

Abstract
Six3 exerts multiple functions in the development of anterior neural tissue of vertebrate embryos. Whereas complete loss of Six3 function in the mouse results in failure of forebrain formation, its hypomorphic mutations in human and mouse can promote holoprosencephaly (HPE), a forebrain malformation that results, at least in part, from abnormal telencephalon development. However, the roles of Six3 in telencephalon patterning and differentiation are not well understood. To address the role of Six3 in telencephalon development, we analyzed zebrafish embryos deficient in two out of three Six3-related genes, six3b and six7, representing a partial loss of Six3 function. We found that telencephalon forms in six3b;six7-deficient embryos; however, ventral telencephalic domains are smaller and dorsal domains are larger. Decreased cell proliferation or excess apoptosis cannot account for the ventral deficiency. Instead, six3b and six7 are required during early segmentation for specification of ventral progenitors, similar to the role of Hedgehog (Hh) signaling in telencephalon development. Unlike in mice, we observe that Hh signaling is not disrupted in embryos with reduced Six3 function. Furthermore, six3b overexpression is sufficient to compensate for loss of Hh signaling in isl1- but not nkx2.1b-positive cells, suggesting a novel Hh-independent role for Six3 in telencephalon patterning. We further find that Six3 promotes ventral telencephalic fates through transient regulation of foxg1a expression and repression of the Wnt/β-catenin pathway.
Keywords: Six3, Telencephalon, Zebrafish
INTRODUCTION
The telencephalon, located in the anterior-most region of the embryonic neural tube, develops into the cerebral cortex and basal ganglia in mammals, and the pallium and subpallium in other vertebrates. During early segmentation stages of vertebrate embryogenesis, the telencephalon becomes patterned along its dorsoventral (DV) axis, as evidenced by restricted expression domains of numerous genes. Dorsally located progenitors generate cortical projection neurons, whereas ventral progenitors generate striatal projection neurons, as well as interneurons and oligodendrocytes (Wilson and Rubenstein, 2000). Cooperation between many molecules, including ligands secreted from local signaling centers and regionally expressed transcription factors, determines the size and fate of telencephalic progenitor domains (Wilson and Rubenstein, 2000; Hebert and Fishell, 2008). However, the exact functions of genes involved in DV patterning of the telencephalon and the interactions between these genes are still not well understood.
The homeodomain transcription factor Six3 has been shown to regulate a number of events that are involved in telencephalon development in several vertebrates. Beginning at late gastrulation, Six3 is expressed broadly in the anterior neuroectoderm (Oliver et al., 1995; Bovolenta et al., 1998; Kobayashi et al., 1998; Loosli et al., 1998; Seo et al., 1998a; Seo et al., 1998b; Zhou et al., 2000), where it may function in controlling the expression of extracellular ligands that influence telencephalic development, as well as providing competence to respond to such signals (Kobayashi et al., 2002; Lagutin et al., 2003; Gestri et al., 2005; Jeong et al., 2008). Telencephalon induction is severely impaired in Six3-null mouse embryos, as well as medaka fish embryos in which Six3 activity is blocked by antisense morpholino oligonucleotides (MOs) (Carl et al., 2002; Lagutin et al., 2003; Lavado et al., 2008). By contrast, dominant mutations in human SIX3 have been linked to holoprosencephaly (HPE) (Wallis et al., 1999; Lacbawan et al., 2009), a congenital malformation in which the telencephalon forms, but forebrain midline structures are disrupted, resulting in ventrally biased neuropathologies and failure of telencephalic hemispheres to separate (Dubourg et al., 2007; Monuki, 2007). A study with a recently generated mouse model of Six3-mediated HPE suggested that reduced Six3 function disrupts a positive-feedback loop between Six3 and Sonic Hedgehog (Shh) (Geng et al., 2008), thereby linking Six3 with Hedgehog (Hh) signaling pathway activity, which is crucial for normal telencephalon DV patterning (Chiang et al., 1996). The regulation of the SHH gene by SIX3 is conserved in humans, as shown by the ability of mouse Six3 to bind to a conserved enhancer element upstream of the human SHH gene and directly activate transcription from this element (Jeong et al., 2008). Together, studies in human, mouse and zebrafish demonstrate that most SIX3 mutations associated with HPE are hypomorphic alleles, that can become haploinsufficient when Shh activity is reduced by other mutations (Domene et al., 2008; Geng et al., 2008; Jeong et al., 2008; Lacbawan et al., 2009). However, it remains unknown whether regulation of Shh expression is the only mechanism by which Six3 influences DV telencephalic development.
In addition to Hh signaling, several Wnt ligands are expressed posterior to the telencephalon anlage, and some have been shown to affect telencephalic DV patterning (Ciani and Salinas, 2005). Six3 has been shown to repress directly the expression of both Wnt1 and Wnt8b in mouse embryos, thereby affecting telencephalon induction and patterning of the eye field, respectively (Lagutin et al., 2003; Liu et al., 2010). However, a link between Six3 and Wnt signaling in telencephalon DV patterning has not been established.
Here, we use the zebrafish Danio rerio, which has three orthologs of the mammalian Six3 gene in its genome, six3a, six3b and six7 (Seo et al., 1998a; Seo et al., 1998b), to dissect the role of Six3 in telencephalon patterning. Zebrafish embryos that are deficient in both six3b and six7 function exhibit severely reduced eye size and abnormalities in left-right brain asymmetry, yet have largely normal anterior-posterior patterning of the central nervous system (CNS) (Inbal et al., 2007). Our current work shows that six3b;six7-deficient embryos have a telencephalon, but with DV patterning defects similar to those found in HPE. Unlike in Six3 mutant mice, the reduction of ventral cell fates is not mediated by reduced Hh signaling, but may be due to reduced expression during early segmentation of foxg1a, which is required for telencephalic DV patterning downstream of Hh signaling. Analysis of discrete cell populations in the ventral telencephalon revealed that the telencephalic nkx2.1b expression domain requires function of Six3, Foxg1a and Hh signaling, whereas six3b overexpression can compensate for loss of Foxg1a or Hh signaling in the more dorsolateral isl1 domain. We further show that loss of Six3 function leads to expanded Wnt/β-catenin pathway activity in the telencephalon anlage, which could contribute to the DV patterning defects. Our results lend support to the notion that Six3 provides competence for anterior neural tissue to respond to Hh signaling, and uncover new Shh-independent mechanisms through which Six3 mediates telencephalon development.
