

Abstract
Gastrulation movements place endodermal precursors, mesodermal precursors and primordial germ cells (PGCs) into the interior of the embryo. Somatic cell gastrulation movements are regulated by transcription factors that also control cell fate, coupling cell identity and position. By contrast, PGCs in many species are transcriptionally quiescent, suggesting that they might use alternative gastrulation strategies. Here, we show that C. elegans PGCs internalize by attaching to internal endodermal cells, which undergo morphogenetic movements that pull the PGCs into the embryo. We show that PGCs enrich HMR-1/E-cadherin at their surfaces to stick to endoderm. HMR-1 expression in PGCs is necessary and sufficient to ensure internalization, suggesting that HMR-1 can promote PGC-endoderm adhesion through a mechanism other than homotypic trans interactions between the two cell groups. Finally, we demonstrate that the hmr-1 3′ untranslated region promotes increased HMR-1 translation in PGCs. Our findings reveal that quiescent PGCs employ a post-transcriptionally regulated hitchhiking mechanism to internalize during gastrulation, and demonstrate a morphogenetic role for the conserved association of PGCs with the endoderm.
Keywords: Gastrulation, Primordial germ cell (PGC), Endoderm, Cadherin, 3′ UTR, Adhesion
INTRODUCTION
Gastrulation is a crucial morphogenetic event when cells that will give rise to internal tissues and organs move to interior regions of the embryo. Although both somatic cells (endodermal and mesodermal precursors) and primordial germ cells (PGCs) internalize during gastrulation, most studies of gastrulation have focused on somatic cells because they comprise the vast majority of internalizing cells. Somatic cells and PGCs are set apart during the initial stages of embryogenesis and follow distinct developmental trajectories. Soon after somatic cells are born, they activate specific transcription factors that begin to restrict their developmental potential. Cells that acquire endodermal or mesodermal fates then move into the interior of the embryo during gastrulation. Cell fate specification and gastrulation are often coordinated by the same transcription factors, which regulate both cell-identity genes and genes that control gastrulation movements. For example, the C. elegans endodermal precursor E is specified through the combined action of GATA transcription factors END-1 and END-3 (Maduro, 2006). The daughters of E (Ea and Ep) initiate gastrulation when they constrict their apical surfaces and ingress into the interior of the embryo (Nance and Priess, 2002; Lee and Goldstein, 2003). Disrupting function of the end genes prevents the E lineage from producing endoderm and also blocks Ea and Ep ingression (Nance and Priess, 2002; Lee et al., 2006; Owraghi et al., 2010). An analogous coupling between cell fate specification and gastrulation is well documented in vertebrate mesendodermal cells (for example, by Nodal signaling) and fly mesodermal cells (by Snail and Twist) (Leptin, 2005; Heisenberg and Solnica-Krezel, 2008), suggesting that this is a conserved strategy that embryos use to place cells of the appropriate fate in the proper position within the embryo.
In contrast to somatic cells, PGCs in many animals (including C. elegans, Drosophila, ascidians, and frogs) repress transcription during early embryogenesis to remain undifferentiated (Seydoux and Braun, 2006; Venkatarama et al., 2010; Shirae-Kurabayashi et al., 2011). For example, C. elegans germline precursor cells and PGCs use several different mechanisms, including inhibition of proteins needed for transcription and repressive chromatin modifications, to remain largely or completely transcriptionally silent during early embryogenesis; activating transcription in these cells induces somatic differentiation programs (Mello et al., 1996; Seydoux et al., 1996; Schaner et al., 2003; Guven-Ozkan et al., 2008). Therefore, unless PGCs use post-transcriptional mechanisms to trigger the same gastrulation program used by somatic cells, PGCs are likely to use distinct internalization mechanisms. Although elegant genetic and live-imaging studies have identified genes required for the guidance and migration of PGCs to the gonad after gastrulation is complete (Richardson and Lehmann, 2010), the molecular mechanisms that trigger and execute PGC internalization during gastrulation are largely unexplored.
The C. elegans embryo contains only two PGCs, called Z2 and Z3, which internalize during the middle stages of gastrulation by ingressing from the ventral surface (Nance and Priess, 2002). Here, we investigate the physical and molecular mechanisms that promote the internalization of C. elegans PGCs during gastrulation. We show that PGC ingression relies on regulated adhesive interactions with internal endodermal cells, which pull the PGCs into the embryo. PGC internalization and adhesion to endoderm is mediated by HMR-1/E-cadherin, which is post-transcriptionally upregulated in PGCs and whose expression specifically in PGCs is sufficient to promote their internalization. Our findings define a post-transcriptional gastrulation strategy that is used by quiescent PGCs, and reveal a role for the conserved association between endoderm and PGCs in promoting PGC internalization.
MATERIALS AND METHODS
Strains
All hmr-1 mutants were hmr-1(zu389), which contains a nonsense mutation prior to sequences encoding the transmembrane domain (Broadbent and Pettitt, 2002). All unc-119 mutants were unc-119(ed3). The following strains were used (all FT strains were created in this study): N2 (wild type); CX2993, sax-7(ky146); kyIs4 (Zallen et al., 1999); FT233, hmr-1; xnEx42 [hmr-1(+), dpy-7P::RFP, end-1P::GFP]; FT598, xnSi1 [mex-5P::GFP-PHPLC∂::nos-2UTR]; xnIs91 [end-1P::mCherry-PHPLC∂]; FT669, xnIs91; pie-1P::GFP-PAR-2; FT696, hmr-1; xnEx42; xnSi1; xnIs99 [end-1P::mCherry-PHPLC∂]; FT741, xnSi6 [mex-5P::HMR-1-GFP::hmr-1UTR]; unc-119(ed3); FT774, xnSi7 [mex-5P::GFP-PHPLC∂::hmr-1UTR]; zuIs244 [nmy-2P::PGL-RFP]; unc-119; FT776, hmr-1; xnEx42; xnSi6; FT834, xnSi13 [mex-5P::GFP-PHPLC∂::tbb-2UTR]; zuIs244; unc-119; FT850, xnSi18 [mex-5P::HMR-1-GFP-ZF1::hmr-1UTR]; unc-119; FT853, zuIs60 [pie-1P::secGFP]; zuIs244; ltIs44 [pie-1P::mCherry-PHPLC∂]; FT925, qtIs12 [pal-1P::YFP]; xnIs91; FT1040, hmr-1; xnIs375 [hmr-1P::HMR-1-ZF1-GFP::hmr-1UTR]; and MS1248, end-1(ok558) end-3(ok1448); irEx568 [end-1(+), end-3(+), sur-5::RFP] (Owraghi et al., 2010).
Transgene construction
mex-5P::HMR-1-GFP::hmr-1UTR, mex-5P::HMR-1-GFP-ZF1::hmr-1UTR, mex-5P::GFP-PHPLC∂::nos-2UTR, mex-5P::GFP-PHPLC∂::hmr-1UTR and mex-5P::GFP-PHPLC∂::tbb-2UTR were created using Multisite Gateway (Invitrogen), the pCFJ150 destination vector (Frokjaer-Jensen et al., 2008) and the following entry clones: 5′, pJA252 (mex-5P) and pJA254 (mex-5P::GFP) (Zeiser et al., 2011); middle, pJN527 (HMR-1-GFP), pDC21 (HMR-1-GFP-ZF1) and pDC08 (PHPLC∂); 3′, pDC10 (nos-2UTR), pCM1.36 (tbb-2UTR) (Merritt et al., 2008) and pJN522 (hmr-1UTR).
Gateway entry clones were constructed as follows. pJN527 (HMR-1-GFP): hmr-1a cDNA was cloned into pDONR221 (Invitrogen) and gfp was inserted into a BsiWI site engineered before the stop codon. pDC21 (HMR-1-GFP-ZF1): the pie-1 zf1 domain (Nance et al., 2003) was cloned into the BsiWI site of pJN527. pDC08 (PHPLC∂): rat PHPLC∂1 domain (Audhya et al., 2005) was cloned into pDONR221. pJN522 (hmr-1UTR): 700 bp of sequence downstream of the hmr-1 stop codon was cloned into pDONR P2R-P3 (Invitrogen). pDC10 (nos-2UTR): 844 bp of sequence downstream of the nos-2 stop codon was cloned into pDONR P2R-P3.
