

Abstract
Although the identification of cancer stem cells as therapeutic targets is now actively being pursued in many human malignancies, the leukemic stem cells in acute myeloid leukemia (AML) are a paradigm of such a strategy. Heterogeneity of these cells was suggested by clonal analyses indicating the existence of both leukemias resulting from transformed multipotent CD33− stem cells as well others arising from, or predominantly involving, committed CD33+ myeloid precursors. The latter leukemias, which may be associated with an intrinsically better prognosis, offer a particularly attractive target for stem cell-directed therapies. Targeting the CD33 differentiation antigen with gemtuzumab ozogamicin was the first attempt of such an approach. Emerging clinical data indicate that gemtuzumab ozogamicin is efficacious not only for acute promyelocytic leukemia but, in combination with conventional chemotherapy, also for other favorable- and intermediate-risk AMLs, providing the first proof-of-principle evidence for the validity of this strategy. Herein, we review studies on the nature of stem cells in AML, discuss clinical data on the effectiveness of CD33-directed therapy, and consider the mechanistic basis for success and failure in various AML subsets.
Introduction
Normal human hematopoiesis is hierarchically organized, with tissue-specific, quiescent stem cells at the apex that have the ability to perpetuate themselves through self-renewal and generate more mature, transiently amplifying progeny through differentiation.1 Similar to normal hematopoiesis, acute myeloid leukemia (AML) encompasses functionally diverse cells, and origination from a leukemic stem cell (LSC) was initially suspected many decades ago.2 Observations in AML were indeed instrumental for the model of cancer stem cells as cells within a tumor that possess the capacity to self-renew and to cause the heterogeneous lineages of cancer cells that compose the tumor.3
This model has important clinical implications as it predicts that the inability to eradicate cancer stem cells represents the cause of relapse and therapeutic failure; in turn, effective tumor therapy will require eradication of these cells.2,3 Interest in AML has thus long focused on the nature of LSCs and their specific qualities that predict therapeutic response. The cellular origin of AMLs, however, remains unclear, with ongoing controversy as to whether they arise from transformed hematopoietic stem cells (HSCs) or emerge as a result of genetic events occurring in more mature progenitor cells.2,4–7 The nature of the cells giving rise to AML may have important biologic, therapeutic, and prognostic implications. Indeed, early recognition that some AMLs may predominantly or entirely involve committed myeloid progenitors led to efforts targeting underlying LSCs with antibodies recognizing the CD33 (SIGLEC-3) differentiation antigen, as exemplified by the development of the immunoconjugate, gemtuzumab ozogamicin (GO; Mylotarg).8 In this review, we summarize studies on stem cells in AML indicating heterogeneous involvement of stem/progenitor populations, discuss emerging data on the effectiveness of CD33-directed therapy, and consider the mechanistic basis for success or failure against individual AML subsets.
Heterogeneity of stem/progenitor cells in human AML
There may be no single, unifying cellular origin across the entire spectrum of human AML. Rather, research conducted over the last several decades indicates that AML may arise in (or predominantly involve) either multipotent HSCs or more mature committed myeloid precursors downstream of HSCs. The first hint to this heterogeneity came from early studies on X chromosome inactivation patterns, which identify the clonal cell population in females based on discrimination of the active from the inactive X chromosome and differentiation of each X chromosome's parental origin.9 In some leukemias, these investigations showed dominance of the clonal process in multiple cell lineages (granulocytes, monocytes, erythrocytes, platelets, and occasionally B lymphocytes), reflecting AML origination and expansion at the level of pluripotent stem/progenitor cells.10,11 In other cases, clonal dominance was limited to granulocytes and monocytes,10,11 suggesting that expansion of the malignant clone could occur at the level of committed myeloid precursors.
In the latter leukemias, we hypothesized that CD33− precursors (Figure 1) would be predominantly or completely normal. To test this assumption, we removed CD33+ cells in vitro via CD33-directed complement-mediated lysis or FACS in a small number of patients with such leukemias and placed the remaining CD33− cells in long-term culture together with irradiated allogeneic stroma cells.12,13 Over time, CD33− precursors from some patients indeed generated colony-forming cells (CFCs) with X chromosome inactivation patterns consistent with predominantly nonclonal hematopoiesis12,13; because of the inherent limitations of X chromosome inactivation studies,9 we could not distinguish complete from predominant nonclonal derivation. These findings were suggestive of transformation of lineage-committed myeloid precursors; however, restriction of the clonal dominance to granulocytes and monocytes could alternatively result from a mutated pluripotent stem cell that either lost the capacity for differentiation along the other cell lineages14 or only displayed dysregulated growth once single lineage commitment had occurred. Indeed, initial xenotransplantation assays, where only rare, immature CD34+/CD38− cells initiated and sustained leukemia growth in all AML subtypes except possibly acute promyelocytic leukemia (APL) suggested that pluripotent HSCs were generally the target of leukemic transformation.15,16 However, immunophenotypic differences in differentiation markers between normal HSCs and LSCs were noted by several investigators,17–19 and more recent xenotransplantation studies indicate that the transformation process may occur in precursor cells beyond the stem cell stage.20–22 In these investigations, AML was reconstituted in severely immunodeficient mice from cells that appeared phenotypically more mature than pluripotent HSCs and resided in the CD34+/CD38+ or CD34− compartment in some cases.20,21 Moreover, in a recent study suggesting that leukemia cells that engraft in immunodeficient mice and recapitulate human AML resemble hematopoietic progenitor cells phenotypically more closely than HSCs, the LSC-containing cell fraction also most closely resembled normal progenitor populations by gene expression profiling.22 It remains to be determined, however, to what degree this profile reflects the gene expression of the subset of LSCs contained in this cell population. Nevertheless, these more recent studies overall imply a greater variation in the cellular origin of transplantable AML cells than initially perceived and support the concept of LSC heterogeneity.
Figure 1.

CD33 and CD34 as differentiation antigens. Simplified hypothetical model of stem and progenitor cells in the human hematopoietic system, showing expression patterns of CD33 and CD34.
Xenotransplantation assays have provided essential insight into AML biology and may currently be considered the gold standard to investigate LSCs on a functional level.2 There are important caveats, however: in particular, many patient specimens fail to engraft and are thus not amenable to study; there is also a bias toward specimens with large numbers of available cells. Thus, it is possible that xenotransplantation models underestimate LSC diversity and provide a skewed, incomplete assessment of these cells. Furthermore, although poorly understood, it is a recurrent observation that the ability to engraft in immunodeficient mice is a nonrandom, inherent property of specimens from patients with worse-prognosis AML (eg, those with unfavorable cytogenetics or aberrations in FLT3).23–29 Finally, some observations have raised the concern that cells without bona fide human-relevant stem cell properties may be transplantable in such model systems. This possibility is suggested by the recent finding that CD33+ cord blood cells can engraft with multilineage hematopoiesis.30 This is unexpected because previous studies on normal bone marrow showed CD33 expression primarily on multipotent myeloid precursors, unipotent CFCs, as well as maturing granulocytes, and monocytes but, unlike CD34, not on pluripotent HSCs (Figure 1).31–34 These in vitro investigations were consistent with clinical studies demonstrating delayed but durable multilineage engraftment after transplantation of CD33-depleted autografts in patients with AML,35,36 similarly indicating that normal HSCs lack CD33. Thus, although xenotransplantation models are widely considered the most stringent approach to study the nature of stem cells, some of the findings may need to be interpreted cautiously.
