom L. S.; Leftin A.; Kamp F.; Haass C.; Brown M. F.; Beyer K. (2010) The N-Terminus of the Intrinsically Disordered Protein α-Synuclein Triggers Membrane Binding and Helix Folding. Biophys. J. 99, 2116–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trexler A. J.; Rhoades E. (2012) N-terminal acetylation is critical for forming α-helical oligomer of α-synuclein. Protein Sci. 21, 601–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M.; Coulton A. T.; Geeves M. A.; Mulvihill D. P. (2010) Targeted Amino-Terminal Acetylation of Recombinant Proteins in E. coli. PLoS One 5, e15801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altieri A. S.; Hinton D. P.; Byrd R. A. (1995) Association of Biomolecular Systems Via Pulsed-Field Gradient NMR Self-Diffusion Measurements. J. Am. Chem. Soc. 117, 7566–7567. [Google Scholar]
- Ying J. F.; Chill J. H.; Louis J. M.; Bax A. (2007) Mixed-time parallel evolution in multiple quantum NMR experiments: Sensitivity and resolution enhancement in heteronuclear NMR. J. Biomol. NMR 37, 195–204. [DOI] [PubMed] [Google Scholar]
- Hyberts S. G.; Milbradt A. G.; Wagner A. B.; Arthanari H.; Wagner G. (2012) Application of iterative soft thresholding for fast reconstruction of NMR data non-uniformly sampled with multidimensional Poisson Gap scheduling. J. Biomol. NMR 52, 315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. (1995) NMRpipe: A multidimensional spectral processing system based on Unix pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Goddard T. D., and Kneller D. G. (2010) Sparky 3, University of California, San Francisco. [Google Scholar]
- Kuboniwa H.; Grzesiek S.; Delaglio F.; Bax A. (1994) Measurement of HN-Ha J couplings in calcium-free calmodulin using new 2D and 3D water-flip-back methods. J. Biomol. NMR 4, 871–878. [DOI] [PubMed] [Google Scholar]
- Bodner C. R.; Maltsev A. S.; Dobson C. M.; Bax A. (2010) Differential Phospholipid Binding of α-Synuclein Variants Implicated in Parkinson’s Disease Revealed by Solution NMR Spectroscopy. Biochemistry 49, 862–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjaergaard M.; Brander S.; Poulsen F. M. (2011) Random coil chemical shift for intrinsically disordered proteins: Effects of temperature and pH. J. Biomol. NMR 49, 139–149. [DOI] [PubMed] [Google Scholar]
- Camilloni C.; De Simone A.; Vranken W. F.; Vendruscolo M. (2012) Determination of Secondary Structure Populations in Disordered States of Proteins Using Nuclear Magnetic Resonance Chemical Shifts. Biochemistry 51, 2224–2231. [DOI] [PubMed] [Google Scholar]
- Bertoncini C. W.; Jung Y. S.; Fernandez C. O.; Hoyer W.; Griesinger C.; Jovin T. M.; Zweckstetter M. (2005) Release of long-range tertiary interactions potentiates aggregation of natively unstructured α-synuclein. Proc. Natl. Acad. Sci. U.S.A. 102, 1430–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruschweiler R.; Case D. A. (1994) Adding Harmonic Motion To The Karplus Relation For Spin-Spin Coupling. J. Am. Chem. Soc. 116, 11199–11200. [Google Scholar]
- Yao L.; Vogeli B.; Torchia D. A.; Bax A. (2008) Simultaneous NMR study of protein structure and dynamics using conservative mutagenesis. J. Phys. Chem. B 112, 6045–6056. [DOI] [PubMed] [Google Scholar]
- Gagne S. M.; Tsuda S.; Li M. X.; Chandra M.; Smillie L. B.; Sykes B. D. (1994) Quantification of the calcium-induced secondary structural changes in the regulatory domain of troponin-C. Protein Sci. 3, 1961–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltsev A. S.; Grishaev A.; Bax A. (2012) Monomeric α-Synuclein Binds Congo Red Micelles in a Disordered Manner. Biochemistry 51, 631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabartty A.; Doig A. J.; Baldwin R. L. (1993) Helix capping propensities in peptides parallel those in proteins. Proc. Natl. Acad. Sci. U.S.A. 90, 11332–11336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis J. A.; Ryan M. T.; Hoogenraad N. J.; Craik D. J.; Hoj P. B. (1995) Solution structure of the acetylated and noncleavable mitochondrial targeting signal of rat chaperonin-10. J. Biol. Chem. 270, 1323–1331. [DOI] [PubMed] [Google Scholar]
- Varkey J.; Isas J. M.; Mizuno N.; Jensen M. B.; Bhatia V. K.; Jao C. C.; Petrlova J.; Voss J. C.; Stamou D. G.; Steven A. C.; Langen R. (2010) Membrane Curvature Induction and Tubulation Are Common Features of Synucleins and Apolipoproteins. J. Biol. Chem. 285, 32486–32493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drescher M.; Veldhuis G.; van Rooijen B. D.; Milikisyants S.; Subramaniam V.; Huber M. (2008) Antiparallel arrangement of the helices of vesicle-bound α-synuclein. J. Am. Chem. Soc. 130, 7796–7797. [DOI] [PubMed] [Google Scholar]
- Robotta M.; Braun P.; van Rooijen B.; Subramaniam V.; Huber M.; Drescher M. (2011) Direct Evidence of Coexisting Horseshoe and Extended Helix Conformations of Membrane-Bound α-Synuclein. ChemPhysChem 12, 267–269. [DOI] [PubMed] [Google Scholar]
- Drescher M.; van Rooijen B. D.; Veldhuis G.; Subramaniam V.; Huber M. (2010) A Stable Lipid-Induced Aggregate of α-Synuclein. J. Am. Chem. Soc. 132, 4080–4082. [DOI] [PubMed] [Google Scholar]
- Lokappa S. B.; Ulmer T. S. (2011) α-Synuclein Populates Both Elongated and Broken Helix States on Small Unilamellar Vesicles. J. Biol. Chem. 286, 21450–21457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S.; Holt M.; Stenius K.; Lemke E. A.; Gronborg M.; Riedel D.; Urlaub H.; Schenck S.; Brugger B.; Ringler P.; Muller S. A.; Rammner B.; Grater F.; Hub J. S.; De Groot B. L.; Mieskes G.; Moriyama Y.; Klingauf J.; Grubmuller H.; Heuser J.; Wieland F.; Jahn R. (2006) Molecular anatomy of a trafficking organelle. Cell 127, 831–846. [DOI] [PubMed] [Google Scholar]
- Koo H. J.; Lee H. J.; Im H. (2008) Sequence determinants regulating fibrillation of human α-synuclein. Biochem. Biophys. Res. Commun. 368, 772–778. [DOI] [PubMed] [Google Scholar]
- Giasson B. I.; Murray I. V. J.; Trojanowski J. Q.; Lee V. M. Y. (2001) A hydrophobic stretch of 12 amino acid residues in the middle of α-synuclein is essential for filament assembly. J. Biol. Chem. 276, 2380–2386. [DOI] [PubMed] [Google Scholar]
- Pfefferkorn C. M.; Heinrich F.; Sodt A. J.; Maltsev A. S.; Pastor R. W.; Lee J. C. (2012) Depth of α-Synuclein in a Bilayer Determined by Fluorescence, Neutron Reflectometry, and Computation. Biophys. J. 102, 613–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.