Non-Gateway plasmids were constructed as follows. hmr-1P::HMR-1-GFP-ZF1 was created from hmr-1P::HMR-1-GFP::unc-54UTR (Achilleos et al., 2010) digested with ApaI to remove gfp and the unc-54 3′ UTR. gfp, zf1 and the hmr-1 3′ UTR were inserted using Gibson end-joining (Gibson et al., 2009). end-1P::mCherry-PHPLC∂ was created by cloning the end-1 promoter from end-1P::GFP (Nance et al., 2003) into mCherry plasmid pGC326 (a gift from E. J. Hubbard, Skirball Institute, New York, USA) digested with HindIII and AgeI, then inserting PCR-amplified rat PLC1∂1 PH domain (Audhya et al., 2005) into the AgeI site. pie-1::sec-GFP was created by digesting pie-1::GFP::ACTIN plasmid pJH4.64 (a gift from Geraldine Seydoux, Johns Hopkins School of Medicine, Baltimore, MD, USA) with BamHI to excise GFP::ACTIN and inserting secreted GFP (sec-GFP) from plasmid pPD95.85 (a gift from Andy Fire, Stanford School of Medicine, CA, USA). unc-119 from plasmid pJN254 (Nance et al., 2003) was cloned into the NotI site.
Worm transformation
Gateway plasmids were integrated using MosSCI and the EG4322 strain (Frokjaer-Jensen et al., 2008). xnEx42 was created by microinjecting a genomic clone containing hmr-1a (pW02-21) (Broadbent and Pettitt, 2002), with end-1P::GFP and dpy-7P::RFP co-transformation markers. All other transgene insertions or arrays were created by microparticle bombardment (Praitis et al., 2001).
DIC microscopy and ingressions
Four-dimensional DIC movies were acquired using a Zeiss AxioImager and 40× 1.3NA or 63× 1.4 NA objective. Z-stacks (1 μm) were captured every minute and embryos were lineaged. Ingressions were scored when a cell moved permanently into the interior of the embryo. Representative cells in somatic lineages were chosen for analysis [E, Ea; MSw, MSpppp; MSc, MSpappp; D, Dpp; C, Cpppa]. PGC ingressions were scored only if both Z2 and Z3 sank into the embryo. Ingression times were normalized to 20°C cell division timings (Sulston et al., 1983) using the MSpp to MSppp interval. In C-irradiation experiments, dorsal movements of endodermal cells and PGCs were measured by determining the dorsal distance traveled by Epla and Z2 nuclei. For statistical comparisons, PGC ingression was scored as succeeding if it occurred before 210 minutes (after the first embryonic cleavage) and as failing if it had not.
Laser irradiation
Laser irradiation was performed on a Zeiss AxioImager using a 100× 1.3 NA objective and MicroPoint laser with Coumarin dye cell. Embryos were mounted on 4% agarose and the targeted nucleus was pulse-irradiated until refractile debris appeared. Founder cells were laser-irradiated in each targeted lineage, except for the Cxp lineage where Cap and Cpp were targeted. Irradiated embryos were analyzed only if irradiated cell(s) ceased dividing and cell divisions in non-targeted lineages were normal.
Fluorescence microscopy and analysis of cell adhesion and movements
Fluorescence time-lapse movies of embryos expressing endoderm and PGC surface markers (strain FT669) were acquired using a Leica SP5 confocal microscope, 488 nm and 594 nm lasers, and 63× 1.3 NA water-immersion objective. Embryos were suspended from the coverslip in water. Dorsal shift was measured by calculating the distance from the dorsal-most endodermal or Z2 cell surface to the eggshell. To analyze cell contacts and separations in wild-type embryos, strain FT853 was used. Separations between PGCs and endoderm were examined using strain FT598 (wild type) and strain FT696 (hmr-1).
RNAi
hmr-1 RNAi was performed by the feeding method as described (Totong et al., 2007), using empty vector pPD129.36 in HT115 as a negative control.
Immunostaining
Embryos were fixed, stained and imaged as described (Anderson et al., 2008). The following antibodies were used: mouse OIC1D4 ‘P granules’ 1:5 (DSHB); rabbit anti-HMR-1 1:50 (Costa et al., 1998); rabbit anti-SAX-7 1:200 (Chen et al., 2001); and rabbit anti-GFP 1:2000 (Abcam).
FRAP and image analysis
Images of embryos expressing membrane and P-granule markers (strains FT774 and FT834) were acquired using a Zeiss AxioImager and 63× 1.4 NA objective. Twenty-four-cell stage embryos were photobleached and images were taken before, immediately after and 3 hours after photobleaching. GFP expression levels at a PGC-D cell contact and control somatic (AB-AB cell) contact were analyzed. Intensities were measured by averaging the maximum intensities along three membranes using ImageJ (NIH), and background autofluorescence was subtracted.
In situ hybridization
In situ hybridization was performed as described previously (Seydoux and Fire, 1995), except single-stranded RNA probes were used and hybridization was performed at 42°C. Slides were allowed to develop until embryos hybridized with nos-2 probe showed staining whereas those incubated with hmr-1 sense probe did not. Specificity of the hmr-1 antisense probe was determined by the presence of specific staining of the adult gonad and oocytes, which were not stained by hmr-1 sense probe. DNA templates for in situ hybridization probes were generated by PCR using genomic DNA and the following primers: hmr-1 antisense, CATCGGAACGTATCGTTGGTC, T7-CACCTCCTTCCACACCATAC; hmr-1 sense, T7-CATCGGAACGTATCGTTGGTC, CACCTCCTTCCACACCATAC; nos-2 antisense, CCACCTGCCGAATATCTTC, T7-GTCTCGGTGTGATTCATTTCAC.
RESULTS
Movements of PGCs and surrounding cells during gastrulation
To determine whether PGC gastrulation movements are active or whether PGC internalization requires forces provided by other cells, we asked if the corpse of a laser-irradiated PGC could internalize. Laser irradiation of somatic cells or the PGC P4, parent of Z2 and Z3, prevented their further division. As noted previously (Junkersdorf and Schierenberg, 1992; Nance and Priess, 2002), most laser-irradiated somatic cells failed to internalize (Fig. 1E). By contrast, the corpse of an equivalently irradiated P4 cell always internalized at the time when its daughters Z2 and Z3 would normally do so (Fig. 1E,F; supplementary material Movie S1), suggesting that PGC ingression is assisted by other cells.
Fig. 1.

PGC ingression and interacting cells. Embryos (~50 μm in length) are oriented anterior towards the left and are shown from the indicated perspective. (A-C) Time-lapse stills of a wild-type embryo. Time is in minutes after first cleavage. Cell lineages are color-coded (PGCs, cyan; MSw, red; MSc, magenta; E, yellow; D, orange; Cxp, green). Ingressing cells in deeper focal planes are shown in more transparent colors, and out-of-focus internalized cells are indicated by dashed borders. (A) An ~100-cell stage embryo, just before PGC ingression; asterisks mark the two PGCs. (B) An ~190-cell stage embryo, PGC ingression. (C) ~200-cell stage, ingression of PGCs and cells in MS, D, E and Cxp lineages is complete. (D) Quantification of ingression times of the indicated lineage in wild-type embryos (n=10). The median (line in box), 25th and 75th percentiles (box boundary), and s.d. (error bars) are shown. (E) Ingression of cells in indicated lineages following laser irradiation (E wild type, n=17; E irradiated, n=21; D wild type, n=10; D irradiated n=10; MS wild type, n=10; MS irradiated, n=10; P4 wild type, n=10; P4 irradiated, n=11). In this and subsequent figures, circles indicate cell ingression times in individual embryos; circles above the dashed line indicate that cells failed to ingress by 210 minutes, when movies were stopped. Asterisks indicate a significant difference in whether or not PGCs ingressed relative to unirradiated controls (Fisher's exact test, *P<0.05, **P<0.01; NS, not significant). (F) PGC ingression following P4 laser irradiation. The position of the internalized P4 corpse is indicated with dashed cyan outlines.
To identify candidate force-generating cells that could push or pull the PGCs into the embryo, we captured three-dimensional time-lapse (4D) differential interference contrast (DIC) movies of gastrulating embryos (Fig. 1A-C; supplementary material Movie S2). At the time of their ingression, PGCs were surrounded on the ventral surface by a ring of mesodermal cells descended from the MS and D blastomeres, and sat atop internal endodermal cells descended from the E blastomere (Fig. 1A). The four D descendants flanked the lateral and posterior sides of the PGCs and ingressed just after the PGCs (Fig. 1A,B,D, orange cells). A subset of MS descendants called wishbone cells (‘MSw’) also contacted the lateral sides of the PGCs, and ingressed just before the PGCs (Fig. 1A,D, red cells). The remaining MS descendants, named central cells (‘MSc’), were positioned immediately anterior to the PGCs and ingressed 30 minutes later (Fig. 1A,D, magenta cells). In addition to these cells that contacted the PGCs, the mesodermal descendants of the C blastomere (Cxp lineage) migrated towards the site of PGC ingression (Fig. 1B, green cells). Based on these observations, we considered the possibility that movements of the D, MS or Cxp descendants could push the PGCs into the embryo, or that the internal E descendants could pull the PGCs into the embryo.