Congruent with studies of human AMLs, experimental AML models in mice demonstrate that HSCs and committed myeloid precursors can be transformed into LSCs in the appropriate context. This was initially suggested by viral transduction studies with AML-associated oncogenic fusion proteins,37–41 although, extrapolation of these data might be limited because oncogene overexpression could lead to transformation in a nonphysiologic manner; recent findings using Mll-AF9 have indeed highlighted a critical role of oncogene dosage in hematopoietic cell transformation, with thresholds depending on the developmental stage of the progenitor cell.42 Nevertheless, data from knockin and transgenic mice suggest that committed myeloid progenitors can acquire stem cell properties even at physiologic expression levels of a mutated gene.43,44 The leukemias seen in such models are associated with enhanced self-renewal of committed myeloid progenitors and are transplantable via committed myeloid progenitors.43,44
Leukemic and preleukemic stem cells
Relevant to any discussion of LSCs is the hypothesis that AML is generally the consequence of a multistep process rather than a single catastrophic event.45,46 Support for this assumption comes from mouse models in which single AML-associated somatic mutations lead to AML after long latency or exposure to chemical mutagens, whereas malignancies mimicking human AML can develop after short latencies when such mutations are coexpressed.43,45–49 In humans, the requirement for cooperating mutations is most directly suggested by patients with rare germline mutations in myeloid transcription factors who develop AML only after long (up to several decades) latency.50–52 Also informative are cases of core-binding factor (CBF) AMLs, that is, leukemias that are defined by the presence of chromosomal aberrations disrupting one of the CBF transcription factor genes, most commonly t(8;21)(q22;q22) and inv(16)(p13q22), which result in the generation of the RUNX1/RUNX1T1 (also known as AML1-ETO) and CBFB/MYH11 fusion proteins, respectively.53 The need for complementing mutations is indicated by the detection of AML1-ETO transcripts in neonatal Guthrie blood spots in some patients with nonfamilial CBF AML who only much later develop leukemia.54 Finally, epidemiologic data from sporadic AML demonstrate that many patients have more than one recurring nonrandom genetic abnormality, further suggesting that single mutations are insufficient to cause AML.46
To date, the relative significance of causative mutations and target cell(s) of transformation are unclear, except for a few situations. Examples for the latter are patients with familial leukemia syndromes and probably those with nonfamilial CBF AML in whom AML1-ETO is detected many years before development of AML, suggesting its role as a founder mutation. Also consistent with the notion of a founder mutation, or at least a very early event, is the demonstration of AML1-ETO in other patients in long-term remission after therapy for CBF AML.55–58 Furthermore, the observation that aberrant CEBPA, DNMT3A, IDH1/2, and possibly NPM1 alleles are typically present at diagnosis and relapse suggests that these could be very early, possibly preleukemic, mutational events.59–62 In contrast, the mutation status of FLT3, NRAS/KRAS, and WT1 often changes between diagnosis and relapse, and mutations may only be present in a subset of leukemic blasts, indicating a role as secondary events.63–65 This role is supported by our recent studies on specimens from patients with CBF leukemias and concomitant mutations in either FLT3 or KIT, in which we found that many CFCs only harbor the CBF but not tyrosine kinase aberration.66
Patients with familial leukemia syndromes or in utero origin of nonfamilial AMLs exemplify that complementing mutations leading to AML transformation can be acquired over prolonged time periods. Initial genetic event(s) may thus generate one or more long-lived, “preleukemic” stem cell(s) with establishment of preleukemic clones with self-renewal capacity. Such clones will require acquisition of additional mutation(s) for progression to a leukemic clone and the development of overt AML. The “indolent” persistence of preleukemic clones over time suggests origination in relatively quiescent stem cells with long-term repopulating capacity, with the mutational event affecting cellular differentiation rather than proliferation. It is plausible that one or several subsequent mutation(s) are then acquired in more mature progeny of the preleukemic stem cells. The detection of AML1-ETO transcripts in myeloid cells as well as B lymphocytes in some patients, indisputably demonstrating mutation acquisition at the level of pluripotent HSCs, is consistent with this notion.18 Thus, preleukemic stem cells and LSCs may coexist and vary in their differentiative and proliferative properties and perhaps their response to therapy, that is, persistence of preleukemic stem cells but eradication of fully transformed LSCs. This could explain why AML1-ETO transcripts remain detectable in the blood and, at a low frequency, in various clonogenic progenitors in many patients with AML1-ETO AML in long-term remission,55–58 whereas a second mutation (eg, KIT) becomes undetectable despite similar detection sensitivities.67 The persistence of preleukemic stem cells is similarly supported by X chromosome inactivation studies showing persistently clonal hematopoiesis in some patients in morphologic remission after treatment for AML.11,68 Although rare in the absence of constitutional skewing of hematopoietic cells, clonal remissions have commonly been found in association with dysplastic changes, consistent with the notion of persistence of a preleukemic stem cells (clone).69–71
Implications of models of stem cell heterogeneity for stem/progenitor cell-targeted therapy in AML
Despite their limitations, observations to date suggest 3 possibly simplified but testable scenarios of AML development (Figure 2). In the first, both the initial and subsequent mutational events occur at the level of pluripotent CD33− precursors; for reasons of simplicity, we will refer to those as “immature” leukemias. In these, clonal dominance could develop in multiple cell lineages or be confined to granulocytes and monocytes if the capacity for differentiation along the other cell lineages is lost, or if unregulated growth is displayed only once single lineage commitment has occurred because of cell context-specific effects of the mutational event(s).11,14 Many of these leukemias may be identifiable in xenotransplantation assays by the ability of CD34+/CD38− cells to recapitulate the disease. In contrast, in “mature” leukemias, at least one relevant mutational event occurs at the level of committed myeloid cells. Specifically, in the second scenario, the initial mutation is acquired in pluripotent HSCs, whereas the collaborating mutational event(s) leading to full AML transformation and subsequent clonal expansion only occur at a later stage, possibly at the level of committed CD33+ myeloid precursors. As discussed in the previous section, at least some of the CBF leukemias may follow this pattern of transformation. Finally, in the third scenario, all mutational events as well as clonal expansion occur at the level of committed CD33+ myeloid precursors. An example may be APL, as small studies indicate expression of APL mainly in the granulocytic/monocytic lineage and predominant involvement of committed CD33+ myeloid progenitors.72
Figure 2.

Proposed models of AML transformation. Three proposed simplified scenarios of step-wise transformation in human AML, leading from a normal cell (green) to a premalignant cell (light red) and, eventually, to a malignant cell (dark red) with clonal expansion: Scenario 1, both the initial transforming event and subsequent mutations leading to clonal expansion occur at the level of multipotent, CD33− precursors; Scenario 2, the initial transforming event occurs at the level of multipotent, CD33− precursors, whereas the collaborating mutational event leading to clonal expansion occurs at the level of CD33+ committed myeloid progenitors; and Scenario 3, both initial and subsequent mutations occur at the level of CD33+ committed myeloid precursors. Similar models have been proposed by others.129
The nature of LSCs likely has significant prognostic implications. For example, the ability of leukemias to engraft in immunodeficient mice correlates with adverse prognosis in most series.24,26–29 Consistently, an LSC-specific signature derived from engrafting leukemia specimens identifies AML patients with poor long-term survival.73 In turn, a distinct type of LSC may underlie those up to 50% of human AML that fail to engraft in immunodeficient mice and may be more responsive to current therapies, with a paradigm, again, being APL.
Our LSC models emphasize the importance of the differentiative and proliferative potential of the target cells in which the genetic events occur rather than the specific mutation per se; indeed, limited data indicate that similar mutations may lead to distinct outcomes depending on the proliferative potential of the cell in which they arose. For example, Pollard et al demonstrated in a set of FLT3/ITD AMLs that leukemias in which the FLT3 abnormality could be detected in CD34+/CD33− progenitors had a higher relapse risk and worse outcome than leukemias in which FLT3/ITD was only detectable in more mature CD34+/CD33+ cells.74 More recently, Bachas et al demonstrated, among patients initially presenting with various recognized mutations, that the continued presence of these mutations at relapse was associated with a shorter time to relapse, whereas loss of such mutations correlated with a longer time to relapse.65 These observations possibly indicate a worse prognosis if the AML-associated mutations arose in a chemotherapy-resistant, presumably less mature, cellular compartment. The notion that the nature of the stem/progenitor cells acquiring the mutation, rather than the particulars of the mutation itself, determines outcome could explain why prognosis can be similar for many leukemias with dissimilar mutations but different for many leukemias with similar mutations. An alternative explanation, of course, invokes the existence of important yet-to-be identified mutations.
Targeting AML with anti–CD33 antibodies
“Mature” leukemias may be particularly suited for stem cell-directed therapies as underlying LSCs may be distinct from normal HSCs and intrinsically less resistant to therapy. LSC-targeted therapy could then (partially) replace other therapeutics and spare patients from some nonspecific toxicity in such lower-risk diseases. As a first attempt to eliminate LSCs in these leukemias, we focused on the myeloid differentiation antigen, CD33, which is expressed on leukemic blasts from 85% to 90% of AML patients.32,75 The utility of CD33-targeted therapeutics was suggested by the observation that ablation of CD33+ cells could restore normal hematopoiesis in vitro in some leukemias with clonal dominance limited to granulocytes/monocytes.8 However, unconjugated anti–CD33 antibodies were largely ineffective in patients with overt CD33+ non-APL AML.76,77 On the other hand, radiolabeled anti–CD33 antibodies were selectively taken up by AML cells and rapidly saturated leukemic blast cells in peripheral blood and bone marrow at intravenous doses of more than or equal to 5 mg/m2,76,78,79 suggesting that addition of a toxic payload might produce better results. In collaboration with industry, we selected N-acetyl γ-calicheamicin dimethyl hydrazide, a stable derivative of a natural enediyne anti–tumor antibiotic initially identified in a screen for potent DNA-damaging agents,80,81 for this purpose. Subsequently, the antibody was humanized to minimize immune responses and conjugated with the calichemicin derivative via an acid-labile linker that provided good potency and selectivity because of its stability in the circulation but rapid toxin release under acidic conditions, such as those in lysosomes (Figure 3).80,82 Preclinically, GO showed selective cytotoxicity against CD33+ AML cells, effectively inhibited CFCs in human AML specimens, and caused regression of CD33+ AML cell line xenografts in athymic mice.82
Figure 3.
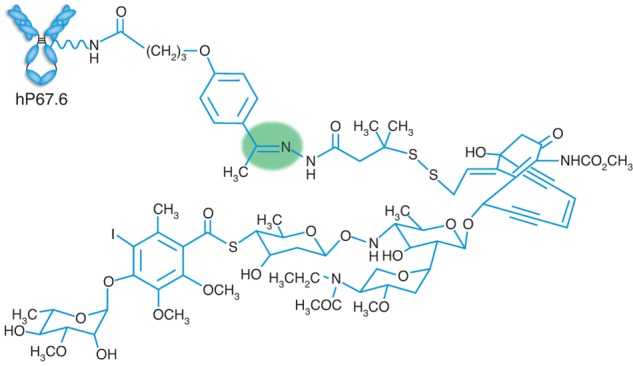
Schematic structure of GO. The humanized IgG4 anti–CD33 antibody (hP67.6) contains amino acid sequences that are approximately 98.3% of human origin. Lysine residues on hP67.6 are linked to N-acetyl γ-calicheamicin dimethyl hydrazide via a hybrid 4-(4′-acetylphenoxy)butanoic acid linker. The labile hydrazone bond leading to drug release under acidic conditions, presumably within lysosomes, is shaded in green. GO has approximately 50% of the antibody loaded with 4 to 6 mol of the toxic moiety per mole of antibody; the remaining 50% of antibody is unbound. Reproduced with permission from Macmillan Publishers Ltd.130
In GO, the unconjugated antibody itself is largely nontoxic83 and primarily facilitates uptake of the calicheamicin derivative into CD33+ cells (Table 1). Once released intracellularly, the enediyne moiety undergoes rearrangement to form a 1,4-benzenoid diradical that initiates single- and double-stranded DNA breaks.81,84 This damage elicits a strong cellular response with cell-cycle arrest and subsequent DNA repair or, if damage is overwhelming, apoptosis and cell death, predominantly via mitochondrial membrane permeabilization and caspase activation.84 The central role of DNA damage in GO-induced cyotoxicity is supported by the observation that cell lines defective in DNA repair are highly sensitive to calicheamicins.85,86
Table 1.