MATERIALS AND METHODS
Zebrafish strains, embryo culture and generation of transgenic fish
Adult zebrafish were maintained according to established methods (Westerfield, 1993). Embryos were obtained from natural matings, grown at 28.5°C and staged according to Kimmel et al. (Kimmel et al., 1995). The following published strains were used and genotyped as previously described: wild-type AB, tp53zdf1 (Berghmans et al., 2005), six3bvu87 (Inbal et al., 2007), smob641 (Varga et al., 2001), Tg(hsp70l:Gal4-VP16)vu22 (Shin et al., 2007), Tg(UAS:six3b)vu156 (Inbal et al., 2007) and Tg(hsp70l:wnt8a-GFP)w34 (Weidinger et al., 2005).
To generate the Tg(UAS:shha-NH-EGFP)vu486 transgenic line, the coding sequence for zebrafish shha was obtained from zShh-T7TS vector (Ekker et al., 1995). A non-helical oligopeptide linker (NH), APAETKAEPMT (George and Heringa, 2002), was inserted upstream of the EGFP-coding sequence isolated from pEGFP-C1 (Clontech). To insert NH-EGFP in frame into the Shha-coding sequence, unique NheI and XhoI restriction sites were introduced between Ala192 and Ala193 of Shha and flanking NH-EGFP. GFP was released from pT2-UAS-GFP-γCry-GM2 (Inbal et al., 2006), followed by replacement with the Shha-NH-EGFP construct using KpnI and ApaI restriction sites. Transgenic fish were generated using the Sleeping Beauty transposon system (Davidson et al., 2003) by co-injecting 15-20 pg pT2-UAS-Shha-NH-EGFP-γCry-GM2 DNA with synthetic RNA encoding SB10 transposase into one-cell stage embryos. Founder fish were identified as previously described (Inbal et al., 2006). Sequences for PCR primers used for cloning pT2-UAS-Shha-NH-EGFP-γCry-GM2 are available upon request.
Morpholino oligonucleotides (MOs), cell cycle inhibition, heat shock and cyclopamine treatment
MOs directed against the translation start site of the foxg1a gene (MO2-foxg1a) (Danesin et al., 2009) and the 5′ untranslated region of the six7 gene (MO1-six7) (Inbal et al., 2007) have been previously described. Embryos were injected at the 1- to 2-cell stage with 2-3 ng MO1-six7 or 1 ng MO2-foxg1a for phenotypic analysis.
To inhibit cellular proliferation, mid-gastrula embryos (80% epiboly) were incubated in 30% Danieau's solution containing 20 mM hydroxyurea (Sigma-Aldrich), 150 μM aphidicolin (Sigma-Aldrich) and 2% dimethyl sulfoxide. Control embryos were incubated with 2% dimethyl sulfoxide alone.
Embryos were heat shocked in prewarmed 30% Danieau's solution at 37°C for 30 minutes, and subsequently developed at 28.5°C.
To inhibit Hh signaling, early gastrula embryos (shield stage) were treated with 30% Danieau's solution containing 10 μM cyclopamine hydrate (Sigma-Aldrich) with 0.1% ethanol. Control embryos were treated with 0.1% ethanol alone.
In situ hybridization, immunohistochemistry and TUNEL
Embryos were fixed in 4% paraformaldehyde in phosphate-buffered saline. Whole-mount in situ hybridization was performed according to standard protocols and developed with BMPurple (Roche) and Fast Red (Roche) or INT (iodonitrotetrazolium chloride, Sigma-Aldrich). Digoxigenin- or fluorescein-labeled probes were generated from cDNA templates: axin2 (Carl et al., 2007), dlx2a (Akimenko et al., 1994), emx3 (Morita et al., 1995), eomesa (Mione et al., 2001), foxg1a (Toresson et al., 1998), isl1 (Inoue et al., 1994), nkx2.1b (Rohr et al., 2001), ptch2 (formerly described as ptc1) (Concordet et al., 1996), six3b (Seo et al., 1998a), six7 (Seo et al., 1998b) and wnt8b (Kelly et al., 1995).
Rabbit polyclonal phospho-Histone H3 antibody (Upstate Biotechnology) and Cy3 conjugated goat anti-rabbit antibody (Jackson ImmunoResearch) were applied at 1:3000 and 1:250, respectively.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) was performed as described (Verduzco and Amatruda, 2011) using an alkaline phosphatase-conjugated anti-digoxigenin antibody (1:5000; Roche) and developed with BMPurple.
Image acquisition, analysis and quantitation
Images were acquired using Zeiss Axiophot, Zeiss Imager Z.1 compound microscope, or Zeiss Discovery.V12 stereomicroscope and an Axiocam digital camera. Images from anti-phospho-Histone H3 and TUNEL labeling were taken in a z-series, and a z-projection was generated using the Extended Focus computation in Axiovision software (Zeiss).
Each individual experiment was performed two to four times and the number of affected and observed embryos was compiled from the total over all experiments. All mutant genotypes were confirmed by PCR or morphology.
Quantification of cell counts and measurements was carried out with Fiji software (NIH). Cells labeled positive by anti-phospho-Histone H3 antibody within an identically sized region of the anterior body axis or anterior neural tube were counted from z-stacks using the cell counter plug-in for Fiji software (supplementary material Fig. S1). One-dimensional measurements along the DV telencephalon axis in vehicle- and hydroxyurea/aphidicolin-treated embryos were also taken with Fiji software. For each individual experiment, experimental measurements were determined as a fraction of the average control measurement. Statistical significance was determined by a Student's unpaired t-test with a two-tailed distribution.