PGC ingression requires endodermal cells
To determine whether PGC ingression requires morphogenetic movements of the D, MS, Cxp or E blastomere descendants, we laser-irradiated each of these blastomeres individually to prevent their internalization, then captured 4D DIC movies of gastrulation. In each experiment, we analyzed the ingression time of PGCs as well as of mesodermal cells within the D and MS lineages, and endodermal cells within the E lineage. We captured movies up to the 210-minute stage, when all of the analyzed cells in unirradiated embryos had completed their ingression (Fig. 1C,D). PGCs that remained on the surface at the 210-minute stage were scored as failing to ingress.
Whereas irradiating the D, MS or Cxp blastomeres did not affect PGC ingression (Fig. 2A,D; supplementary material Movie S3), irradiating the E blastomere prevented the PGCs from ingressing in most embryos (Fig. 2B,D; supplementary material Movie S4). Importantly, irradiating the E blastomere did not disrupt gastrulation broadly, as mesodermal cells ingressed normally after E irradiation (supplementary material Fig. S1). To prevent endodermal cell ingression using a less invasive approach, we analyzed end-1 end-3 double mutant embryos, in which the E cell acquires a mesectodermal fate (Owraghi et al., 2010) and the transformed E daughters remain on the surface or ingress at a later stage (supplementary material Fig. S1C). Similar to embryos with a laser-irradiated E blastomere, most PGCs failed to ingress in end-1 end-3 double mutant embryos (Fig. 2C,D), whereas mesodermal cells ingressed normally (supplementary material Fig. S1). Taken together, these experiments indicate that endodermal cells, which are positioned in the interior of the embryo adjacent to the PGCs, are required specifically for PGC ingression.
Fig. 2.
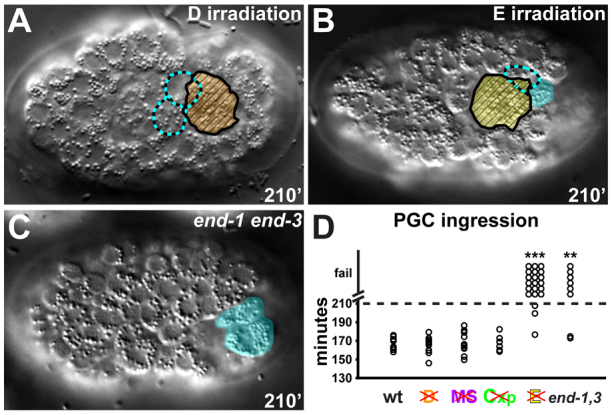
Requirement of interacting cells for PGC ingression. (A-C) Embryos (~50 μm in length) are oriented anterior towards the left and are shown from the ventral perspective. PGC ingression following D irradiation (A), E irradiation (B) and in end-1 end-3 mutant embryos (C). PGCs, cyan; E, yellow; D, orange. The position of internalized PGCs is indicated with dashed cyan outlines, while PGCs that failed to ingress are shaded in cyan. The corpse of the irradiated cell remaining on the surface is indicated with hatched fill. (D) Ingression of PGCs in laser-irradiated and mutant embryos (wild-type, n=10; D irradiation, n=12; MS irradiation, n=11; Cxp irradiation, n=6; E irradiation, n=21; end-1 end-3, n=8). Asterisks indicate a significant difference in whether PGCs ingressed relative to unirradiated controls (Fisher's exact test, **P<0.01, ***P<0.001).
Dorsal movements of endodermal cells are required for PGC ingression
Given that ingression of the endodermal cells Ea and Ep is complete 1 hour before PGC ingression begins (Fig. 1D), Ea and Ep do not pull the PGCs with them as they ingress. Therefore, we considered the possibility that the internal descendants of Ea and Ep might undergo a subsequent morphogenetic movement that pulls the PGCs into the interior of the embryo. To observe movements of endodermal cells and PGCs simultaneously, and to visualize the contacts that these cells make with one another, we created a strain expressing plasma membrane markers that label endodermal cells and PGCs in different colors. In fluorescence time-lapse movies, we observed that PGCs and endodermal cells associated continuously throughout the period of gastrulation [six out of six embryos] (Fig. 3A-C). As PGCs ingressed from the ventral surface into the interior, the internal endodermal cells moved dorsally a similar distance (Fig. 3F). Endodermal cells also partially enveloped the internalized PGCs by wrapping around their surfaces (Fig. 3D,E; Fig. 4E). These observations suggest that dorsal movements of internal endodermal cells might pull the attached PGCs into the interior of the embryo.
Fig. 3.

PGC ingression and dorsal movements of endodermal cells. Embryos (~50 μm in length) are oriented anterior towards the left and are shown from the lateral perspective. (A-E) Time-lapse stills of a wild-type embryo expressing PGC-specific (pie-1P::GFP-PAR-2, cyan) and endoderm-specific (end-1P::mCherry-PHPLC∂, yellow) cell surface markers. Time sequence begins just after the birth of Z2 and Z3 (0 minutes), and continues through the period of PGC ingression (A-C). Broken lines mark the eggshell. (B,C) During PGC ingression. (D,E) After PGC ingression, endodermal cells begin to wrap around PGCs. (F) Quantification of dorsal movements of PGCs and endoderm during PGC ingression; error bars indicate s.d., n=6 embryos. (G,H) Wild-type embryo expressing cytoplasmic YFP in C and D lineages (pal-1P::YFP, green) and endoderm-specific mCherry (end-1P::mCherry-PHPLC∂, magenta) localized to the plasma membrane (G, eight E cells; H, 16 E cells). Double-headed arrow indicates the thickness of the dorsal C-cell layer. (I) Quantification of PGC and endoderm dorsal movements following C irradiation (n=6 wild-type embryos, n=10 C-irradiated embryos); primary data are shown in supplementary material Fig. S2. Asterisks indicate a significant difference in distance traveled (***P<0.001, two-tailed Student's t-test).
Fig. 4.
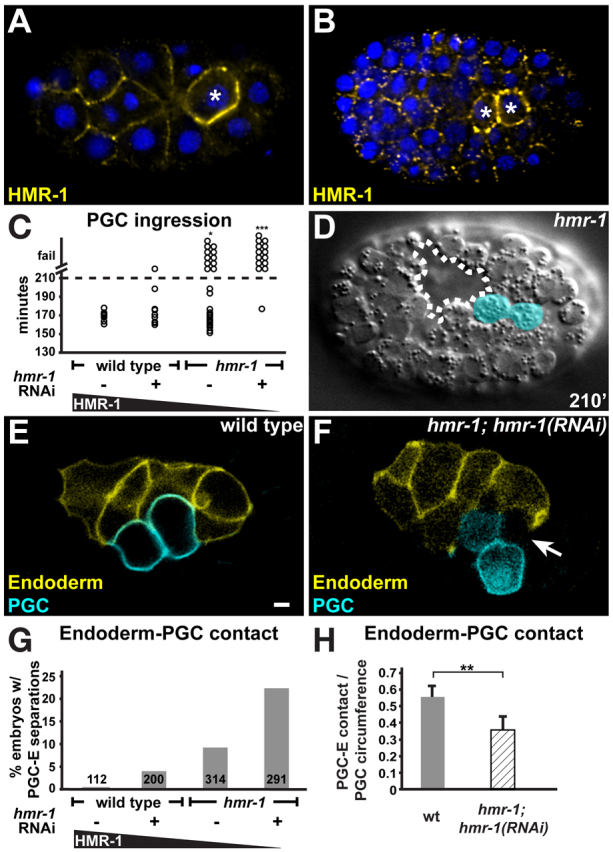
HMR-1 and PGC ingression. Embryos (~50 μm in length) in A,B,D are oriented anterior towards the left and are shown from the ventral perspective. (A,B) Forty-four-cell stage (A) and ~200-cell stage (B) embryos immunostained for HMR-1 (yellow). Nuclei, blue; PGCs, asterisks. (C) PGC ingression in wild-type embryos fed on empty vector RNAi bacteria (n=10), in hmr-1(RNAi) embryos (n=9), in hmr-1; hmr-1Ex embryos (n=29) and in hmr-1; hmr-1Ex hmr-1(RNAi) embryos (n=14). Asterisks indicate a significant difference in whether PGCs ingressed (*P<0.05, ***P<0.001) relative to wild-type controls fed on empty vector RNAi (Fisher's exact test). (D) hmr-1; hmr-1Ex embryo showing superficial PGCs (cyan) at 210 minutes. Dashed line marks the gastrulation cleft. (E,F) Wild-type (E) or hmr-1; hmr-1Ex hmr-1(RNAi) (F) embryos expressing endoderm-specific (end-1P::mCherry-PHPLC∂) and PGC-specific (mex-5P::GFP-PHPLC∂::nos-2UTR) cell surface markers during the stage when PGCs ingress; embryos are shown from the lateral perspective. A gap (arrow) between endodermal cell and PGCs can be seen in hmr-1; hmr-1Ex hmr-1(RNAi) embryos. Scale bar: 2.5 μm. (G) Frequency of spaces detected between endoderm and PGC in wild-type, hmr-1(RNAi), hmr-1; hmr-1Ex and hmr-1; hmr-1Ex hmr-1(RNAi) embryos. Number of embryos examined is indicated. (H) Extent of interface between PGCs and endoderm, represented in fraction of PGC circumference [wild type, n=10; hmr-1; hmr-1Ex hmr-1(RNAi), n=11]. Error bars indicate s.d. Asterisks indicate a significant difference in length of contact between PGCs and endoderm (**P<0.01, two-tailed Student's t-test).