Cellular parameters that affect GO efficacy
| Factor | Comment |
|---|---|
| Uptake of CD33/GO complexes | |
| Receptor-mediated uptake | |
| CD33 expression levels | Good evidence supporting quantitative relationship from experimental and correlative studies83,90,91,125; expression levels associated with cytogenetic risk of AML126 and CD33 SNPs127 |
| CD33 saturation | In vitro evidence linking reduced CD33 saturation to reduced GO cytotoxicity128 |
| CD33 internalization | Relatively slow process, controlled by intracellular tyrosine motifs91 and likely tyrosine phosphorylation status of CD33 |
| Reexpression of CD33 binding sites | Surface CD33 levels return to pretreatment levels within 72 hours after CD33 antibody administration104; could contribute to amount of internalized GO, in particular if GO is administered in fractionated doses. |
| Non–receptor-mediated uptake | Suggested by experimental studies93; clinical role unknown |
| Intracellular trafficking of GO | Hypothetical |
| Activation of GO | Low pH in lysosomes required (R.B.W., unpublished data, December 2004) |
| Extrusion of GO | |
| ABC family of drug transporters | Good evidence from experimental and correlative studies for role of P-glycoprotein and multidrug resistance-associated protein 184,89,90; role of other transporters unknown |
| Induction of cytotoxicity | |
| Generation of SS- and DS-DNA breaks | Hypersensitivity of cell lines with defects in DNA repair to calicheamicins85,86 |
| Mitochondrial pathways of apoptosis | Good experimental evidence for role of pro- and antiapoptotic Bcl-2 protein family members84 |
| Other downstream pro- or antiapoptotic signaling pathways | Not examined in detail |
| Cell-cycle status | Limited in vitro data suggesting that resting cells are relatively less susceptible to GO93 |
SNP indicates single-nucleotide polymorphism; SS, single-stranded; and DS, double-stranded.
This mechanism of action implies a critical role for the intracellular accumulation of the calicheamicin-γ1 derivative as well as the cellular response to the toxin's DNA-damaging effect (Table 1). Conceptually, the amount of intracellular, active calicheamicin is affected by cellular uptake, toxin release and activation, as well as drug inactivation/metabolism or extrusion. On the other hand, the toxicity of the calicheamicin moiety will be modulated by the ability of the cell to repair DNA damage and the activity of downstream pro- and antiapoptotic pathways. Large interindividual differences exist in this respect, as indicated by the observation that the sensitivity to the toxic moiety varies more than 100 000-fold between individual human AML cell specimens.87
Consistent with this mechanism, drug efflux mediated by members of the adenosine triphosphate (ATP) binding cassette (ABC) superfamily of proteins, predominantly P-glycoprotein (ABCB1) and, to a lesser degree, multidrug resistance-associated protein 1 (ABCC1), mediate resistance to GO in vitro and in vivo.84,88–90 Furthermore, experimental studies revealed a striking, quantitative association between CD33 expression/uptake and GO efficacy in engineered human AML cell lines.91,92 In contrast, the clinical impact of DNA repair and downstream pathways has not been examined in detail, but GO efficacy is modulated in vitro by members of the Bcl-2 family of proteins.84 Similarly unknown is optimal timing of the use of GO when combined with other agents. It is conceivable that timing and cell-cycle status matter, however, as in vitro data suggest that resting cells are relatively more resistant to GO.93
Clinical experience with GO
A phase 1 study was conducted in 40 adults with relapsed/refractory CD33+ AML who received up to 3 doses of GO at 2-week intervals.94 Elimination of morphologically detectable leukemia occurred in 8 (20%) of the patients, with 3 (7.5%) achieving a complete remission (CR) as conventionally defined and 2 (5%) achieving a CR with incomplete platelet recovery (CRp). As dose-limiting nonhematologic toxicity was not reached, the highest dose level of 9 mg/m2, which provided almost complete saturation of peripheral blood and marrow CD33 binding sites, was chosen for further study.94 Three open-label, multicenter single-arm phase 2 trials subsequently evaluated GO in a larger cohort of adults with CD33+ de novo AML in first relapse. An interim analysis included 142 patients with a median age of 61 years (range, 22-84 years) who typically received 2 doses of GO 14 days apart; 23 patients (16.2%) achieved a CR, and 19 (13.4%) achieved a CRp, for an overall response rate of 29.6%.95 Based on these results, GO was given accelerated marketing approval in the United States in May 2000 for the treatment of patients older than 60 years with CD33+ AML in first relapse who were not candidates for standard cytotoxic chemotherapy.96 The final report on 277 patients confirmed the early results with demonstration of an overall response rate of 25.6%, although remission durations were relatively short.97
Several additional phase 2 studies investigated GO monotherapy for treatment of newly diagnosed and/or relapsed/refractory disease in unselected patients with non-APL AML. Together, these studies confirmed single-agent activity of GO in a subset of patients; however, overall response rates have generally not exceeded 25% to 35% and have occasionally been quite disappointing, particularly in heavily pretreated patients.98–103 In these studies, GO has usually been given at 2-week intervals. However, early studies indicated that new CD33 binding sites were continuously reexpressed, and surface CD33 levels returned to pretreatment levels within 72 hours after anti–CD33 antibody administration despite antigen internalization and modulation.104,105 This observation suggests that repeated administration of lower, (near-)saturating doses of GO every 3 days may enhance intracellular accumulation of the calicheamicin-γ1 derivative beyond what can be achieved with 2-weekly administration. The Acute Leukemia French Association group has relatively recently reported the use of GO in fractionated, lower doses with promising efficacy and acceptable safety profile,89,106 but comparative data with the traditional administration scheme are currently not available.
Consistent with preclinical predictions that GO would be most effective for leukemias likely originating in a committed progenitor as suggested for APL, the drug appears to be highly active against this entity.107–109 This was emphasized by the observation that GO monotherapy routinely resulted in molecular remissions that could be durable in patients with molecularly relapsed APL.107,108 In newly diagnosed APL, GO adds to the effectiveness of all-trans retinoic acid and can replace anthracyclines in the curative backbone of treatment.110,111
In addition to those in APL, many studies have explored GO together with other therapeutics in non-APL AML.98–103 Emerging data from large, phase 3 randomized studies now indicate that substantial numbers of patients with newly diagnosed AML experience improved survival when GO is combined with conventional chemotherapy (Table 2). Particularly important are findings from the Medical Research Council AML15 trial.112 In this trial, 1113 predominantly adult patients were randomized to receive a single dose of GO (3 mg/m2) on day 1 of the first of 2 induction courses with one of 3 induction regimens (cytarabine/daunorubicin/etoposide, daunorubicin/cytarabine, fludarabine/cytarabine/G-CSF/idarubicin). Patients in remission who were not scheduled for hematopoietic cell transplantation (HCT) were then randomized to consolidation with or without the addition of GO. Although adding GO to either induction or consolidation did not affect overall remission rates or survival, protocol-predefined subgroup analyses showed a highly significant interaction between cytogenetics and use of GO in induction therapy. Specifically, with the addition of GO, there was improved overall survival (OS) at 5 years for patients with favorable cytogenetics (79% vs 51%; P = .0003) but no benefit for patients with unfavorable cytogenetics (8% vs 11%; P = .4). There was also an OS benefit for some patients with intermediate-risk disease, as indicated by an internally validated index using cytogenetics, age, and performance status, which predicted that approximately 70% of all intermediate-risk patients would have a 10% improvement in 5-year OS if given GO during induction; within the intermediate-risk subgroup, those 30% predicted to not benefit were older, had higher white blood cell counts, worse performance status, or secondary disease.112
Table 2.
Phase 3 studies of GO in newly diagnosed non-APL AML
| Study | Disease | N | Age, y (median) | Treatment | Results |
|---|---|---|---|---|---|
| MRC AML15112 | AML | 1113 | 0-71 (49) | ± GO (3 mg/m2) on day 1 of the first of 2 induction courses with either ADE, DA, or FLAG-IDA | No difference in ORR, TRM, relapse, or survival. Improved 5-y OS for favorable-risk subgroup (79% vs 51%; P = .0003) with GO; predicted 10% OS benefit for ∼ 70% of patients with intermediate-risk disease |
| ALFA 0701113 | AML | 278 | 59-66 (62) | ± GO (3 mg/m2) on days 1, 4, and 7 of DA induction and day 1 of each of 2 courses of DA consolidation | No difference in ORR or mortality. Improved 2-y EFS (40.8% vs 17.1%, P = .0003), DFS (50.3% vs 22.7%, P = .0003) and OS (53.2% vs 41.9%, P = .037) with GO. Survival benefit seen in favorable/intermediate- but not adverse-risk disease |
| GOELAMS AML 2006 IR114 | AML (intermediate risk) | 238 | 18-60 (50) | ± GO (6 mg/m2) on day 1 with DA induction and MA consolidation | No difference in ORR, TRM, or 3-y EFS. Improved EFS with GO in patients who did not undergo allogeneic HCT (53.7% vs 27%, P = .0308) |
| NCRI AML16115 | AML, high-risk MDS | 1115 | 51-84 (67) | ± GO (3 mg/m2) on day 1 of the first of 2 induction courses with either DA or DCLo | No difference in TRM; trend towards reduced risk of persistent disease with GO (17% vs 21%, P = .06). Reduced 3-y relapse risk (68% vs 76%; P = .007) and superior DFS (21% vs 16%; P = .04) and OS (25% vs 20%; P = .05) with GO |
| SWOG S0106116,117 | AML | 596 | 18-60 (47) | ± GO (6 mg/m2) on day 4 of the first of up to 2 induction courses with DA* | Increased TRM in GO arm (5.7% vs 1.4%; P = .01). No difference in ORR, DFS, or OS. Possible trend towards improved OS in favorable-risk subgroup with GO (hazard ratio: 0.49 [0.12–2.04]) |
ADE indicates cytarabine/daunorubicin/etoposide; DA, daunorubicin/cytarabine; DClo, daunorubicin/clofarabine; DFS, disease-free survival; EFS, event-free survival; FLAG-Ida, fludarabine/cytarabine/G-CSF/idarubicin; HCT, hematopoietic cell transplantation; MA, mitoxantrone/cytarabine; ORR, overall response rate (CR + CRi); OS, overall survival; and TRM, treatment-related mortality.