RESULTS
The telencephalon of six3b;six7-deficient embryos is dorsalized
To gain insight into the roles of Six3 in telencephalic development, we compared patterning of the telencephalon at 24 hours post-fertilization (hpf) between control zebrafish embryos (six3bvu87/+ and six3bvu87/vu87, identified by PCR and comparable with wild type) and six3b;six7-deficient embryos [six3bvu87/vu87 embryos injected with MO1-six7, which inhibits translation of six7 mRNA (Inbal et al., 2007)]. At this developmental stage, subdomains of telencephalon are evident along the DV axis by distinct gene expression (Rohr et al., 2001). We first examined expression of foxg1a, a pan-telencephalic marker encoding a forkhead transcription factor (Toresson et al., 1998). foxg1a was expressed in six3b;six7-deficient embryos, indicating that telencephalic tissue was specified (Fig. 1A,B). Next, we examined expression of nkx2.1b, encoding a homeodomain transcription factor that is expressed in the most ventromedial domain of the telencephalon and in the hypothalamus (Rohr et al., 2001). In six3b;six7-deficient embryos, telencephalic nkx2.1b expression was strongly reduced (Fig. 1C,D). Similarly, expression of isl1, which encodes a LIM homeodomain transcription factor expressed in a subpopulation of ventrolateral telencephalic cells (Inoue et al., 1994), was strongly reduced in six3b;six7-deficient embryos (Fig. 1E,F). The expression domain of a third ventral telencephalic marker, the homeobox gene dlx2a (Akimenko et al., 1994), was also reduced in six3b;six7-deficient telencephalon (Fig. 1G,H). By contrast, expression domains of dorsally expressed genes were expanded. We observed that telencephalic domains of both emx3 and eomesodermin homolog a (eomesa), which encode homeodomain and T-box transcription factors, respectively, and are normally limited to the dorsal telencephalon (Morita et al., 1995; Mione et al., 2001), were expanded ventrally in six3b;six7-deficient embryos (Fig. 1I-L). Additionally, the diencephalic domains of nkx2.1b, isl1, dlx2a and eomesa expression were mispatterned in six3b;six7-deficient embryos; however, in this work we focused our analysis on telencephalon (Fig. 1C-H,K,L). Collectively, these data suggest that reduced Six3 function leads to an expansion of dorsal telencephalic fates at the expense of ventral ones, and hence to dorsalization of the telencephalon.
Fig. 1.
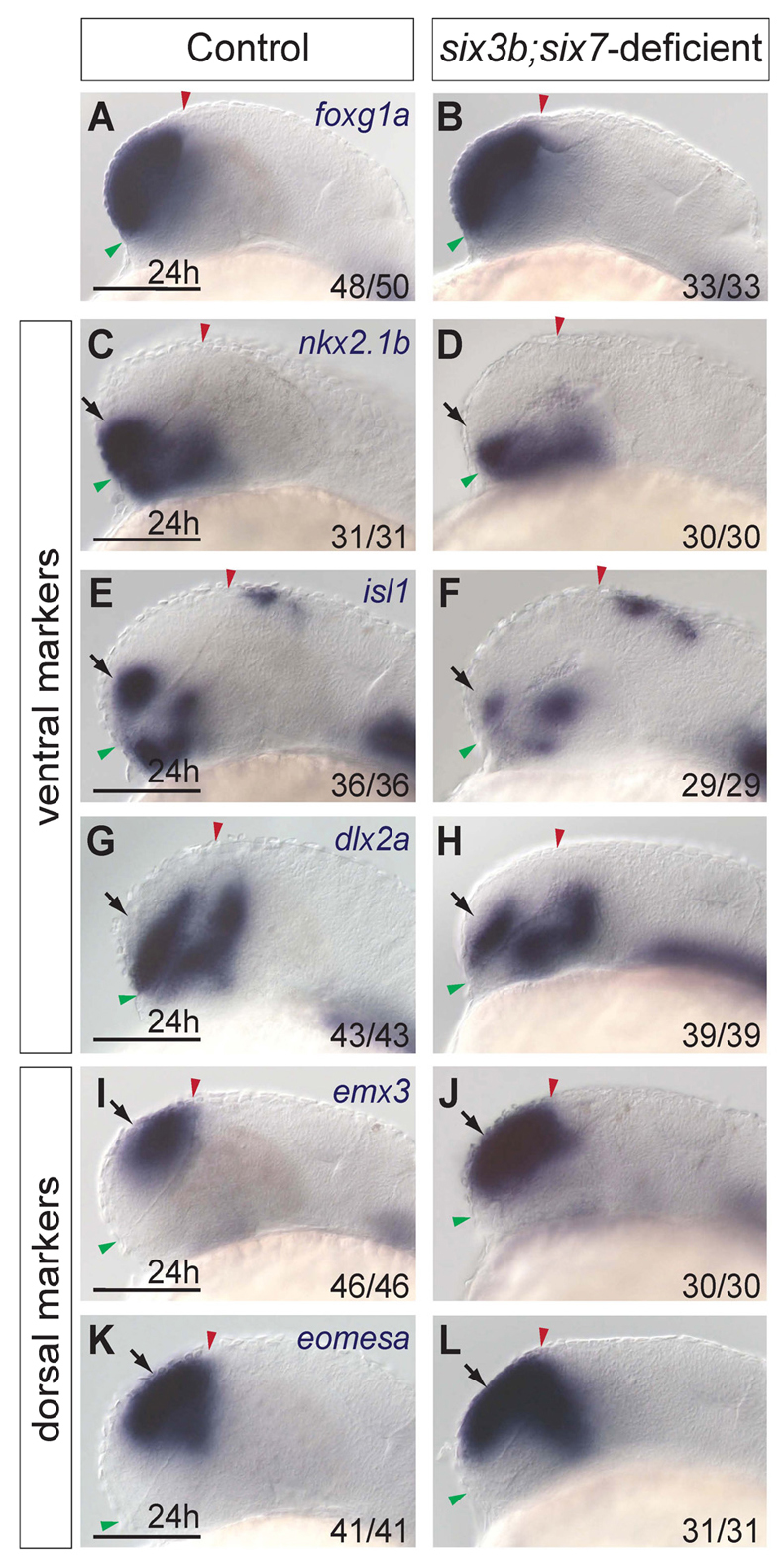
The telencephalon of six3b;six7-deficient embryos is dorsalized. (A,B) foxg1a expression in control (A) and six3b;six7-deficient (B) embryos. (C-H) Ventral telencephalic expression of nkx2.1b (C,D), isl1 (E,F) and dlx2a (G,H) in control (C,E,G) and six3b;six7-deficient (D,F,H) embryos. (I-L) Dorsal telencephalic expression of emx3 (I,J) and eomesa (K,L) in control (I,K) and six3b;six7-deficient (J,L) embryos. All embryos are at 24 hpf. Control embryos are uninjected six3bvu87/+ or six3bvu87/vu87 embryos. Arrows indicate telencephalic expression domains. Embryos are shown in lateral view with anterior towards the left. Red and green arrowheads indicate dorsal and ventral edges of the telencephalon, respectively. Fraction in each panel denotes number of embryos affected over number examined. Scale bars: 100 μm.