To test whether endodermal cell dorsal movements are required for PGC ingression, we sought a means to prevent the dorsal movement of endodermal cells without killing or transforming the cells. Because embryonic cells are tightly packed and surrounded by a confining eggshell, significant movements of one cell group must be accompanied by compensatory movements of another (Nance and Priess, 2002). Therefore, we reasoned that endodermal cell dorsal movements might require a redistribution of more dorsal cells. During gastrulation, a single superficial layer of dorsal cells, derived predominantly from the C blastomere, covered the endodermal cells and separated them from the eggshell (Fig. 3G,H, dorsal green cells). The C descendants divided within the surface plane of the embryo, reducing the thickness of the layer over time (Fig. 3G,H, compare arrows). To test whether preventing the thinning of the dorsal cell layer could block endodermal cell dorsal movements, we laser-irradiated the C blastomere to block its division. The position of the irradiated C corpse was variable, but in many cases remained on the surface immediately dorsal to the endodermal cells. In comparison with control embryos, the distance that PGCs ingressed and that endodermal cells shifted dorsally was greatly diminished following C laser irradiation (Fig. 3I; supplementary material Fig. S2), and in four out of 10 embryos, PGCs remained on the ventral surface. These observations are consistent with the hypothesis that dorsal movements of endodermal cells, rather than signals provided by these cells, are required for PGC ingression.
In order to be pulled into the embryo by endodermal cells, PGCs would need to adhere tightly to endodermal cell surfaces yet be able to break contacts with adjacent mesodermal cells. To examine contacts that the PGCs make with endodermal cells and mesodermal cells, we created a strain expressing transgenes that separately mark all cell surfaces (membrane-localized mCherry), intercellular separations (secreted GFP) and PGCs (PGL-1-RFP, ‘P-granules’). During gastrulation, we failed to detect secreted GFP between PGCs and endodermal cells, indicating that the surfaces of PGCs and endodermal cells were juxtaposed (supplementary material Fig. S3). By contrast, pockets of secreted GFP accumulated between PGCs and mesodermal cells, as well as between endodermal cells and mesodermal cells (supplementary material Fig. S3). We conclude that PGCs and endodermal cells make more contiguous contacts with each other than either group of cells makes with its mesodermal neighbors.
HMR-1/E-cadherin mediates PGC-endoderm adhesion
We wondered how PGCs adhere to the dorsally shifting endodermal cells during gastrulation. SAX-7/L1CAM and HMR-1/E-cadherin are known to mediate adhesion between C. elegans early embryonic cells; removing SAX-7 and HMR-1 together, but not individually, causes blastomeres to become more rounded and prevents endodermal cells from ingressing (Grana et al., 2010). In early embryos, both SAX-7 and HMR-1 are found at sites of cell-cell contact (Costa et al., 1998; Chen et al., 2001; Grana et al., 2010). We examined SAX-7 and HMR-1 localization during PGC gastrulation to determine whether either protein is found at contacts between the PGCs and endodermal cells. As in early embryos, SAX-7 localized uniformly to all sites of cell-cell contact (supplementary material Fig. S4A). However, HMR-1 was markedly enriched at the surfaces of the PGCs relative to all other cells (Fig. 4A,B). HMR-1 enrichment was first evident in P4 and persisted in Z2 and Z3 throughout gastrulation stages, suggesting that HMR-1 may have a specific function in the PGCs.
sax-7 null mutants are viable and fertile, and PGCs ingressed normally in sax-7 mutant embryos (supplementary material Fig. S4B). Embryos zygotically mutant for hmr-1 arrest at late stages of embryogenesis and gastrulation phenotypes have not been reported, although mutant embryos still contain maternal HMR-1 protein (Costa et al., 1998; Grana et al., 2010). We were able to reduce but not to eliminate both maternal and zygotic HMR-1 using RNAi (supplementary material Fig. S4C,D), and a small number of hmr-1(RNAi) embryos displayed PGC ingression defects (Fig. 4C). To deplete HMR-1 levels further, we rescued hmr-1 mutants with an extrachromosomal array (xnEx42, hereafter hmr-1Ex) that robustly expressed zygotic HMR-1 (supplementary material Fig. S5A) but expressed maternal HMR-1 only at very low levels (due to germ-line transgene silencing; supplementary material Fig. S4E). In 11 out of 29 hmr-1; hmr-1Ex embryos, PGCs failed to ingress (Fig. 4C,D). By treating hmr-1; hmr-1Ex embryos with hmr-1 RNAi, we depleted HMR-1 below the level of detection by immunostaining (supplementary material Fig. S4F), and PGC ingression was blocked (13 of 14 embryos) (Fig. 4C). Endodermal and mesodermal cells ingressed normally in embryos lacking detectable HMR-1 (supplementary material Fig. S4G-I), although we observed later defects in closure of the gastrulation cleft, which normally occurs well after PGC ingression is complete (Fig. 4D, dashed area) (Grana et al., 2010). Therefore, failed PGC ingression in hmr-1 mutants is not caused by a general defect in cell adhesion or gastrulation, suggesting a specific role for HMR-1 in PGC internalization.
To determine whether HMR-1 is required for adhesion between PGCs and endodermal cells, we depleted HMR-1 in embryos expressing endoderm and PGC cell surface markers. We examined embryos just after the stage when PGC ingression is normally complete and determined whether PGCs and endodermal cells were in contact or were visibly separated. In contrast to control embryos, in which PGCs were always partially wrapped by endodermal cells (Fig. 4E), we detected separations between PGCs and endoderm with increasing frequency as HMR-1 levels were reduced (Fig. 4G). When examined at higher resolution by acquiring confocal z-stacks, at least one of the PGCs in hmr-1 embryos was partially or fully detached from endodermal cells and remained on the surface (Fig. 4F, 10 of 11 embryos), and the contact interface between PGCs and endodermal cells was significantly reduced (Fig. 4H, seven out of 11 hmr-1 embryos had a PGC-endoderm interface falling below the 95% confidence interval of the interface in wild-type embryos). Reducing HMR-1 levels did not cause PGCs to dissociate from one another, although in a small number of embryos the PGC-PGC contact interface was reduced (data not shown). We conclude that HMR-1 is needed for PGCs to adhere to endodermal cells during gastrulation.
HMR-1 is required in PGCs but not somatic cells
We used transgenes to determine whether HMR-1 is required in PGCs, somatic cells or both cell types. HMR-1 in the embryo arises from two sources – a maternally inherited pool that is present in both somatic cells and PGCs, and a zygotically expressed pool that is probably found only in somatic cells (because of transcriptional inhibition in PGCs) (Costa et al., 1998; Achilleos et al., 2010). Therefore, we first asked whether maternal HMR-1 is essential for PGC ingression, as this is the likely source of HMR-1 protein in PGCs. To test the requirement for maternal HMR-1, we allowed hmr-1; hmr-1Ex hermaphrodites to self-fertilize and compared PGC ingression in embryos that inherited the hmr-1Ex extrachromosomal array with those that did not. Both classes of embryos contained very low levels of maternal HMR-1 derived from the partially silenced hmr-1Ex transgene (supplementary material Fig. S4E), whereas embryos that inherited hmr-1Ex also expressed zygotic HMR-1 in somatic cells (supplementary material Fig. S5A). Embryos showed equivalent defects in PGC ingression irrespective of whether they inherited hmr-1Ex, suggesting that maternal HMR-1 is required for PGC ingression (Fig. 5A, compare 2nd and 3rd columns). To determine whether supplying maternal HMR-1 is sufficient for PGC ingression, we created a transgene expressing HMR-1-GFP from the maternal mex-5 promoter and the hmr-1 3′ UTR (mex-5P::HMR-1-GFP::hmr-1UTR), and crossed it into hmr-1 mutant embryos. HMR-1-GFP was present in all cells, but, like endogenous HMR-1, was markedly enriched in PGCs (Fig. 6A) and PGC ingression was rescued completely (Fig. 5A, compare 2nd and 4th columns). We conclude that maternal HMR-1 is necessary and sufficient for PGC ingression. Additionally, the finding that expressing hmr-1 zygotically in somatic cells (from hmr-1Ex) was insufficient to rescue PGC internalization suggests that HMR-1 is required in PGCs or in both PGCs and somatic cells, but argues against a requirement solely in somatic cells.