Daunorubicin was used at 60 mg/m2/d in the control arm and 45 mg/m2/d in the GO arm.
Results from 3 additional European studies support the conclusions from AML15 and extend those findings to older adults. In the Acute Leukemia French Association 0701 trial, 278 evaluable patients with primary non-APL AML 50 to 70 years of age received daunorubicin/cytarabine with or without GO (3 mg/m2) on days 1, 4, and 7; a second course of daunorubicin/cytarabine was given for residual disease on day 15. Patients in remission then received 2 courses of daunorubicin/cytarabine with or without GO (3 mg/m2) on day 1 of each cycle.113 Although there was no statistically significant difference in remission rates (CR + CRp: 81% vs 75%, P = .25) and treatment-related mortality (6% vs 4%, P = .41) between the +GO and −GO arms, the event-free survival at 2 years was significantly superior in the GO arm (40.8% vs 17.1%, P = .0003), as was disease-free survival (50.3% vs 22.7%, P = .0003) and OS (53.2% vs 41.9%, P = .037). Subgroup analysis showed that the event-free survival benefit was associated with favorable/intermediate but not adverse karyotype.113 In the GOELAMS AML 2006 IR study, adults 18 to 60 years of age with de novo AML and intermediate karyotype received daunorubicin/cytarabine with or without GO (6 mg/m2) on day 4; GO was also added to consolidation therapy with mitoxantrone/cytarabine according to the initial randomization.114 Among 238 analyzed patients, there was no difference in remission rates or treatment-related mortality between the 2 treatment arms. Overall, there was also no statistically significant difference in event-free survival (GO vs control: 51% vs 33%) or OS (53% vs 46%) at 3 years. However, subgroup analyses showed that, in patients who did not undergo allogeneic HCT, event-free survival was significantly higher in the GO group (53.7% vs 27%, P = .0308). Finally, in the NCRI AML16 trial, 1115 older adults (51-84 years of age) with AML or high-risk MDS (> 10% marrow blasts) received either daunorubicin/cytarabine or daunorubicin/clofarabine for 2 cycles with or without GO (3 mg/m2) on day 1 of the first induction course; patients in CR were then randomized to receive, or not, a third course of daunorubicin/cytarabine with or without maintenance therapy with azacitidine.115 Similar to the other trials, there was no statistically significant difference in remission rates and treatment-related mortality between the +GO and −GO arms, although the patients in the −GO arm tended to have a higher likelihood of persistent disease (21% vs 17%, P = .06). However, patients receiving GO had a reduced relapse risk (at 3 years: 68% vs 76%; P = .007), resulting in a superior disease-free survival (21% vs 16%; P = .04) and OS (25% vs 20%; P = .05); the benefit of GO appeared greater in patients with de novo disease than those with secondary disease.115 Finally, a meta-analysis of AML15 and AML16 on 2224 patients showed a significant benefit of GO for risk of relapse (odds ratio [OR] = 0.82; 95% CI, 0.72-0.93, P = .002) and survival (OR = 0.88; 95% CI, 0.79-0.98, P = .02); the survival benefit was seen in patients with favorable-risk (OR = 0.47; 95% CI, 0.28-0.77) and intermediate-risk (OR = 0.84; 95% CI, 0.73-0.97) but not adverse-risk (OR = 1.02; 95% CI, 0.81-1.27) disease.115
The results from these European studies differ from a trial conducted by the Southwest Oncology Group (trial S0106),116 which was developed with the drug sponsor to fulfill their postapproval commitment to the FDA. A total of 637 patients 18 to 60 years of age with de novo AML were accrued to receive up to 2 cycles of induction chemotherapy with daunorubicin/cytarabine with or without a single dose of GO (6 mg/m2) on day 4 of the first induction.116,117 Unlike the European studies in which identical doses of conventional chemotherapeutics were used in both arms, S0106 used a lower daunorubicin dose with GO (45 mg/m2 vs 60 mg/m2) with the intent of evaluating 2 similarly toxic treatment arms.116 After consolidation therapy, patients in remission were eligible for a second randomization (stratified by cytogenetic risk category at diagnosis and use of GO during induction) between GO or observation only. Among 596 evaluable patients, S0106 showed no overall difference in response or survival in induction with the addition of GO, but a trend toward improved OS was seen for patients with CBF leukemias (hazard ratio = 0.49; 0.12-2.04); there was also no benefit for GO in consolidation.116,117 It should be noted that S0106 was not powered to detect important outcome differences in the favorable-risk patients. Given the attempt to achieve equitoxicity with a 25% lower dose of daunorubicin in the GO arm,116 the S0106 trial may thus be confounded toward the null as the dose reduction of daunorubicin might mask a greater benefit of GO. Based on the lack of prespecified overall improvement in outcome, S0106 was prematurely terminated before planned accrual was reached.117 After discussions with the FDA, the failure to detect improved survival together with a trend toward increased treatment-related mortality in S0106 led Pfizer to discontinue commercial availability of GO in the United States in October 2010.118
Of note, the early studies on the clonal origin of AML indicated that diseases limited primarily to the granulocytic/monocytic lineage occurred predominantly in younger adults and children,11 suggesting that GO-based therapies might be particularly effective for some pediatric patients. So far, only a few clinical studies have explored the use of GO in pediatric AML, primarily in patients with relapsed/refractory disease, and the drug has no established role in childhood AML.102,103 A large trial testing the addition of GO to induction chemotherapy in a randomized fashion was led by the Children's Oncology Group (AAML0531) and has recently completed accrual of more than 1000 participants, with preliminary outcome results expected for 2012 or 2013.
Synthesis and conclusion
The trials conducted over the last decade demonstrate that GO improves survival for many, but not all, AML patients, supporting the preclinical conclusion that CD33 is a valid target for some disease subforms. The long-term benefit achieved with GO in APL and CBF leukemias and some patients with less favorable prognoses, in particular those with intermediate-risk diseases, is consistent with an ablative effect on disease-relevant CD33+ cells (ie, AML stem or progenitor cells). As discussed previously, we hypothesized that CD33-targeting therapeutics would be most beneficial for patients with “mature” LSCs. The most compelling data to support the notion that CD33-directed therapy might indeed effectively target malignant stem cells in some “mature” leukemias come from the treatment of APL.107,108 Although the high and homogeneous expression of CD33 and low activity of P-glycoprotein may partially explain the exquisite sensitivity of APL toward GO,92,119,120 even monotherapy with an unconjugated humanized anti–CD33 antibody (HuM195) leads to a molecular remissions in approximately half of the APL patients with molecular minimal residual disease after induction chemotherapy.121
The situation is likely different for CBF leukemias and other AML subforms, in which founder mutations may arise at the level of multipotent CD33− stem cells in at least some of the cases. Although non–receptor-mediated drug uptake of GO has been suggested by small in vitro studies with CD33− acute lymphoblastic leukemia cell lines,93 its contribution to clinical GO efficacy is unknown, and CD33− LSCs are unlikely effective targets for GO. Nevertheless, the clinical benefit of CD33-targeted therapy in these leukemias could be understood if the preleukemic CD33− cells were relatively nonprolific and additional mutations leading to frank leukemic transformation were acquired only at a later stage when the cell expressed CD33 (scenario 2 of the proposed AML transformation models). In this scenario, successful depletion of CD33+ cells would eradicate the fully transformed, proliferative clone and leave the CD33− pre-LSCs behind (ie, reset the clock to a premalignant stage). For a cure, such preleukemic cells may need to be controlled or eliminated by other means (eg, immunologic mechanisms), be it via the patient's own immunosurveillance or via graft-versus-leukemia effects after allogeneic HCT. This may be most successful if the preleukemic cells were undetectable or present at very minimal levels at the time of HCT.
It is important to emphasize that studies to date have not determined whether GO, besides acting on more mature CD33+ progeny, can indeed directly kill CD33+ LSCs in vivo, and whether long-term benefit from GO is related to successful targeting of LSCs. Instead, it is imaginable that the observed clinical long-term efficacy in some leukemias could result from effects on AML cells without stem cell properties. This possibility needs to be considered because “intensification” of the chemotherapeutic regimens via addition of GO (eg, as done in the recent European phase 3 trials) could lead to more robust non-LSC AML cell debulking. The long-term benefit in this situation is conceivable even if successful depletion of CD33+ cells only eliminates the most proliferative cells and leaves fully transformed LSCs behind; the latter might then be eradicated by other chemotherapeutic, radiotherapeutic, or immunologic means, especially if at minimal levels and not overly prolific. This scenario could explain a benefit of GO in leukemias that harbor CD33− LSCs (scheme 1 of the proposed AML transformation models). As an alternative for those leukemias, CD33− LSCs remaining after chemotherapy-induced bulk reduction might enter cell cycle and acquire CD33 and thus be susceptible to CD33 targeting. Finally, the (short-lasting) remissions seen in unselected patients with relapsed/refractory AML could result from debulking of more mature CD33+ AML blasts by GO, even if underlying LSCs remained CD33− and could not be eradicated. There is thus a critical need for further studies to determine the relationship between the origin of LSCs and efficacy of CD33-directed therapy, and formally test our hypothesis that benefit from this therapeutic strategy is mostly confined to patients with “mature” leukemias. These investigations will need to use functional characterizations of cells (eg, via long-term in vitro culture) and carefully consider the possibility of aberrant cell surface marker expression that may limit the conclusions that can be drawn with regard to cell of origin from immunophenotypic studies.