Abnormal proliferation and apoptosis do not significantly contribute to deficiency of ventral telencephalic fates
Decreased proliferation, increased apoptosis or altered fate specification could contribute to the reduction of ventral telencephalon in six3b;six7-deficient embryos. Indeed, Six3 was demonstrated to affect each of these processes (Carl et al., 2002; Lagutin et al., 2003; Del Bene et al., 2004; Gestri et al., 2005; Appolloni et al., 2008). Thus, we first investigated whether decreased proliferation of ventral telencephalic precursors contributed to the reduction in the nkx2.1b telencephalic expression domain. Because the exact location of these progenitors at early segmentation stages is not well defined, we could not directly assess their proliferation. We therefore asked whether global inhibition of cellular proliferation affects telencephalic DV patterning in wild-type embryos. To block proliferation, wild-type embryos were treated with DNA replication inhibitors hydroxyurea (20 mM) and aphidicolin (150 μM) from mid-gastrulation (80% epiboly; 8 hpf) onwards. Treatment at this time does not interfere with telencephalon induction and correlates with the onset of six3b and six7 expression in the forebrain anlage (Grinblat et al., 1998; Seo et al., 1998a; Seo et al., 1998b). We counted proliferating cells located within a defined anterior region of the embryos (supplementary material Fig. S1), where ventral telencephalon progenitors are known to reside (Woo and Fraser, 1995). Hydroxyurea/aphidicolin treatment effectively inhibited proliferation within 2 hours (tailbud stage, 10 hpf), as evidenced by an 81% reduction in the number of cells positively labeled with phospho-Histone H3 antibody, a marker of late G2-M phase (supplementary material Fig. S1A,B,E). Inhibition of proliferation was maintained until the 20-somite stage when we observed a 65% reduction in the number of phospho-Histone H3-positive cells in the anterior neural tube (supplementary material Fig. S1C-E). In 20-somite stage hydroxyurea/aphidicolin-treated embryos, we observed a 9.5% reduction of the DV length of the telencephalon, as defined by foxg1a expression, compared with vehicle-treated embryos (Fig. 2A,B,G; P<0.01). Notably, emx3 and nkx2.1b expression domains appeared relatively normal in the telencephalon in hydroxyurea/aphidicolin-treated embryos (Fig. 2C-F). Quantification of the length of these domains along the telencephalic DV axis showed a slight but statistically insignificant reduction (~5%) compared with vehicle-treated embryos (Fig. 2G; P>0.27 for both markers). This discrepancy between the reduction of the telencephalon size and the smaller reduction of its ventral and dorsal domains may resolve from analyzing additional embryos. As a severe reduction in proliferation did not significantly affect the domain size of ventral telencephalic progenitors in wild-type embryos, we conclude that cellular proliferation does not play a prominent role in generating ventral telencephalon fates during the time when Six3 function is required.
Fig. 2.
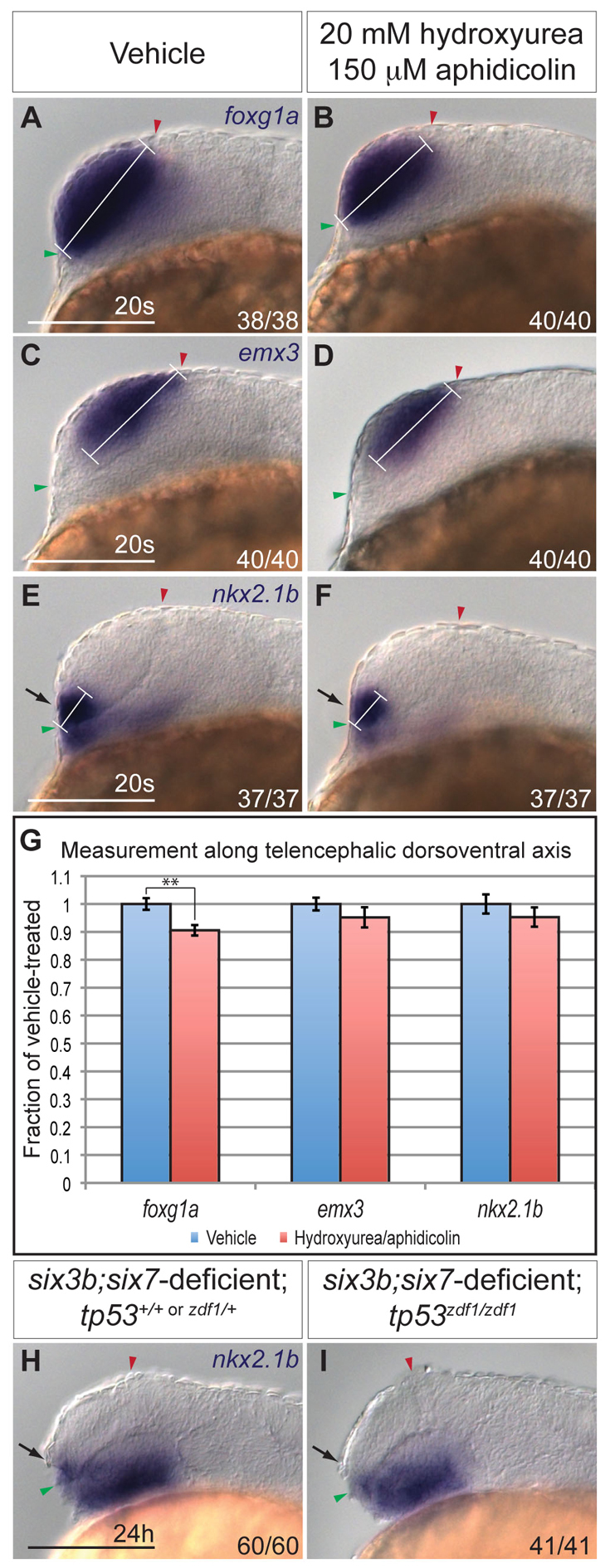
Cellular proliferation and apoptosis do not significantly contribute to reduction of ventral telencephalon. (A-F) Expression of foxg1a (A,B), emx3 (C,D), and nkx2.1b (E,F) at the 20-somite stage in wild-type embryos treated with 2% dimethyl sulfoxide alone (A,C,E) or 20 mM hydroxyurea and 150 μM aphidicolin at 80% epiboly (B,D,F). White bracket indicates length of DV domain measured for quantification. Embryos are shown in lateral view with anterior towards the left. Red and green arrowheads indicate dorsal and ventral edges of the telencephalon, respectively. (G) Graph shows expression domain length along the DV telencephalic axis divided by average DV domain length of vehicle-treated embryos. For each sample, n=11 embryos. Blue and red columns denote vehicle- and hydroxyurea/aphidicolin-treated embryos, respectively. Error bars denote s.e.m. **P<0.01. (H,I) Expression of nkx2.1b at 24 hpf in six3b;six7-deficient embryos (H) that are also tp53zdf1/zdf1 (I). Arrows in E,F,H,I indicate ventral telencephalon. Scale bars: 100 μm.