Fig. 5.
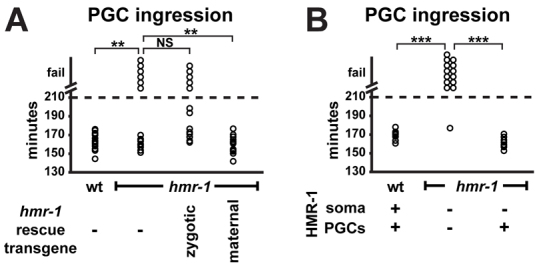
HMR-1 mosaic analysis. (A) PGC ingression in wild type (1st column, n=17), in embryos with low maternal and no zygotic hmr-1 expression (2nd column, progeny of hmr-1; hmr-1Ex mothers that do not inherit hmr-1Ex, n=16), in embryos with low maternal and high zygotic hmr-1 expression (3rd column, progeny of hmr-1; hmr-1Ex mothers that inherit hmr-1Ex, n=13), and in embryos with high maternal and no zygotic hmr-1 expression (4th column, progeny of hmr-1; hmr-1Ex; mex-5P:HMR-1-GFP::hmr-1UTR mothers that do not inherit hmr-1Ex, n=18). Asterisks indicate a significant difference in whether PGCs ingressed (**P<0.01, NS, not significant; Fisher's exact test). (B) PGC ingression in wild-type embryos fed on empty vector RNAi bacteria (1st column, n=10), hmr-1; hmr-1Ex hmr-1(RNAi) embryos (2nd column, n=14) and embryos from hmr-1; hmr-1P:HMR-1-ZF1-GFP::hmr-1UTR/ + mothers that did not inherit hmr-1P:HMR-1-ZF1-GFP::hmr-1UTR (3rd column, n=12) and therefore express HMR-1 only in PGCs. Asterisks indicate a significant difference in whether PGCs ingressed (***P<0.001, Fisher's exact test).
Fig. 6.

Regulation of HMR-1 enrichment in PGCs. Embryos (~50 μm in length) are oriented anterior towards the left and are shown from the indicated perspective. (A-C) GFP expression from the indicated transgenes. PGCs, asterisks. (D-F) GFP in embryos expressing mex-5P::GFP-PHPLC∂::hmr-1UTR immediately before (D) and after (E) photobleaching, and following recovery (F). Photobleaching was terminated before completion to prevent embryo lethality. (G-I) Quantification of fluorescence recovery after photobleaching (FRAP). AU, arbitrary units; error bars indicate s.d. (mex-5P::GFP-PHPLC∂::hmr-1UTR, n=9; mex-5P::GFP-PHPLC∂::tbb-2UTR, n=7). Asterisks indicate a significant difference in expression levels between PGC and control somatic cell contacts (**P<0.01, two-tailed Student's t-test). (I) Fold difference in expression level between PGC and somatic cell contacts.
To determine whether PGC ingression requires that HMR-1 be present in PGCs and somatic cells, or just in PGCs, we created a transgene that would allow us to supply maternal HMR-1 specifically to PGCs. Maternally expressed proteins tagged with the zinc finger 1 (ZF1) domain from PIE-1 are degraded rapidly in somatic cells of early embryos, but are protected in germ-line precursor cells and PGCs (Reese et al., 2000; Nance et al., 2003; Anderson et al., 2008; Wehman et al., 2011). We tested whether the ZF1 tag could be used to restrict maternal HMR-1 to PGCs by inserting the tag into mex-5P::HMR-1-GFP::hmr-1UTR. mex-5-driven HMR-1-GFP-ZF1 degraded rapidly from somatic cells and by gastrulation stages was detectable only in PGCs (supplementary material Fig. S5B). To create a functional HMR-1-ZF1-GFP transgene capable of rescuing the lethality of hmr-1 mutants, and that therefore could serve as the sole source of HMR-1 in embryos, we tagged a genomic hmr-1 construct with zf1 and gfp. hmr-1 mutants expressing hmr-1P::HMR-1-ZF1-GFP::hmr-1UTR from an integrated transgene were viable and fertile, contained low levels of maternal HMR-1-ZF1-GFP that degraded from somatic cells prior to gastrulation and was protected in PGCs (supplementary material Fig. S5C,D), and expressed zygotic HMR-1-ZF1-GFP robustly from the 50-cell stage onwards. By self-fertilizing hmr-1 mutants heterozygous for hmr-1P::HMR-1-ZF1-GFP::hmr-1UTR and examining progeny that did not inherit the transgene, we obtained embryos where the only source of HMR-1 was maternal HMR-1-GFP-ZF1. Maternal HMR-1-ZF1-GFP rescued the PGC gastrulation defects of hmr-1 mutant embryos completely (Fig. 5B). In combination with the rescue experiments described above, this finding strongly suggests that PGC ingression requires HMR-1 in PGCs but not in somatic cells (see Discussion).
HMR-1 enrichment in PGCs occurs through 3′ UTR-mediated translational control
How does HMR-1 become enriched in PGCs? Laser irradiation of the E cell did not prevent HMR-1-GFP enrichment in the PGCs (n=7), indicating that endodermal cells do not induce HMR-1 upregulation and suggesting that HMR-1 enrichment might be an autonomous property of the PGCs. We considered it unlikely that HMR-1 enrichment resulted from zygotic hmr-1 transcription, given that PGCs are thought to be transcriptionally inactive, and because we observed that HMR-1-GFP driven by the heterologous mex-5 promoter also became enriched in PGCs (Fig. 6A). To confirm that mex-5P::HMR-1-GFP::hmr-1UTR does not contain a cryptic promoter that drives zygotic expression in PGCs, we introduced the transgene at fertilization by crossing wild-type hermaphrodites with mex-5P::HMR-1-GFP::hmr-1 UTR males. None of the outcross embryos expressed HMR-1-GFP in PGCs (0/54 embryos), in contrast to embryos produced from transgenic mothers (57/66 embryos). In addition, although laser irradiation of the E nucleus prevented zygotic expression of a pes-10P::GFP reporter (data not shown), we were unable to prevent enrichment of HMR-1-GFP in P4 by laser-irradiating the P4 nucleus (seven out of seven embryos), potentially explaining why the irradiated cells were internalized (Fig. 1E,F). We conclude that HMR-1 enrichment in PGCs arises from maternal hmr-1 mRNA or protein rather than from zygotic transcription of hmr-1 in PGCs.
To determine whether hmr-1 mRNA is preferentially inherited or stabilized in PGCs, we performed in situ hybridization. A hmr-1 antisense probe labeled all early embryonic cells uniformly and was not enriched in P4 (107/107 embryos) (supplementary material Fig. S6). By contrast, most control embryos probed for nos-2 mRNA, which was previously shown to concentrate in PGCs (Subramaniam and Seydoux, 1999), showed a visible enrichment of probe staining in P4 (33/45 embryos) (supplementary material Fig. S6). Therefore, at a stage when HMR-1 protein is visibly enriched in PGCs (see Fig. 4A), the hmr-1 mRNA is present at comparable levels in somatic cells and PGCs.
We next tested whether there is increased stability or translation of the HMR-1 protein in PGCs relative to somatic cells. To address this possibility, we replaced hmr-1 coding sequences in the mex-5P::HMR-1-GFP::hmr-1UTR transgene with sequences encoding the PHPLC∂ membrane localization domain. GFP-PHPLC∂ expressed from the mex-5 promoter and hmr-1 3′ UTR showed PGC enrichment similar to HMR-1 (Fig. 6B). This finding suggests that HMR-1 enrichment is mediated by the hmr-1 3′ UTR, which is the only hmr-1 regulatory or coding element remaining in the transgene. Indeed, replacing the hmr-1 3′ UTR with that of the housekeeping gene tbb-2 (β-tubulin) (mex-5P::GFP-PHPLC∂::tbb-2UTR) caused GFP-PHPLC∂ to be expressed uniformly in both soma and germ line (Fig. 6C). Together, these experiments indicate that PGC enrichment of HMR-1 protein occurs post-transcriptionally, and that the hmr-1 3′ UTR is sufficient to mediate PGC-enriched expression.