Response to GO parallels sensitivity to conventional chemotherapy, as patients deriving most long-term benefit from GO also have a better response to standard chemotherapeutics. Thus, GO may be used with different goals in mind: first, as best exemplified by the European phase 3 trials, GO could be added to conventional chemotherapy to improve the outcome of standard therapy alone with relatively little increase in toxicity. On the other hand, as suggested by the experience in APL, GO could at least partially replace nontargeted therapeutics in low-risk patients with preservation of treatment efficacy but possibly reduced nonspecific toxicity.
It is plausible that not all CD33+ AML cells, and in particular CD33+ LSCs, may be immediately susceptible to GO. CD33 is challenging to target with immunotoxins because of antigen expression at relatively low abundance (∼ 104 CD33 molecules/cell)122,123 and slow conjugate internalization.91 Furthermore, GO's efficacy is significantly reduced by drug efflux, a concern for any stem cell-directed therapy given increasing evidence linking ABC drug transporters to protection of cancer stem cells.124 Rational strategies to improve immunotherapy with GO would therefore include maneuvers that increase intracellular toxin accumulation, for example by manipulating CD33 expression/turnover or inhibiting drug efflux.92 In addition, approaches that interfere with DNA repair and/or downstream pro-/antiapoptotic signaling may lead to a lowering of the apoptotic threshold and increased cellular toxicity of calicheamicins. An alternative entails the development of novel immunoconjugates using more potent toxins that are not (or less) susceptible to ABC transporter activity. Substitution of the calicheamicin-γ1 derivative with a different toxin may have the potential to reduce the risk of significant liver toxicity, specifically veno-occlusive disease, a risk that appears to have decreased but not entirely disappeared with the lower doses of GO used in the recent phase 3 trials.
The experience with GO is a reminder of the intrinsic heterogeneity in AML. Conceivably reflecting differences in the cellular origin of individual leukemias, this diversity is of particular significance for therapies that aim at eradicating stem cells in AML and implies that no such approach may be universally effective for all leukemias. Nevertheless, the clinical results with GO strongly support the utility of CD33-targeting therapeutics in AML and suggest that the current unavailability of GO is unfortunate. With the emerging data on the benefit of GO for some subsets of AML, reconsideration of the drug's value by the manufacturer and regulatory authorities is warranted. As we improve our abilities to identify those AMLs likely to respond to specific treatments, clinical trials testing these treatments may restrict eligibility to patients with these leukemias. In the future, we may thus have to place greater emphasis on collaborative, perhaps multinational studies, or more likely, accept higher false-positive and false-negative rates or detection of only relatively large differences between treatment and controls.
Acknowledgments
This work was supported by the National Cancer Institute/National Institutes of Health (grants P30-CA015704-35S6 and U10-CA098543), the Leukemia & Lymphoma Society (Specialized Center of Research grant 7008-08), and the St Baldrick's Foundation.
Authorship
Contribution: R.B.W. and I.D.B. conceived and wrote the manuscript; and F.R.A. and E.H.E. assisted with writing the manuscript.
Conflict-of-interest disclosure: R.B.W. received research funding from Seattle Genetics Inc. R.B.W. and I.D.B. were consultants for Seattle Genetics Inc. The remaining authors declare no competing financial interests.
Correspondence: Roland B. Walter, Clinical Research Division, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave N, D2-190, Seattle, WA 98109-1024; e-mail: rwalter@fhcrc.org.
References
- 1.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 2.Dick JE. Stem cell concepts renew cancer research. Blood. 2008;112(13):4793–4807. doi: 10.1182/blood-2008-08-077941. [DOI] [PubMed] [Google Scholar]
- 3.Clarke MF, Dick JE, Dirks PB, et al. Cancer stem cells. Perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66(19):9339–9344. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 4.Stubbs MC, Armstrong SA. Therapeutic implications of leukemia stem cell development. Clin Cancer Res. 2007;13(12):3439–3442. doi: 10.1158/1078-0432.CCR-06-3090. [DOI] [PubMed] [Google Scholar]
- 5.Lane SW, Gilliland DG. Leukemia stem cells. Semin Cancer Biol. 2010;20(2):71–76. doi: 10.1016/j.semcancer.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 6.Majeti R. Monoclonal antibody therapy directed against human acute myeloid leukemia stem cells. Oncogene. 2011;30(9):1009–1019. doi: 10.1038/onc.2010.511. [DOI] [PubMed] [Google Scholar]
- 7.Passegué E, Jamieson CH, Ailles LE, Weissman IL. Normal and leukemic hematopoiesis: are leukemias a stem cell disorder or a reacquisition of stem cell characteristics? Proc Natl Acad Sci U S A. 2003;100(Suppl 1):11842–11849. doi: 10.1073/pnas.2034201100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernstein ID. Monoclonal antibodies to the myeloid stem cells: therapeutic implications of CMA-676, a humanized anti–CD33 antibody calicheamicin conjugate. Leukemia. 2000;14(3):474–475. doi: 10.1038/sj.leu.2401663. [DOI] [PubMed] [Google Scholar]
- 9.Chen GL, Prchal JT. X-linked clonality testing: interpretation and limitations. Blood. 2007;110(5):1411–1419. doi: 10.1182/blood-2006-09-018655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fialkow PJ, Singer JW, Adamson JW, et al. Acute nonlymphocytic leukemia: heterogeneity of stem cell origin. Blood. 1981;57(6):1068–1073. [PubMed] [Google Scholar]
- 11.Fialkow PJ, Singer JW, Raskind WH, et al. Clonal development, stem-cell differentiation, and clinical remissions in acute nonlymphocytic leukemia. N Engl J Med. 1987;317(8):468–473. doi: 10.1056/NEJM198708203170802. [DOI] [PubMed] [Google Scholar]
- 12.Bernstein ID, Singer JW, Andrews RG, et al. Treatment of acute myeloid leukemia cells in vitro with a monoclonal antibody recognizing a myeloid differentiation antigen allows normal progenitor cells to be expressed. J Clin Invest. 1987;79(4):1153–1159. doi: 10.1172/JCI112932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bernstein ID, Singer JW, Smith FO, et al. Differences in the frequency of normal and clonal precursors of colony-forming cells in chronic myelogenous leukemia and acute myelogenous leukemia. Blood. 1992;79(7):1811–1816. [PubMed] [Google Scholar]
- 14.McCulloch EA. Stem cells in normal and leukemic hemopoiesis (Henry Stratton Lecture, 1982). Blood. 1983;62(1):1–13. [PubMed] [Google Scholar]
- 15.Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 16.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 17.Blair A, Hogge DE, Ailles LE, Lansdorp PM, Sutherland HJ. Lack of expression of Thy-1 (CD90) on acute myeloid leukemia cells with long-term proliferative ability in vitro and in vivo. Blood. 1997;89(9):3104–3112. [PubMed] [Google Scholar]
- 18.Miyamoto T, Weissman IL, Akashi K. AML1/ETO-expressing nonleukemic stem cells in acute myelogenous leukemia with 8;21 chromosomal translocation. Proc Natl Acad Sci U S A. 2000;97(13):7521–7526. doi: 10.1073/pnas.97.13.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chao MP, Seita J, Weissman IL. Establishment of a normal hematopoietic and leukemia stem cell hierarchy. Cold Spring Harb Symp Quant Biol. 2008;73:439–449. doi: 10.1101/sqb.2008.73.031. [DOI] [PubMed] [Google Scholar]
- 20.Taussig DC, Miraki-Moud F, Anjos-Afonso F, et al. Anti–CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood. 2008;112(3):568–575. doi: 10.1182/blood-2007-10-118331. [DOI] [PubMed] [Google Scholar]
- 21.Taussig DC, Vargaftig J, Miraki-Moud F, et al. Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34− fraction. Blood. 2010;115(10):1976–1984. doi: 10.1182/blood-2009-02-206565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goardon N, Marchi E, Atzberger A, et al. Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell. 2011;19(1):138–152. doi: 10.1016/j.ccr.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 23.Ailles LE, Gerhard B, Kawagoe H, Hogge DE. Growth characteristics of acute myelogenous leukemia progenitors that initiate malignant hematopoiesis in nonobese diabetic/severe combined immunodeficient mice. Blood. 1999;94(5):1761–1772. [PubMed] [Google Scholar]
- 24.Rombouts WJC, Martens ACM, Ploemacher RE. Identification of variables determining the engraftment potential of human acute myeloid leukemia in the immunodeficient NOD/SCID human chimera model. Leukemia. 2000;14(5):889–897. doi: 10.1038/sj.leu.2401777. [DOI] [PubMed] [Google Scholar]