To test whether increased apoptosis is responsible for the reduction of ventral telencephalic progenitors in six3b;six7-deficient embryos, we first analyzed apoptosis using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL). No apparent increase in TUNEL was observed in the anterior neural tube at either the eight-somite or 12-somite stage in six3b;six7-deficient embryos (supplementary material Fig. S2A,B,D,E). To address more directly whether apoptosis plays a role in the reduction of ventral telencephalic fates observed in six3b;six7-deficient embryos, we introduced the tp53zdf1 allele, a mutation in the DNA-binding domain of tp53 (tumor protein 53), into six3b;six7-deficient embryos to interfere with apoptosis genetically. Similar to tp53zdf1/zdf1 embryos, which are characterized by a dramatic global reduction of apoptosis (Berghmans et al., 2005), six3b;six7;tp53-deficient embryos showed a strong reduction or absence of apoptotic cells at the 8-somite stage, as evidenced by TUNEL (supplementary material Fig. S2C). However, global reduction of tp53-dependent apoptosis failed to suppress the smaller size of the telencephalic nkx2.1b domain in six3b;six7-deficient embryos (Fig. 2H,I). Collectively, these data support the conclusion that increased apoptosis and reduced proliferation are not major contributing mechanisms to the reduction of ventral telencephalon cell fates in six3b;six7-deficient embryos.
Ventral telencephalon is not properly specified in six3b;six7-deficient embryos
A third potential mechanism for Six3 function in DV telencephalon patterning is through specification of ventral telencephalic progenitors. If Six3 specifies ventral fate in the telencephalon, then lack of ventral progenitors in six3b;six7-deficient embryos should be evident near the onset of specific marker gene expression. DV polarity in the telencephalon can first be recognized by specific gene expression at the 12-somite stage (Danesin et al., 2009). We examined six3b;six7-deficient embryos at the 16-somite stage when expression of dorsal and ventral markers can be unambiguously detected. At this early stage, telencephalic DV patterning was already perturbed, as evidenced by reduced nkx2.1b expression domain (Fig. 3A,B). Consistent with this result, emx3 expression, which normally becomes restricted to dorsal progenitors by mid-segmentation, was observed throughout the telencephalon in 12-somite stage six3b;six7-deficient embryos (Fig. 3C,D). This early patterning defect supports the notion that ventral telencephalic progenitors are not specified properly in six3b;six7-deficient embryos.
Fig. 3.
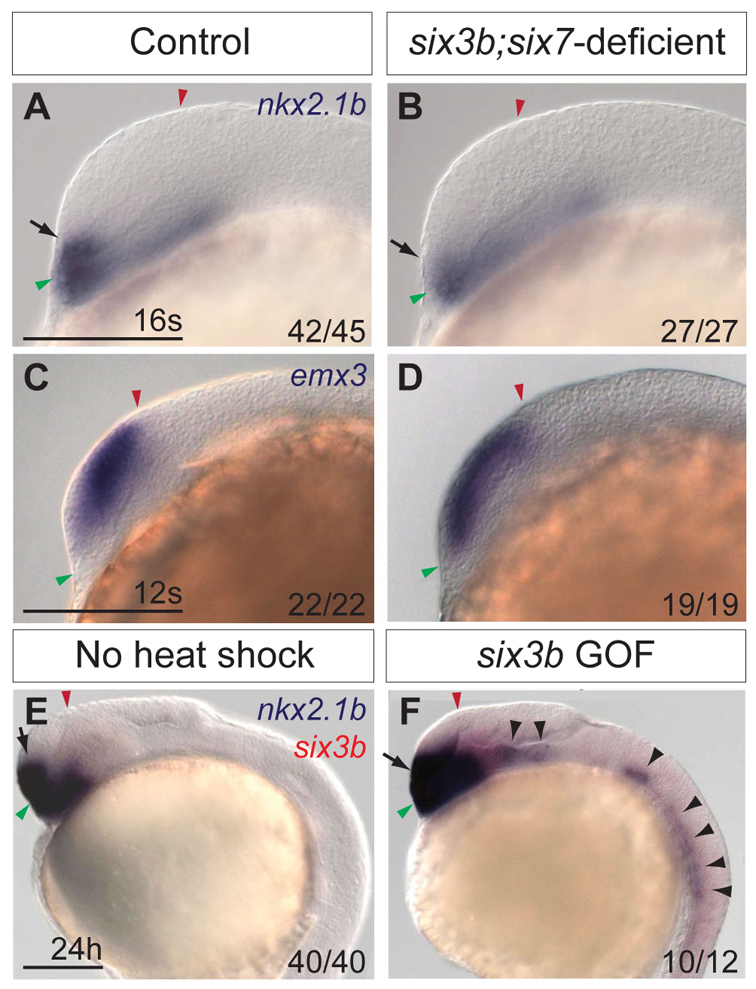
Six3b is required for specification of ventral telencephalon. (A,B) nkx2.1b expression in telencephalon (arrows) of control (A) and six3b;six7-deficient (B) embryos at the 16-somite stage. (C,D) emx3 expression in control (C) and six3b;six7-deficient (D) embryos at the 12-somite stage. (E,F) nkx2.1b (purple) expression in telencephalon (arrows) and ectopic expression (black arrowheads) at 24 hpf in Tg(hsp70l:Gal4-VP16); Tg(UAS:six3b) embryos globally misexpressing six3b (red) (F) compared with embryos not subjected to heat shock (E). GOF denotes gain of function. Embryos are shown in lateral view with anterior towards the left. Red and green arrowheads indicate dorsal and ventral edges of the telencephalon, respectively. Scale bars: 100 μm.