To test whether the hmr-1 3′ UTR affects HMR-1 translation in PGCs, we performed fluorescence recovery after photobleaching (FRAP) experiments on embryos expressing mex-5P::GFP-PHPLC∂::hmr-1UTR. Just after the birth of P4, somatic cells and P4 expressed equivalent levels of GFP-PHPLC∂ (Fig. 6D,G,I). We photobleached whole embryos at this stage to quench most GFP fluorescence (Fig. 6E), then assayed expression levels again after the birth of Z2 and Z3 to measure nascent translation of the maternally supplied transgene mRNA. New GFP expression occurred in both somatic cells and PGCs, but was significantly higher in PGCs (Fig. 6F,G,I). By contrast, in control experiments performed using the mex-5P::GFP-PHPLC∂::tbb-2UTR transgene, both cell types showed an equivalent recovery following photobleaching (Fig. 6H,I). We conclude that the hmr-1 3′ UTR mediates HMR-1 enrichment in the PGCs by promoting increased translation of the hmr-1 mRNA in PGCs relative to somatic cells.
DISCUSSION
Our findings indicate that PGCs ingress using a hitchhiking mechanism that is enabled by post-transcriptionally regulated adhesion. We have shown that endodermal cells are required for PGCs to internalize and contact the PGCs directly throughout gastrulation. As the PGCs ingress, endodermal cells shift dorsally a similar distance, and blocking the dorsal shift prevents PGC ingression. Therefore, after endodermal cells internalize during gastrulation, they undergo a second morphogenetic movement that pulls the attached PGCs into the embryo (Fig. 7). PGCs express high levels of the cell-adhesion protein HMR-1/E-cadherin, which we have shown is upregulated post-transcriptionally in PGCs and functions to promote their robust adhesion to endoderm and subsequent internalization (Fig. 7). Thus, although the PGCs move into the embryo using a hitchhiking mechanism, they play an active role in their internalization by raising the levels of HMR-1/E-cadherin – an adhesion protein that enables their morphogenetic movements.
Fig. 7.
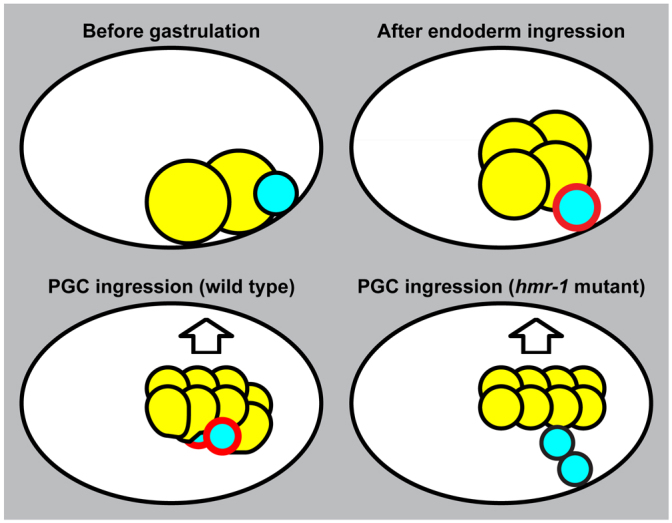
Model for PGC ingression. Schematic of embryos, dorsal upwards, anterior leftwards. Endoderm, yellow; PGCs, cyan; HMR-1, red. See Discussion for details.
E-cadherin regulation has been shown to be crucial for gastrulation in many species, although in these instances E-cadherin is downregulated in internalizing cells. For example, murine cells inhibit E-cadherin expression as they migrate through the primitive streak (Ciruna and Rossant, 2001; Zohn et al., 2006), and sea urchin primary mesenchyme cells rapidly internalize surface E-cadherin as they ingress into the blastocoel (Miller and McClay, 1997). E-cadherin downregulation is needed for these ingressing cells to delaminate from epithelial sheets as they migrate to form a new germ layer. The findings we present here demonstrate that E-cadherin upregulation can also promote ingression, by facilitating adhesion to other cells whose movements provide forces for internalization.
Although we found that the most severe depletion of HMR-1 resulted in an almost complete failure in PGC ingression, we detected a smaller percentage of embryos that showed clear PGC-endoderm separation (compare Fig. 4C,G). This discrepancy probably results from two factors. First, we scored PGC ingression as successful only if both PGCs dropped from the surface of the embryo; therefore, one PGC could still remain associated with endoderm whereas the other detached. Second, separations between the two cell groups are difficult to detect unless they are viewed from a lateral perspective – an orientation embryos adopt infrequently when standard mounting methods are used. Indeed, when we examined only those embryos in a lateral orientation at high resolution by acquiring confocal z-stacks, at least one of the two PGCs were partially or fully detached from endoderm in 10 of 11 embryos, and the remaining contact interface with endoderm was significantly reduced (Fig. 4H).
E-cadherin mediates adhesion largely through homotypic interactions with cadherins on the adjacent cell (Borghi and Nelson, 2009; Harris and Tepass, 2010). HMR-1 is expressed in both PGCs and endoderm (see Fig. 4), and trans-interactions between HMR-1 on each of these cell groups could contribute to their adhesion. How, though, can we reconcile our finding that PGC ingression can occur when HMR-1 is detectable only in PGCs? Although we cannot exclude the possibility that trace levels of somatic HMR-1 below our limit of detection are sufficient to promote PGC ingression, we envision two models that could explain a PGC-specific role for HMR-1. One possibility is that HMR-1 interacts with a heterotypic ligand, such as SAX-7, that is present on the surfaces of endodermal cells. Heterotypic trans interactions between classic cadherins and other adhesion proteins have been described in a few cell types and shown to promote adhesion. For example, direct interactions between E-cadherin on epithelial cells and integrins on lymphocyte cell surfaces mediate adhesion between the two cell types (Cepek et al., 1994; Higgins et al., 1998). A second model that can explain our findings is that HMR-1 mediates robust PGC-endoderm adhesion by promoting changes in PGC cell surface tension, which in turn induces endodermal cells to spread over and ‘hug’ the PGCs. Experiments with cultured cells and tissues have shown that overexpression of cadherins within a cell group increases the surface tension of the tissue (Foty and Steinberg, 2005). Tissues with different surface tensions placed adjacent one another behave like immiscible liquids, wherein the tissue with lower surface tension wraps around that with higher surface tension, irrespective of the type of adhesion protein that each tissue expresses (Foty et al., 1996; Foty and Steinberg, 2005; Lecuit and Lenne, 2007). High levels of HMR-1 expression in PGCs could increase their surface tension, promoting a spreading behavior by the adjacent endodermal cells and resulting in a tight association between the two cell groups. Ubiquitously expressed adhesion proteins such as SAX-7 could facilitate adhesion and spreading between the two cell groups. In support of this model, we observed that endodermal cells not only adhere to PGCs, but also wrap dramatically around the PGC surfaces (see, for example, Fig. 3E, Fig. 4E).
Why do PGCs preferentially adhere to endodermal cells rather than the ring of mesodermal cells that surround them on the embryo surface? Our ability to rescue PGC internalization by providing HMR-1 solely to PGCs indicates that differences in HMR-1 levels between mesodermal and endodermal cells cannot explain the preferential association of PGCs and endoderm. Rather, we propose that preferential association of PGCs with endodermal cells may reflect differences in the relative stability of adhesive interactions that endodermal cells and mesodermal cells make with their neighbors. Mesodermal cells are highly mobile as they ingress from the ventral surface and in many cases migrate extensively within the embryo (Schnabel et al., 1997; Viveiros et al., 2011). Therefore, mesodermal cells behave like mesenchymal cells and must be able to make and break new contacts rapidly; by contrast, endodermal cells remain as a unified cell group (Schnabel et al., 1997). Our analysis of cell separations supports this view, as we detected separations between mesodermal cells and their neighbors, but not between the two PGCs, between endodermal cells, or at the PGC-endoderm interface (supplementary material Fig. S3).