- 25.Rombouts WJC, Blokland I, Löwenberg B, Ploemacher RE. Biological characteristics and prognosis of adult acute myeloid leukemia with internal tandem duplications in the Flt3 gene. Leukemia. 2000;14(4):675–683. doi: 10.1038/sj.leu.2401731. [DOI] [PubMed] [Google Scholar]
- 26.Lumkul R, Gorin NC, Malehorn MT, et al. Human AML cells in NOD/SCID mice: engraftment potential and gene expression. Leukemia. 2002;16(9):1818–1826. doi: 10.1038/sj.leu.2402632. [DOI] [PubMed] [Google Scholar]
- 27.Monaco G, Konopleva M, Munsell M, et al. Engraftment of acute myeloid leukemia in NOD/SCID mice is independent of CXCR4 and predicts poor patient survival. Stem Cells. 2004;22(2):188–201. doi: 10.1634/stemcells.22-2-188. [DOI] [PubMed] [Google Scholar]
- 28.Pearce DJ, Taussig D, Zibara K, et al. AML engraftment in the NOD/SCID assay reflects the outcome of AML: implications for our understanding of the heterogeneity of AML. Blood. 2006;107(3):1166–1173. doi: 10.1182/blood-2005-06-2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanchez PV, Perry RL, Sarry JE, et al. A robust xenotransplantation model for acute myeloid leukemia. Leukemia. 2009;23(11):2109–2117. doi: 10.1038/leu.2009.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taussig DC, Pearce DJ, Simpson C, et al. Hematopoietic stem cells express multiple myeloid markers: implications for the origin and targeted therapy of acute myeloid leukemia. Blood. 2005;106(13):4086–4092. doi: 10.1182/blood-2005-03-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andrews RG, Torok-Storb B, Bernstein ID. Myeloid-associated differentiation antigens on stem cells and their progeny identified by monoclonal antibodies. Blood. 1983;62(1):124–132. [PubMed] [Google Scholar]
- 32.Griffin JD, Linch D, Sabbath K, Larcom P, Schlossman SF. A monoclonal antibody reactive with normal and leukemic human myeloid progenitor cells. Leuk Res. 1984;8(4):521–534. doi: 10.1016/0145-2126(84)90001-8. [DOI] [PubMed] [Google Scholar]
- 33.Andrews RG, Takahashi M, Segal GM, Powell JS, Bernstein ID, Singer JW. The L4F3 antigen is expressed by unipotent and multipotent colony-forming cells but not by their precursors. Blood. 1986;68(5):1030–1035. [PubMed] [Google Scholar]
- 34.Andrews RG, Singer JW, Bernstein ID. Precursors of colony-forming cells in humans can be distinguished from colony-forming cells by expression of the CD33 and CD34 antigens and light scatter properties. J Exp Med. 1989;169(5):1721–1731. doi: 10.1084/jem.169.5.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robertson MJ, Soiffer RJ, Freedman AS, et al. Human bone marrow depleted of CD33-positive cells mediates delayed but durable reconstitution of hematopoiesis: clinical trial of MY9 monoclonal antibody-purged autografts for the treatment of acute myeloid leukemia. Blood. 1992;79(9):2229–2236. [PubMed] [Google Scholar]
- 36.Bernstein ID, Andrews RG, Berenson R, Bensinger W, Singer JW, Buckner CD. Isolation of human hematopoietic stem cells. In: Champlin RE, Gale RP, editors. New Strategies in Bone Marrow Transplantation. New York, NY: Wiley-Liss; 1991. pp. 201–207. [Google Scholar]
- 37.Cozzio A, Passegué E, Ayton PM, Karsunky H, Cleary ML, Weissman IL. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 2003;17(24):3029–3035. doi: 10.1101/gad.1143403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huntly BJ, Shigematsu H, Deguchi K, et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell. 2004;6(6):587–596. doi: 10.1016/j.ccr.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 39.Krivtsov AV, Twomey D, Feng Z, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442(7104):818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- 40.Somervaille TC, Cleary ML. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell. 2006;10(4):257–268. doi: 10.1016/j.ccr.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 41.So CW, Karsunky H, Passegue E, Cozzio A, Weissman IL, Cleary ML. MLL-GAS7 transforms multipotent hematopoietic progenitors and induces mixed lineage leukemias in mice. Cancer Cell. 2003;3(2):161–171. doi: 10.1016/s1535-6108(03)00019-9. [DOI] [PubMed] [Google Scholar]
- 42.Chen W, Kumar AR, Hudson WA, et al. Malignant transformation initiated by Mll-AF9: gene dosage and critical target cells. Cancer Cell. 2008;13(5):432–440. doi: 10.1016/j.ccr.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kirstetter P, Schuster MB, Bereshchenko O, et al. Modeling of C/EBPalpha mutant acute myeloid leukemia reveals a common expression signature of committed myeloid leukemia-initiating cells. Cancer Cell. 2008;13(4):299–310. doi: 10.1016/j.ccr.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 44.Guibal FC, Alberich-Jorda M, Hirai H, et al. Identification of a myeloid committed progenitor as the cancer-initiating cell in acute promyelocytic leukemia. Blood. 2009;114(27):5415–5425. doi: 10.1182/blood-2008-10-182071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kelly LM, Gilliland DG. Genetics of myeloid leukemias. Annu Rev Genomics Hum Genet. 2002;3:179–198. doi: 10.1146/annurev.genom.3.032802.115046. [DOI] [PubMed] [Google Scholar]
- 46.Fröhling S, Scholl C, Gilliland DG, Levine RL. Genetics of myeloid malignancies: pathogenetic and clinical implications. J Clin Oncol. 2005;23(26):6285–6295. doi: 10.1200/JCO.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 47.Vassiliou GS, Cooper JL, Rad R, et al. Mutant nucleophosmin and cooperating pathways drive leukemia initiation and progression in mice. Nat Genet. 2011;43(5):470–475. doi: 10.1038/ng.796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Corral J, Lavenir I, Impey H, et al. An Mll-AF9 fusion gene made by homologous recombination causes acute leukemia in chimeric mice: a method to create fusion oncogenes. Cell. 1996;85(6):853–861. doi: 10.1016/s0092-8674(00)81269-6. [DOI] [PubMed] [Google Scholar]
- 49.Chen W, Li Q, Hudson WA, Kumar A, Kirchhof N, Kersey JH. A murine Mll-AF4 knock-in model results in lymphoid and myeloid deregulation and hematologic malignancy. Blood. 2006;108(2):669–677. doi: 10.1182/blood-2005-08-3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Song WJ, Sullivan MG, Legare RD, et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet. 1999;23(2):166–175. doi: 10.1038/13793. [DOI] [PubMed] [Google Scholar]
- 51.Smith ML, Cavenagh JD, Lister TA, Fitzgibbon J. Mutation of CEBPA in familial acute myeloid leukemia. N Engl J Med. 2004;351(23):2403–2407. doi: 10.1056/NEJMoa041331. [DOI] [PubMed] [Google Scholar]
- 52.Nanri T, Uike N, Kawakita T, Iwanaga E, Mitsuya H, Asou N. A family harboring a germ-line N-terminal C/EBPalpha mutation and development of acute myeloid leukemia with an additional somatic C-terminal C/EBPalpha mutation. Genes Chromosomes Cancer. 2010;49(3):237–241. doi: 10.1002/gcc.20734. [DOI] [PubMed] [Google Scholar]
- 53.Müller AMS, Duque J, Shizuru JA, Lübbert M. Complementing mutations in core binding factor leukemias: from mouse models to clinical applications. Oncogene. 2008;27(44):5759–5773. doi: 10.1038/onc.2008.196. [DOI] [PubMed] [Google Scholar]
- 54.Wiemels JL, Xiao Z, Buffler PA, et al. In utero origin of t(8;21) AML1-ETO translocations in childhood acute myeloid leukemia. Blood. 2002;99(10):3801–3805. doi: 10.1182/blood.v99.10.3801. [DOI] [PubMed] [Google Scholar]
- 55.Nucifora G, Larson RA, Rowley JD. Persistence of the 8;21 translocation in patients with acute myeloid leukemia type M2 in long-term remission. Blood. 1993;82(3):712–715. [PubMed] [Google Scholar]
- 56.Jurlander J, Caligiuri MA, Ruutu T, et al. Persistence of the AML1/ETO fusion transcript in patients treated with allogeneic bone marrow transplantation for t(8;21) leukemia. Blood. 1996;88(6):2183–2191. [PubMed] [Google Scholar]
- 57.Miyamoto T, Nagafuji K, Akashi K, et al. Persistence of multipotent progenitors expressing AML1/ETO transcripts in long-term remission patients with t(8;21) acute myelogenous leukemia. Blood. 1996;87(11):4789–4796. [PubMed] [Google Scholar]
- 58.Saunders MJ, Brereton ML, Adams JA, Tobal K, Liu Yin JA. Expression of AML1/MTG8 transcripts in clonogenic cells grown from bone marrow of patients in remission of acute myeloid leukaemia with t(8;21). Br J Haematol. 1997;99(4):921–924. doi: 10.1046/j.1365-2141.1997.4673271.x. [DOI] [PubMed] [Google Scholar]
- 59.Shih LY, Liang DC, Huang CF, et al. AML patients with CEBPalpha mutations mostly retain identical mutant patterns but frequently change in allelic distribution at relapse: a comparative analysis on paired diagnosis and relapse samples. Leukemia. 2006;20(4):604–609. doi: 10.1038/sj.leu.2404124. [DOI] [PubMed] [Google Scholar]
- 60.Falini B, Martelli MP, Bolli N, et al. Acute myeloid leukemia with mutated nucleophosmin (NPM1): is it a distinct entity? Blood. 2011;117(4):1109–1120. doi: 10.1182/blood-2010-08-299990. [DOI] [PubMed] [Google Scholar]
- 61.Hou HA, Kuo YY, Liu CY, et al. DNMT3A mutations in acute myeloid leukemia: stability during disease evolution and clinical implications. Blood. 2012;119(2):559–568. doi: 10.1182/blood-2011-07-369934. [DOI] [PubMed] [Google Scholar]