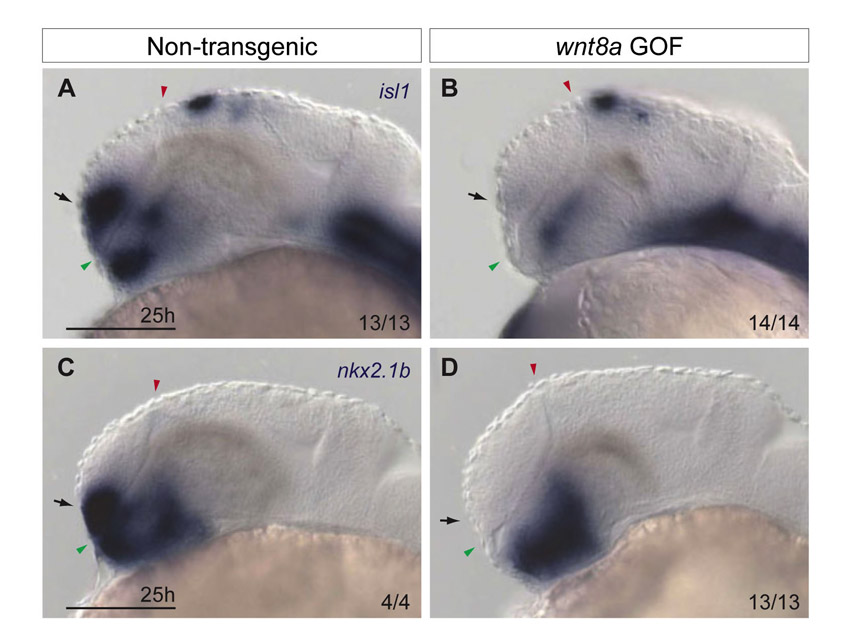
To test further the idea that Six3 controls fate specification of ventral progenitors, we assessed the ability of six3b misexpression to induce ectopic nkx2.1b expression. Six3 overexpression in chick embryos is capable of inducing ectopic Nkx2.1 expression in more posterior brain regions (Kobayashi et al., 2002). To understand whether Six3 regulates expression of nkx2.1b similarly in zebrafish, we analyzed Tg(hsp70l:Gal4-VP16); Tg(UAS:six3b) embryos that were subjected to heat shock at the tailbud stage to globally misexpress six3b. At 24 hpf, these embryos exhibited a dorsally expanded nkx2.1b expression domain within the telencephalon, as well as ectopic nkx2.1b expression in more posterior regions of the brain and ventral spinal cord (Fig. 3E,F). These data provide strong support for the ability of Six3 to promote specification of nkx2.1b-expressing cells, and suggest that the ventral telencephalic deficits observed in six3b;six7-deficient embryos are due to impaired cell fate specification.
Six3 function is required during early segmentation for establishing ventral telencephalic cell fates
six3b and six7 are expressed in the anterior neuroectoderm, including the prospective telencephalon from late-gastrulation (9 hpf) (Kobayashi et al., 1998; Seo et al., 1998a; Seo et al., 1998b), whereas ventral telencephalic progenitors are affected by the 16-somite stage (17 hpf) in six3b;six7-deficient embryos. This temporal discrepancy raises the issue of when Six3 function is required for specification of ventral telencephalic cell fates. To address this, we induced six3b expression at different time points and examined when it was capable of rescuing the reduced nkx2.1b- and isl1-positive telencephalic cell populations in six3b;six7-deficient embryos. To misexpress six3b globally in a six3b;six7-deficient background, we crossed Tg(hsp70l:Gal4-VP16); six3bvu87/+ and Tg(UAS:six3b); six3bvu87/+ lines, injected resulting embryos with MO1-six7, and subjected these embryos to heat shock at late gastrulation or early segmentation stages. Telencephalic expression domains of isl1 and nkx2.1b were analyzed at 24 hpf. We found that inducing six3b expression at late gastrulation (10 hpf) restored nkx2.1b- and isl1-positive ventral telencephalic cell populations in six3b;six7-deficient embryos (Fig. 4C,G). However, when heat shock was applied at the 4-somite stage, nkx2.1b expression was no longer restored (Fig. 4D). Similarly, applying heat shock to embryos at the 6-somite stage could not suppress the reduction in isl1-positive cells (Fig. 4H). Given that strong global six3b mRNA induction was not present until 1.5 hours after onset of heat shock (data not shown), these results suggest that Six3 function is required by the 8-somite and 10-somite stage for generation of nkx2.1b- and isl1-positive telencephalic cell populations, respectively.
Fig. 4.