Our findings reveal for the first time the different molecular strategies that C. elegans PGCs and somatic cells use to internalize during gastrulation. Somatic cells rely on lineage-specific cell fate transcription factors to trigger their ingression movements (Nance and Priess, 2002; Lee et al., 2006; Harrell and Goldstein, 2011). Endodermal cell ingressions, and probably those of mesodermal cells, occur when myosin accumulates at the apical surface and promotes apical constriction (Nance and Priess, 2002; Lee and Goldstein, 2003; Nance et al., 2003). Laser-irradiation experiments show that somatic cell ingression movements are largely autonomous, as killing one group of ingressing cells does not affect the ingression of another (Nance and Priess, 2002). In contrast to somatic cells, we have shown that PGC ingression is regulated post-transcriptionally by 3′ UTR-mediated HMR-1 upregulation and occurs through a hitchhiking mechanism that is dependent on endodermal cell movements. Our data support a role for the hmr-1 3′ UTR in regulating HMR-1 translation, rather than stability or localization of the hmr-1 mRNA, as hmr-1 mRNA appears uniformly distributed in all embryonic blastomeres. 3′ UTR-mediated post-transcriptional regulation is a conserved mechanism that germ cells in both invertebrates and vertebrates use to control levels of proteins important for their development and differentiation (Knaut et al., 2002; Kataoka et al., 2006; Merritt et al., 2008; Rangan et al., 2009; Suzuki et al., 2010). The data we present here show that PGCs also use 3′ UTR control to regulate the level of proteins needed for morphogenesis. Given that PGCs are transcriptionally quiescent in early embryos of many animals, we anticipate that UTR regulation could be a conserved mechanism that PGCs use to control proteins important for gastrulation.
The association between embryonic endoderm and primordial germ cells is observed in a wide variety of species, including worms, flies, sea urchins, frogs and mice (Santos and Lehmann, 2004; Juliano et al., 2006), but the significance of this association is not known. Our analysis of PGC ingression demonstrates a role for endoderm-PGC association in helping to ensure that PGCs are properly internalized during gastrulation. Other species may use similar strategies. For example, in some other nematodes, PGCs are born at a distance from the endoderm and are repositioned to lie adjacent the endoderm before gastrulation commences (Wiegner and Schierenberg, 1998; Lahl et al., 2009). Drosophila PGCs require E-cadherin to adhere to endodermal cells as both cell groups are internalized during gastrulation by extension of the germ band (DeGennaro et al., 2011). Sea urchin PGCs might also rely on association with endoderm to become internalized, as they are found at the tip of the invaginating archenteron (primitive gut) during gastrulation (Juliano et al., 2006; Yajima and Wessel, 2011). PGCs in frogs also associate with the presumptive endoderm as it involutes during gastrulation (Whitington and Dixon, 1975; Nishiumi et al., 2005). Thus, the ancient association between PGCs and endoderm may reflect a morphogenetic role for endoderm in helping place PGCs in the proper position within the embryo.
Supplementary Material
Acknowledgements
We thank Julie Ahringer, Oliver Hobert, Jane Hubbard, Craig Hunter, Morris Maduro, Jonathan Pettitt, Jim Priess, Geraldine Seydoux, Kuppuswamy Subramaniam, Ann Wehman and Eva Zeiser for their generous gifts of strains and reagents; and Lionel Christiaen, Jane Hubbard, Thomas Hurd, Holger Knaut, Ruth Lehmann and Nance lab members for comments on the manuscript. Some strains and antibodies were provided by the NIH-supported CGC and DSHB.
Footnotes
Funding
This work was funded by grants to J.N. from the National Institutes of Health [R01GM078341, R21HD058953], NYSTEM [C024301] and the Irma T. Hirschl Charitable Trust. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.079863/-/DC1
References
- Achilleos A., Wehman A. M., Nance J. (2010). PAR-3 mediates the initial clustering and apical localization of junction and polarity proteins during C. elegans intestinal epithelial cell polarization. Development 137, 1833-1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson D. C., Gill J. S., Cinalli R. M., Nance J. (2008). Polarization of the C. elegans embryo by RhoGAP-mediated exclusion of PAR-6 from cell contacts. Science 320, 1771-1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audhya A., Hyndman F., McLeod I. X., Maddox A. S., Yates J. R., 3rd, Desai A., Oegema K. (2005). A complex containing the Sm protein CAR-1 and the RNA helicase CGH-1 is required for embryonic cytokinesis in Caenorhabditis elegans. J. Cell Biol. 171, 267-279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghi N., Nelson W. J. (2009). Intercellular adhesion in morphogenesis: molecular and biochysical considerations. Curr. Top. Dev. Biol. 89, 1-32 [DOI] [PubMed] [Google Scholar]
- Broadbent I. D., Pettitt J. (2002). The C. elegans hmr-1 gene can encode a neuronal classic cadherin involved in the regulation of axon fasciculation. Curr. Biol. 12, 59-63 [DOI] [PubMed] [Google Scholar]
- Cepek K. L., Shaw S. K., Parker C. M., Russell G. J., Morrow J. S., Rimm D. L., Brenner M. B. (1994). Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the aEß7 integrin. Nature 372, 190-193 [DOI] [PubMed] [Google Scholar]
- Chen L., Ong B., Bennett V. (2001). LAD-1, the Caenorhabditis elegans L1CAM homologue, participates in embryonic and gonadal morphogenesis and is a substrate for fibroblast growth factor receptor pathway-dependent phosphotyrosine-based signaling. J. Cell Biol. 154, 841-855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciruna B., Rossant J. (2001). FGF signaling regulates mesoderm cell fate specification and morphogenetic movement at the primitive streak. Dev. Cell 1, 37-49 [DOI] [PubMed] [Google Scholar]
- Costa M., Raich W., Agbunag C., Leung B., Hardin J., Priess J. R. (1998). A putative catenin-cadherin system mediates morphogenesis of the Caenorhabditis elegans embryo. J. Cell Biol. 141, 297-308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGennaro M., Hurd T. R., Siekhaus D. E., Biteau B., Jasper H., Lehmann R. (2011). Peroxiredoxin stabilization of DE-cadherin promotes primordial germ cell adhesion. Dev. Cell 20, 233-243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foty R. A., Steinberg M. S. (2005). The differential adhesion hypothesis: a direct evaluation. Dev. Biol. 278, 255-263 [DOI] [PubMed] [Google Scholar]
- Foty R. A., Pfleger C. M., Forgacs G., Steinberg M. S. (1996). Surface tensions of embryonic tissues predict their mutual envelopment behavior. Development 122, 1611-1620 [DOI] [PubMed] [Google Scholar]
- Frokjaer-Jensen C., Davis M. W., Hopkins C. E., Newman B. J., Thummel J. M., Olesen S. P., Grunnet M., Jorgensen E. M. (2008). Single-copy insertion of transgenes in Caenorhabditis elegans. Nat. Genet. 40, 1375-1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson D. G., Young L., Chuang R. Y., Venter J. C., Hutchison C. A., 3rd, Smith H. O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343-345 [DOI] [PubMed] [Google Scholar]
- Grana T. M., Cox E. A., Lynch A. M., Hardin J. (2010). SAX-7/L1CAM and HMR-1/cadherin function redundantly in blastomere compaction and non-muscle myosin accumulation during Caenorhabditis elegans gastrulation. Dev. Biol. 344, 731-744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guven-Ozkan T., Nishi Y., Robertson S. M., Lin R. (2008). Global transcriptional repression in C. elegans germline precursors by regulated sequestration of TAF-4. Cell 135, 149-160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrell J. R., Goldstein B. (2011). Internalization of multiple cells during C. elegans gastrulation depends on common cytoskeletal mechanisms but different cell polarity and cell fate regulators. Dev. Biol. 350, 1-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris T. J., Tepass U. (2010). Adherens junctions: from molecules to morphogenesis. Nat. Rev. Mol. Cell Biol. 11, 502-514 [DOI] [PubMed] [Google Scholar]
- Heisenberg C. P., Solnica-Krezel L. (2008). Back and forth between cell fate specification and movement during vertebrate gastrulation. Curr. Opin. Genet. Dev. 18, 311-316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins J. M., Mandlebrot D. A., Shaw S. K., Russell G. J., Murphy E. A., Chen Y. T., Nelson W. J., Parker C. M., Brenner M. B. (1998). Direct and regulated interaction of integrin aEß7 with E-cadherin. J. Cell Biol. 140, 197-210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliano C. E., Voronina E., Stack C., Aldrich M., Cameron A. R., Wessel G. M. (2006). Germ line determinants are not localized early in sea urchin development, but do accumulate in the small micromere lineage. Dev. Biol. 300, 406-415 [DOI] [PubMed] [Google Scholar]