- 62.Kihara R, Hoshino H, Kiyoi H, Naoe T. DNMT3A and IDH1/2 mutations are stable during the progression of acute myeloid leukemia [abstract]. Blood (ASH Annual Meeting Abstracts) 2011;118(21):637. [Google Scholar]
- 63.Kottaridis PD, Gale RE, Langabeer SE, Frew ME, Bowen DT, Linch DC. Studies of FLT3 mutations in paired presentation and relapse samples from patients with acute myeloid leukemia: implications for the role of FLT3 mutations in leukemogenesis, minimal residual disease detection, and possible therapy with FLT3 inhibitors. Blood. 2002;100(7):2393–2398. doi: 10.1182/blood-2002-02-0420. [DOI] [PubMed] [Google Scholar]
- 64.Shih LY, Huang CF, Wu JH, et al. Heterogeneous patterns of FLT3 Asp(835) mutations in relapsed de novo acute myeloid leukemia: a comparative analysis of 120 paired diagnostic and relapse bone marrow samples. Clin Cancer Res. 2004;10(4):1326–1332. doi: 10.1158/1078-0432.ccr-0835-03. [DOI] [PubMed] [Google Scholar]
- 65.Bachas C, Schuurhuis GJ, Hollink IH, et al. High-frequency type I/II mutational shifts between diagnosis and relapse are associated with outcome in pediatric AML: implications for personalized medicine. Blood. 2010;116(15):2752–2758. doi: 10.1182/blood-2010-03-276519. [DOI] [PubMed] [Google Scholar]
- 66.Pollard JA, Meshinchi S, Harrington KH, Zeng R, Bernstein ID, Walter RB. Sequential acquisition of somatic mutations in progenitors with differential proliferative potential in human acute myeloid leukemia [abstract]. Blood (ASH Annual Meeting Abstracts) 2010;116(21):332. [Google Scholar]
- 67.Wang YY, Zhou GB, Yin T, et al. AML1-ETO and C-KIT mutation/overexpression in t(8;21) leukemia: implication in stepwise leukemogenesis and response to Gleevec. Proc Natl Acad Sci U S A. 2005;102(4):1104–1109. doi: 10.1073/pnas.0408831102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fearon ER, Burke PJ, Schiffer CA, Zehnbauer BA, Vogelstein B. Differentiation of leukemia cells to polymorphonuclear leukocytes in patients with acute nonlymphocytic leukemia. N Engl J Med. 1986;315(1):15–24. doi: 10.1056/NEJM198607033150103. [DOI] [PubMed] [Google Scholar]
- 69.Gale RE, Wheadon H, Goldstone AH, Burnett AK, Linch DC. Frequency of clonal remission in acute myeloid leukaemia. Lancet. 1993;341(8838):138–142. doi: 10.1016/0140-6736(93)90004-z. [DOI] [PubMed] [Google Scholar]
- 70.Jowitt SN, Liu Yin JA, Saunders MJ, Lucas GS. Clonal remissions in acute myeloid leukaemia are commonly associated with features of trilineage myelodysplasia during remission. Br J Haematol. 1993;85(4):698–705. doi: 10.1111/j.1365-2141.1993.tb03211.x. [DOI] [PubMed] [Google Scholar]
- 71.Jinnai I, Nagai K, Yoshida S, et al. Incidence and characteristics of clonal hematopoiesis in remission of acute myeloid leukemia in relation to morphological dysplasia. Leukemia. 1995;9(10):1756–1761. [PubMed] [Google Scholar]
- 72.Grimwade D, Enver T. Acute promyelocytic leukemia: where does it stem from? Leukemia. 2004;18(3):375–384. doi: 10.1038/sj.leu.2403234. [DOI] [PubMed] [Google Scholar]
- 73.Eppert K, Takenaka K, Lechman ER, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17(9):1086–1093. doi: 10.1038/nm.2415. [DOI] [PubMed] [Google Scholar]
- 74.Pollard JA, Alonzo TA, Gerbing RB, et al. FLT3 internal tandem duplication in CD34+/CD33− precursors predicts poor outcome in acute myeloid leukemia. Blood. 2006;108(8):2764–2769. doi: 10.1182/blood-2006-04-012260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dinndorf PA, Andrews RG, Benjamin D, Ridgway D, Wolff L, Bernstein ID. Expression of normal myeloid-associated antigens by acute leukemia cells. Blood. 1986;67(4):1048–1053. [PubMed] [Google Scholar]
- 76.Scheinberg DA, Lovett D, Divgi CR, et al. A phase I trial of monoclonal antibody M195 in acute myelogenous leukemia: specific bone marrow targeting and internalization of radionuclide. J Clin Oncol. 1991;9(3):478–490. doi: 10.1200/JCO.1991.9.3.478. [DOI] [PubMed] [Google Scholar]
- 77.Caron PC, Dumont L, Scheinberg DA. Supersaturating infusional humanized anti–CD33 monoclonal antibody HuM195 in myelogenous leukemia. Clin Cancer Res. 1998;4(6):1421–1428. [PubMed] [Google Scholar]
- 78.Appelbaum FR, Matthews DC, Eary JF, et al. The use of radiolabeled anti–CD33 antibody to augment marrow irradiation prior to marrow transplantation for acute myelogenous leukemia. Transplantation. 1992;54(5):829–833. doi: 10.1097/00007890-199211000-00012. [DOI] [PubMed] [Google Scholar]
- 79.van der Jagt RHC, Badger CC, Appelbaum FR, et al. Localization of radiolabeled antimyeloid antibodies in a human acute leukemia xenograft tumor model. Cancer Res. 1992;52(1):89–94. [PubMed] [Google Scholar]
- 80.Hamann PR, Hinman LM, Beyer CF, et al. An anti–CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia: choice of linker. Bioconjug Chem. 2002;13(1):40–46. doi: 10.1021/bc0100206. [DOI] [PubMed] [Google Scholar]
- 81.Damle NK, Frost P. Antibody-targeted chemotherapy with immunoconjugates of calicheamicin. Curr Opin Pharmacol. 2003;3(4):386–390. doi: 10.1016/s1471-4892(03)00083-3. [DOI] [PubMed] [Google Scholar]
- 82.Hamann PR, Hinman LM, Hollander I, et al. Gemtuzumab ozogamicin, a potent and selective anti–CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconjug Chem. 2002;13(1):47–58. doi: 10.1021/bc010021y. [DOI] [PubMed] [Google Scholar]
- 83.Walter RB, Fairchild SE, Flowers DA, Hong TC, Bernstein ID. Priming with myeloid growth factors enhances CD33 expression, decreases P-glycoprotein activity, and improves efficacy of gemtuzumab ozogamicin against acute myeloid leukemia (AML) colony forming cells (CFC) [abstract]. Blood (ASH Annual Meeting Abstracts) 2008;112(11):912–913. [Google Scholar]
- 84.Linenberger ML. CD33-directed therapy with gemtuzumab ozogamicin in acute myeloid leukemia: progress in understanding cytotoxicity and potential mechanisms of drug resistance. Leukemia. 2005;19(2):176–182. doi: 10.1038/sj.leu.2403598. [DOI] [PubMed] [Google Scholar]
- 85.Sullivan N, Lyne L. Sensitivity of fibroblasts derived from ataxia-telangiectasia patients to calicheamicin gamma 1I. Mutat Res. 1990;245(3):171–175. doi: 10.1016/0165-7992(90)90046-m. [DOI] [PubMed] [Google Scholar]
- 86.van Duijn-Goedhart A, Zdzienicka MZ, Sankaranarayanan K, van Buul PP. Differential responses of Chinese hamster mutagen sensitive cell lines to low and high concentrations of calicheamicin and neocarzinostatin. Mutat Res. 2000;471(1):95–105. doi: 10.1016/s1383-5718(00)00122-4. [DOI] [PubMed] [Google Scholar]
- 87.Goemans BF, Zwaan CM, Vijverberg SJ, et al. Large interindividual differences in cellular sensitivity to calicheamicin may influence gemtuzumab ozogamicin response in acute myeloid leukemia. Leukemia. 2008;22(12):2284–2285. doi: 10.1038/leu.2008.147. [DOI] [PubMed] [Google Scholar]
- 88.Walter RB, Raden BW, Hong TC, Flowers DA, Bernstein ID, Linenberger ML. Multidrug resistance protein attenuates gemtuzumab ozogamicin-induced cytotoxicity in acute myeloid leukemia cells. Blood. 2003;102(4):1466–1473. doi: 10.1182/blood-2003-02-0396. [DOI] [PubMed] [Google Scholar]
- 89.Taksin AL, Legrand O, Raffoux E, et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia. 2007;21(1):66–71. doi: 10.1038/sj.leu.2404434. [DOI] [PubMed] [Google Scholar]
- 90.Walter RB, Gooley TA, van der Velden VH, et al. CD33 expression and P-glycoprotein-mediated drug efflux inversely correlate and predict clinical outcome in patients with acute myeloid leukemia treated with gemtuzumab ozogamicin monotherapy. Blood. 2007;109(10):4168–4170. doi: 10.1182/blood-2006-09-047399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Walter RB, Raden BW, Kamikura DM, Cooper JA, Bernstein ID. Influence of CD33 expression levels and ITIM-dependent internalization on gemtuzumab ozogamicin-induced cytotoxicity. Blood. 2005;105(3):1295–1302. doi: 10.1182/blood-2004-07-2784. [DOI] [PubMed] [Google Scholar]
- 92.Jager E, van der Velden VHJ, te Marvelde JG, Walter RB, Agur Z, Vainstein V. Targeted drug delivery by gemtuzumab ozogamicin: mechanism-based mathematical model for treatment strategy improvement and therapy individualization. PLoS One. 2011;6(9):e24265. doi: 10.1371/journal.pone.0024265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jedema I, Barge RM, van der Velden VH, et al. Internalization and cell cycle-dependent killing of leukemic cells by gemtuzumab ozogamicin: rationale for efficacy in CD33-negative malignancies with endocytic capacity. Leukemia. 2004;18(2):316–325. doi: 10.1038/sj.leu.2403205. [DOI] [PubMed] [Google Scholar]
- 94.Sievers EL, Appelbaum FR, Spielberger RT, et al. Selective ablation of acute myeloid leukemia using antibody-targeted chemotherapy: a phase I study of an anti–CD33 calicheamicin immunoconjugate. Blood. 1999;93(11):3678–3684. [PubMed] [Google Scholar]