Six3b is required during early segmentation to promote ventral telencephalic fates. (A-D) nkx2.1b (purple) and six3b (red) expression in control embryos (A), six3b;six7-deficient embryos (B), six3b;six7-deficient Tg(hsp70l:Gal4-VP16); Tg(UAS:six3b) embryos induced to misexpress six3b at tailbud stage (C) and six3b;six7-deficient Tg(hsp70l:Gal4-VP16); Tg(UAS:six3b) embryos induced to misexpress six3b at the 4-somite stage (D). (E-H) isl1 (purple) and six3b (red) expression in control embryos (E), six3b;six7-deficient embryos (F), six3b;six7-deficient Tg(hsp70l:Gal4-VP16); Tg(UAS:six3b) embryos induced to misexpress six3b at tailbud stage (G) and six3b;six7-deficient Tg(hsp70l:Gal4-VP16); Tg(UAS:six3b) embryos induced to misexpress six3b at the 6-somite stage (H). Arrows indicate the ventral telencephalon in 24 hpf embryos. Embryos are shown in lateral view with anterior towards the left. Red and green arrowheads indicate dorsal and ventral edges of the telencephalon, respectively. Scale bars: 100 μm.
Six3 and Hh signaling do not regulate each other during early segmentation stages
The expansion of dorsal telencephalic cell fates at the expense of ventral ones, as observed in six3b;six7-deficient embryos, is reminiscent of telencephalic patterning defects observed when Hh signaling is perturbed. For example, zebrafish embryos in which Hh signaling is disrupted due to a mutation in the obligatory Hh pathway mediator smoothened (smo) or treatment with cyclopamine, a small molecule inhibitor of Smo, exhibit complete loss of telencephalic and diencephalic nkx2.1b expression and reduced telencephalic isl1 domain (Fig. 6A,B,J,K,M,N) (Rohr et al., 2001; Danesin et al., 2009). Because Six3 and Shh were reported to positively regulate each other's expression in mice, and simultaneous reduction of function of these two genes results in HPE with reduced ventral and expanded dorsal telencephalic fates (Geng et al., 2008), we asked whether Six3 and Shh also regulate each other in zebrafish. To determine whether Six3 acts upstream of Hh signaling, we examined the expression of patched2 (ptch2, formerly ptc1) (Concordet et al., 1996), a downstream transcriptional target of the Hh signaling pathway, in six3b;six7-deficient embryos. At early segmentation stages (2- to 3- and 8-somite stage), ptch2 expression appeared normal (Fig. 5A,B; data not shown), suggesting that the combined function of six3b and six7 was not required for Hh pathway activity at this time. Similarly, blocking Hh signaling using cyclopamine from early gastrulation (shield stage, 6 hpf) did not affect the expression of six3b or six7 in prechordal mesendoderm at mid-gastrulation (8 hpf) or in anterior neuroectoderm at the 3- and 8-somite stages (Fig. 5C-F; data not shown). Efficacy of cyclopamine treatment was confirmed by absence of ptch2 expression in sibling embryos at the same stage (data not shown). Therefore, six3b and six7 expression during gastrulation and early segmentation are independent of Hh signaling. Together, these data suggest that a positive-feedback loop between Six3 and Hh signaling that is dependent on full function of Six3 does not operate during developmental stages when telencephalic DV patterning is established in zebrafish.
Fig. 6.
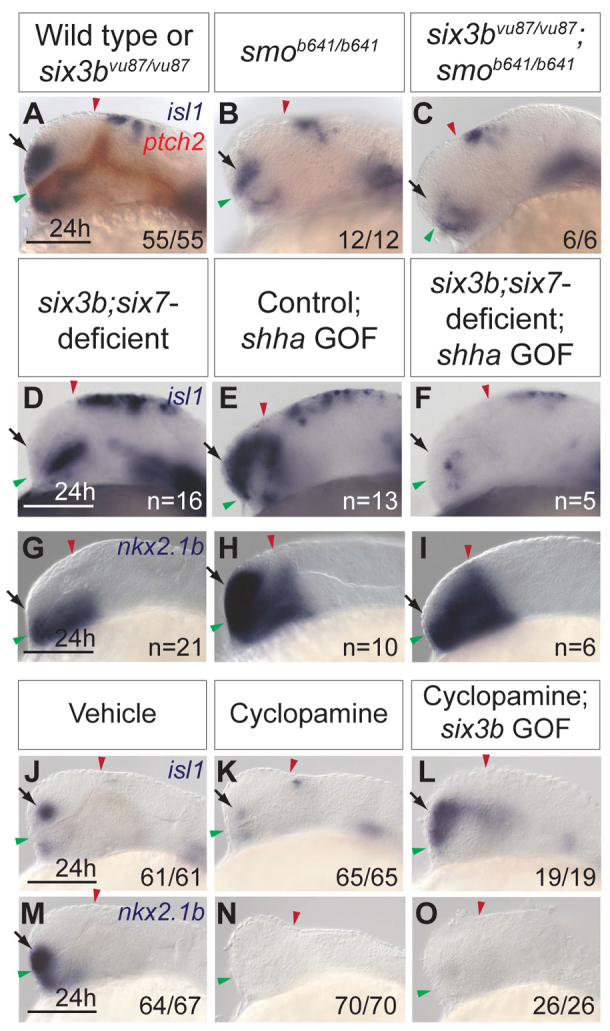
Interactions between Hh signaling and Six3 in ventral telencephalon formation. (A-C) isl1 (purple) and ptch2 (red) expression in wild-type and six3bvu87/vu87 embryos (A), smob641/b641 embryos (B) and six3bvu87/vu87;smob641/b641 embryos (C). (D-I) Expression of isl1 (D-F) and nkx2.1b (G-I) in six3b;six7-deficient embryos (D,G), control Tg(hsp70l:Gal4-VP16); Tg(UAS:shha-NH-EGFP) embryos misexpressing shha-NH-EGFP (E,H) and six3b;six7-deficient Tg(hsp70l:Gal4-VP16); Tg(UAS:shha-NH-EGFP) embryos misexpressing shha-NH-EGFP (F,I). (J-O) isl1 (J-L) and nkx2.1b (M-O) expression in vehicle-treated embryos (J,M), cyclopamine-treated embryos (K,N) and cyclopamine-treated Tg(hsp70l:Gal4-VP16); Tg(UAS:six3b) embryos misexpressing six3b (L,O). All embryos are 24 hpf. Embryos are shown in lateral view with anterior towards the left. Red and green arrowheads indicate dorsal and ventral edges of the telencephalon, respectively. Arrows indicate ventral telencephalon. Scale bars: 100 μm.
Fig. 5.