- Junkersdorf B., Schierenberg E. (1992). Embryogenesis in C. elegans after eliminatin of individual blastomeres or induced alteration of the cell division order. Roux's Arch. Dev. Biol. 202, 17-22 [DOI] [PubMed] [Google Scholar]
- Kataoka K., Yamaguchi T., Orii H., Tazaki A., Watanabe K., Mochii M. (2006). Visualization of the Xenopus primordial germ cells using a green fluorescent protein controlled by cis elements of the 3′ untranslated region of the DEADSouth gene. Mech. Dev. 123, 746-760 [DOI] [PubMed] [Google Scholar]
- Knaut H., Steinbeisser H., Schwarz H., Nusslein-Volhard C. (2002). An evolutionary conserved region in the vasa 3′UTR targets RNA translation to the germ cells in the zebrafish. Curr. Biol. 12, 454-466 [DOI] [PubMed] [Google Scholar]
- Lahl V., Schulze J., Schierenberg E. (2009). Differences in embryonic pattern formation between Caenorhabditis elegans and its close parthenogenetic relative Diploscapter coronatus. Int. J. Dev. Biol. 53, 507-515 [DOI] [PubMed] [Google Scholar]
- Lecuit T., Lenne P. F. (2007). Cell surface mechanics and the control of cell shape, tissue patterns and morphogenesis. Nat. Rev. Mol. Cell Biol. 8, 633-644 [DOI] [PubMed] [Google Scholar]
- Lee J. Y., Goldstein B. (2003). Mechanisms of cell positioning during C. elegans gastrulation. Development 130, 307-320 [DOI] [PubMed] [Google Scholar]
- Lee J. Y., Marston D. J., Walston T., Hardin J., Halberstadt A., Goldstein B. (2006). Wnt/Frizzled signaling controls C. elegans gastrulation by activating actomyosin contractility. Curr. Biol. 16, 1986-1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leptin M. (2005). Gastrulation movements: the logic and the nuts and bolts. Dev. Cell 8, 305-320 [DOI] [PubMed] [Google Scholar]
- Maduro M. F. (2006). Endomesoderm specification in Caenorhabditis elegans and other nematodes. BioEssays 28, 1010-1022 [DOI] [PubMed] [Google Scholar]
- Mello C. C., Schubert C., Draper B., Zhang W., Lobel R., Priess J. R. (1996). The PIE-1 protein and germline specification in C. elegans embryos. Nature 382, 710-712 [DOI] [PubMed] [Google Scholar]
- Merritt C., Rasoloson D., Ko D., Seydoux G. (2008). 3′ UTRs are the primary regulators of gene expression in the C. elegans germline. Curr. Biol. 18, 1476-1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J. R., McClay D. R. (1997). Characterization of the role of cadherin in regulating cell adhesion during sea urchin development. Dev. Biol. 192, 323-339 [DOI] [PubMed] [Google Scholar]
- Nance J., Priess J. R. (2002). Cell polarity and gastrulation in C. elegans. Development 129, 387-397 [DOI] [PubMed] [Google Scholar]
- Nance J., Munro E. M., Priess J. R. (2003). C. elegans PAR-3 and PAR-6 are required for apicobasal asymmetries associated with cell adhesion and gastrulation. Development 130, 5339-5350 [DOI] [PubMed] [Google Scholar]
- Nishiumi F., Komiya T., Ikenishi K. (2005). The mode and molecular mechanisms of the migration of presumptive PGC in the endoderm cell mass of Xenopus embryos. Dev. Growth Differ. 47, 37-48 [DOI] [PubMed] [Google Scholar]
- Owraghi M., Broitman-Maduro G., Luu T., Roberson H., Maduro M. F. (2010). Roles of the Wnt effector POP-1/TCF in the C. elegans endomesoderm specification gene network. Dev. Biol. 340, 209-221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praitis V., Casey E., Collar D., Austin J. (2001). Creation of low-copy integrated transgenic lines in Caenorhabditis elegans. Genetics 157, 1217-1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangan P., DeGennaro M., Jaime-Bustamante K., Coux R. X., Martinho R. G., Lehmann R. (2009). Temporal and spatial control of germ-plasm RNAs. Curr. Biol. 19, 72-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese K. J., Dunn M. A., Waddle J. A., Seydoux G. (2000). Asymmetric segregation of PIE-1 in C. elegans is mediated by two complementary mechanisms that act through separate PIE-1 protein domains. Mol. Cell 6, 445-455 [DOI] [PubMed] [Google Scholar]
- Richardson B. E., Lehmann R. (2010). Mechanisms guiding primordial germ cell migration: strategies from different organisms. Nat. Rev. Mol. Cell Biol. 11, 37-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos A. C., Lehmann R. (2004). Germ cell specification and migration in Drosophila and beyond. Curr. Biol. 14, R578-R589 [DOI] [PubMed] [Google Scholar]
- Schaner C. E., Deshpande G., Schedl P. D., Kelly W. G. (2003). A conserved chromatin architecture marks and maintains the restricted germ cell lineage in worms and flies. Dev. Cell 5, 747-757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnabel R., Hutter H., Moerman D., Schnabel H. (1997). Assessing normal embryogenesis in Caenorhabditis elegans using a 4D microscope: variability of development and regional specification. Dev. Biol. 184, 234-265 [DOI] [PubMed] [Google Scholar]
- Seydoux G., Fire A. (1995). Whole-mount in situ hybridization for the detection of RNA in Caenorhabditis elegans embryos. Methods Cell Biol. 48, 323-337 [DOI] [PubMed] [Google Scholar]
- Seydoux G., Braun R. E. (2006). Pathway to totipotency: lessons from germ cells. Cell 127, 891-904 [DOI] [PubMed] [Google Scholar]
- Seydoux G., Mello C. C., Pettitt J., Wood W. B., Priess J. R., Fire A. (1996). Repression of gene expression in the embryonic germ lineage of C. elegans. Nature 382, 713-716 [DOI] [PubMed] [Google Scholar]
- Shirae-Kurabayashi M., Matsuda K., Nakamura A. (2011). Ci-Pem-1 localizes to the nucleus and represses somatic gene transcription in the germline of Ciona intestinalis embryos. Development 138, 2871-2881 [DOI] [PubMed] [Google Scholar]
- Subramaniam K., Seydoux G. (1999). nos-1 and nos-2, two genes related to Drosophila nanos, regulate primordial germ cell development and survival in Caenorhabditis elegans. Development 126, 4861-4871 [DOI] [PubMed] [Google Scholar]
- Sulston J. E., Schierenberg E., White J. G., Thomson J. N. (1983). The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol. 100, 64-119 [DOI] [PubMed] [Google Scholar]
- Suzuki H., Saba R., Sada A., Saga Y. (2010). The Nanos3-3′UTR is required for germ cell specific NANOS3 expression in mouse embryos. PLoS ONE 5, e9300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totong R., Achilleos A., Nance J. (2007). PAR-6 is required for junction formation but not apicobasal polarization in C. elegans embryonic epithelial cells. Development 134, 1259-1268 [DOI] [PubMed] [Google Scholar]
- Venkatarama T., Lai F., Luo X., Zhou Y., Newman K., King M. L. (2010). Repression of zygotic gene expression in the Xenopus germline. Development 137, 651-660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viveiros R., Hutter H., Moerman D. G. (2011). Membrane extensions are associated with proper anterior migration of muscle cells during Caenorhabditis elegans embryogenesis. Dev. Biol. 358, 189-200 [DOI] [PubMed] [Google Scholar]
- Wehman A. M., Poggioli C., Schweinsberg P., Grant B. D., Nance J. (2011). The P4-ATPase TAT-5 inhibits the budding of extracellular vesicles in C. elegans embryos. Curr. Biol. 21, 1951-1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitington P. M., Dixon K. E. (1975). Quantitative studies of germ plasm and germ cells during early embryogenesis of Xenopus laevis. J. Embryol. Exp. Morphol. 33, 57-74 [PubMed] [Google Scholar]
- Wiegner O., Schierenberg E. (1998). Specification of gut cell fate differs significantly between the nematodes Acrobeloides nanus and Caenorhabditis elegans. Dev. Biol. 204, 3-14 [DOI] [PubMed] [Google Scholar]
- Yajima M., Wessel G. M. (2011). Small micromeres contribute to the germline in the sea urchin. Development 138, 237-243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallen J. A., Kirch S. A., Bargmann C. I. (1999). Genes required for axon pathfinding and extension in the C. elegans nerve ring. Development 126, 3679-3692 [DOI] [PubMed] [Google Scholar]
- Zeiser E., Frokjaer-Jensen C., Jorgensen E., Ahringer J. (2011). MosSCI and gateway compatible plasmid toolkit for constitutive and inducible expression of transgenes in the C. elegans germline. PLoS ONE 6, e20082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zohn I. E., Li Y., Skolnik E. Y., Anderson K. V., Han J., Niswander L. (2006). p38 and a p38-interacting protein are critical for downregulation of E-cadherin during mouse gastrulation. Cell 125, 957-969 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.