- 95.Sievers EL, Larson RA, Stadtmauer EA, et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J Clin Oncol. 2001;19(13):3244–3254. doi: 10.1200/JCO.2001.19.13.3244. [DOI] [PubMed] [Google Scholar]
- 96.Bross PF, Beitz J, Chen G, et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res. 2001;7(6):1490–1496. [PubMed] [Google Scholar]
- 97.Larson RA, Sievers EL, Stadtmauer EA, et al. Final report of the efficacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33-positive acute myeloid leukemia in first recurrence. Cancer. 2005;104(7):1442–1452. doi: 10.1002/cncr.21326. [DOI] [PubMed] [Google Scholar]
- 98.Giles F, Estey E, O'Brien S. Gemtuzumab ozogamicin in the treatment of acute myeloid leukemia. Cancer. 2003;98(10):2095–2104. doi: 10.1002/cncr.11791. [DOI] [PubMed] [Google Scholar]
- 99.Fenton C, Perry CM. Gemtuzumab ozogamicin: a review of its use in acute myeloid leukaemia. Drugs. 2005;65(16):2405–2427. doi: 10.2165/00003495-200565160-00014. [DOI] [PubMed] [Google Scholar]
- 100.Tsimberidou AM, Giles FJ, Estey E, O'Brien S, Keating MJ, Kantarjian HM. The role of gemtuzumab ozogamicin in acute leukaemia therapy. Br J Haematol. 2006;132(4):398–409. doi: 10.1111/j.1365-2141.2005.05872.x. [DOI] [PubMed] [Google Scholar]
- 101.Abutalib SA, Tallman MS. Monoclonal antibodies for the treatment of acute myeloid leukemia. Curr Pharm Biotechnol. 2006;7(5):343–369. doi: 10.2174/138920106778521578. [DOI] [PubMed] [Google Scholar]
- 102.Pagano L, Fianchi L, Caira M, Rutella S, Leone G. The role of Gemtuzumab Ozogamicin in the treatment of acute myeloid leukemia patients. Oncogene. 2007;26(25):3679–3690. doi: 10.1038/sj.onc.1210364. [DOI] [PubMed] [Google Scholar]
- 103.Stasi R, Evangelista ML, Buccisano F, Venditti A, Amadori S. Gemtuzumab ozogamicin in the treatment of acute myeloid leukemia. Cancer Treat Rev. 2008;34(1):49–60. doi: 10.1016/j.ctrv.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 104.Caron PC, Jurcic JG, Scott AM, et al. A phase 1B trial of humanized monoclonal antibody M195 (anti–CD33) in myeloid leukemia: specific targeting without immunogenicity. Blood. 1994;83(7):1760–1768. [PubMed] [Google Scholar]
- 105.van der Velden VHJ, te Marvelde JG, Hoogeveen PG, et al. Targeting of the CD33-calicheamicin immunoconjugate Mylotarg (CMA-676) in acute myeloid leukemia: in vivo and in vitro saturation and internalization by leukemic and normal myeloid cells. Blood. 2001;97(10):3197–3204. doi: 10.1182/blood.v97.10.3197. [DOI] [PubMed] [Google Scholar]
- 106.Farhat H, Reman O, Raffoux E, et al. Fractionated doses of gemtuzumab ozogamicin with escalated doses of daunorubicin and cytarabine as first acute myeloid leukemia salvage in patients aged 50-70-year old: a phase 1/2 study of the Acute Leukemia French Association. Am J Hematol. 2012;87(1):62–65. doi: 10.1002/ajh.22201. [DOI] [PubMed] [Google Scholar]
- 107.Lo-Coco F, Cimino G, Breccia M, et al. Gemtuzumab ozogamicin (Mylotarg) as a single agent for molecularly relapsed acute promyelocytic leukemia. Blood. 2004;104(7):1995–1999. doi: 10.1182/blood-2004-04-1550. [DOI] [PubMed] [Google Scholar]
- 108.Breccia M, Cimino G, Diverio D, Gentilini F, Mandelli F, Lo Coco F. Sustained molecular remission after low dose gemtuzumab-ozogamicin in elderly patients with advanced acute promyelocytic leukemia. Haematologica. 2007;92(9):1273–1274. doi: 10.3324/haematol.11329. [DOI] [PubMed] [Google Scholar]
- 109.Takeshita A, Ono T, Kojima Y, et al. Efficacy of gemtuzumab ozogamicin (GO) monotherapy on relapsed/refractory acute promyelocytic leukemia (APL) [abstract]. Blood (ASH Annual Meeting Abstracts) 2011;118(21):665–666. [Google Scholar]
- 110.Estey EH, Giles FJ, Beran M, et al. Experience with gemtuzumab ozogamycin (“mylotarg”) and all-trans retinoic acid in untreated acute promyelocytic leukemia. Blood. 2002;99(11):4222–4224. doi: 10.1182/blood-2001-12-0174. [DOI] [PubMed] [Google Scholar]
- 111.Ravandi F, Estey E, Jones D, et al. Effective treatment of acute promyelocytic leukemia with all-trans-retinoic acid, arsenic trioxide, and gemtuzumab ozogamicin. J Clin Oncol. 2009;27(4):504–510. doi: 10.1200/JCO.2008.18.6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Burnett AK, Hills RK, Milligan D, et al. Identification of patients with acute myeloblastic leukaemia who benefit from the addition of gemtuzumab ozogamicin: results of the MRC AML15 trial. J Clin Oncol. 2011;29(4):369–377. doi: 10.1200/JCO.2010.31.4310. [DOI] [PubMed] [Google Scholar]
- 113.Castaigne S, Pautas C, Terre C, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet. 2012;379(9825):1508–1516. doi: 10.1016/S0140-6736(12)60485-1. [DOI] [PubMed] [Google Scholar]
- 114.Delaunay J, Recher C, Pigneux A, et al. Addition of gemtuzumab ozogamycin to chemotherapy improves event-free survival but not overall survival of AML patients with intermediate cytogenetics not eligible for allogeneic transplantation: results of the GOELAMS AML 2006 IR study [abstract]. Blood. (ASH Annual Meeting Abstracts) 2011;118(21):37–38. [Google Scholar]
- 115.Burnett AK, Hill RK, Hunter AE, et al. The addition of gemtuzumab ozogamicin to induction chemotherapy improves survival in older patients with acute myeloid leukemia. J Clin Oncol. 2012 doi: 10.1200/JCO.2012.42.2964. in press. [DOI] [PubMed] [Google Scholar]
- 116.Petersdorf S, Kopecky K, Stuart RK, et al. Preliminary results of Southwest Oncology Group Study S0106: an international intergroup phase 3 randomized trial comparing the addition of gemtuzumab ozogamicin to standard induction therapy versus standard induction therapy followed by a second randomization to post-consolidation gemtuzumab ozogamicin versus no additional therapy for previously untreated acute myeloid leukemia [abstract]. Blood (ASH Annual Meeting Abstracts) 2009;114(22):326–327. [Google Scholar]
- 117. [Accessed January 23, 2012]. http://www.swogstat.org/ROS/ROSBooks/Spring%202010/Leukemia.pdf.
- 118. [Accessed January 23, 2012]. http://media.pfizer.com/files/products/mylotarg_hcp_letter.pdf.
- 119.Lo Coco F, Ammatuna E, Noguera N. Treatment of acute promyelocytic leukemia with gemtuzumab ozogamicin. Clin Adv Hematol Oncol. 2006;4(1):57–62. [PubMed] [Google Scholar]
- 120.Breccia M, Lo-Coco F. Gemtuzumab ozogamicin for the treatment of acute promyelocytic leukemia: mechanisms of action and resistance, safety and efficacy. Expert Opin Biol Ther. 2011;11(2):225–234. doi: 10.1517/14712598.2011.543895. [DOI] [PubMed] [Google Scholar]
- 121.Jurcic JG, DeBlasio T, Dumont L, Yao TJ, Scheinberg DA. Molecular remission induction with retinoic acid and anti–CD33 monoclonal antibody HuM195 in acute promyelocytic leukemia. Clin Cancer Res. 2000;6(2):372–380. [PubMed] [Google Scholar]
- 122.Jilani I, Estey E, Huh Y, et al. Differences in CD33 intensity between various myeloid neoplasms. Am J Clin Pathol. 2002;118(4):560–566. doi: 10.1309/1WMW-CMXX-4WN4-T55U. [DOI] [PubMed] [Google Scholar]
- 123.Hauswirth AW, Florian S, Printz D, et al. Expression of the target receptor CD33 in CD34+/CD38-/CD123+ AML stem cells. Eur J Clin Invest. 2007;37(1):73–82. doi: 10.1111/j.1365-2362.2007.01746.x. [DOI] [PubMed] [Google Scholar]
- 124.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5(4):275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 125.Jawad M, Seedhouse C, Mony U, Grundy M, Russell NH, Pallis M. Analysis of factors that affect in vitro chemosensitivity of leukaemic stem and progenitor cells to gemtuzumab ozogamicin (Mylotarg) in acute myeloid leukaemia. Leukemia. 2010;24(1):74–80. doi: 10.1038/leu.2009.199. [DOI] [PubMed] [Google Scholar]
- 126.Pollard JA, Alonzo TA, Loken M, et al. Correlation of CD33 expression level with disease characteristics and response to gemtuzumab ozogamicin containing chemotherapy in childhood AML. Blood. 2012;119(16):3705–3711. doi: 10.1182/blood-2011-12-398370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lamba J, Mortland LJ, Mitra A, et al. Clinical significance of CD33 non-synonymous single nucleotide polymorphisms (SNPs) in pediatric patients with acute myeloid leukemia treated with gemtuzumab ozogamicin-containing chemotherapy [abstract]. Blood (ASH Annual Meeting Abstracts) 2011;118(21):1489. doi: 10.1158/1078-0432.CCR-12-3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.van der Velden VHJ, Boeckx N, Jedema I, et al. High CD33-antigen loads in peripheral blood limit the efficacy of gemtuzumab ozogamicin (Mylotarg) treatment in acute myeloid leukemia patients. Leukemia. 2004;18(5):983–988. doi: 10.1038/sj.leu.2403350. [DOI] [PubMed] [Google Scholar]
- 129.Jordan CT, Guzman ML. Mechanisms controlling pathogenesis and survival of leukemic stem cells. Oncogene. 2004;23(43):7178–7187. doi: 10.1038/sj.onc.1207935. [DOI] [PubMed] [Google Scholar]
- 130.Wu AM, Senter PD. Arming antibodies: prospects and challenges for immunoconjugates. Nat Biotechnol. 2005;23(9):1137–1146. doi: 10.1038/nbt1141. [DOI] [PubMed] [Google Scholar]