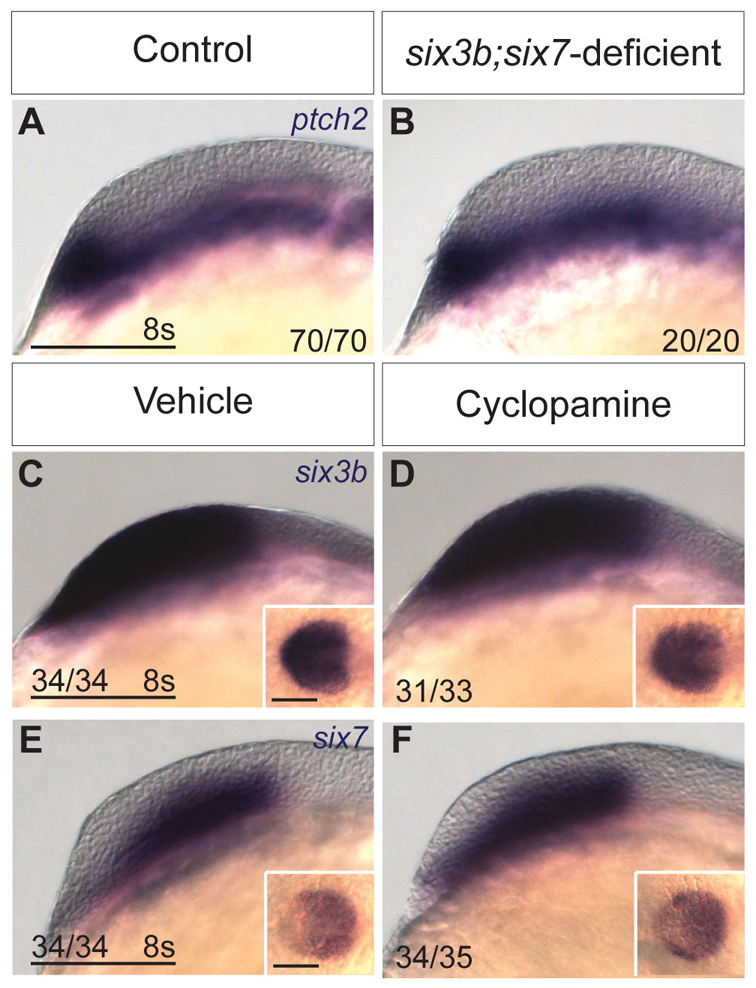
Six3 and Hh signaling do not regulate each other during early segmentation. (A,B) Expression of ptch2 in eight-somite stage control (A) and six3b;six7-deficient (B) embryos. (C-F) Expression of six3b (C,D) and six7 (E,F) in eight-somite stage wild-type embryos treated from 6 hpf with 10 μM cyclopamine (D,F) and embryos treated with 0.1% ethanol alone (C,E). Embryos are shown in lateral view with anterior towards the left. Insets show same embryo as dorsal view with anterior towards the left. Scale bars: 100 μm.
Complex interactions between Six3 and Hh signaling promote ventral telencephalic fates
Although Six3 and Hh signaling appear to promote ventral telencephalic fates independently during early segmentation stages in zebrafish, it is possible that they genetically interact in this process. To test this, we generated six3bvu87/vu87; smob641/b641 embryos and examined isl1 telencephalic expression. The isl1 telencephalic domain appeared similar in wild-type and six3bvu87/vu87 embryos, and was only mildly reduced in smob641/b641 embryos (Fig. 6A,B). However, in six3bvu87/vu87; smob641/b641 embryos, very few isl1-positive telencephalic cells could be detected (Fig. 6C). These results demonstrate a synergistic interaction between six3b function and Hh pathway activity and suggest that Six3 and Hh signaling cooperate to promote isl1-positive telencephalic cells. A similar experiment analyzing the telencephalic domain of nkx2.1b is precluded owing to the complete loss of nkx2.1b expression in Hh signaling-deficient embryos (Fig. 6N), as previously described (Rohr et al., 2001).
To better understand the interactions between Six3 and Hh signaling in the generation of ventral telencephalic cells, we tested whether overactivation of the Hh signaling pathway by misexpression of shha can compensate for the loss of six3b and six7 function. We crossed Tg(hsp70l:Gal4-VP16); six3bvu87/+ and Tg(UAS:shha-NH-EGFP); six3bvu87/+ fish, injected resulting embryos with MO1-six7 and induced shha-NH-EGFP misexpression by heat shock at late gastrulation (10 hpf). Analysis at 24 hpf showed that shha-NH-EGFP misexpression caused strong expansion of isl1 and nkx2.1b expression domains throughout the telencephalon in uninjected embryos, as well as wild-type and six3bvu87/+ embryos injected with MO1-six7 (Fig. 6E,H). By contrast, isl1 and nkx2.1b telencephalic expression domains remained strongly reduced in six3b;six7-deficient embryos overexpressing shha-NH-EGFP (Fig. 6F,I), suggesting that induction of telencephalic isl1- and nkx2.1b-positive fates by Hh signaling depends on Six3 function.
In a set of reciprocal experiments, we treated Tg(hsp70l:Gal4-VP16); Tg(UAS:six3b) embryos with cyclopamine from early gastrulation to block Hh signaling, and induced six3b misexpression by heat shock at the end of gastrulation. All cyclopamine-treated embryos showed identical morphology to smo mutant embryos at 24 hpf, confirming a disruption of Hh signaling (data not shown). Control embryos treated with cyclopamine but not subjected to heat shock had a strongly reduced isl1-positive telencephalic domain at 24 hpf (Fig. 6K). However, in cyclopamine-treated embryos misexpressing six3b, expression of telencephalic isl1 was restored or even expanded (Fig. 6L). The same result was obtained when six3b was misexpressed in smob641/b641 background (not shown). We also analyzed nkx2.1b expression in embryos misexpressing six3b in the absence of Hh signaling. In contrast to its ability to promote isl1-positive cells, six3b misexpression could not restore nkx2.1b expression in Hh signaling-deficient embryos (Fig. 6M-O), suggesting Six3 promotes telencephalic nkx2.1b-positive cell population in an Hh-dependent manner. These results are consistent with the notion that Six3 functions permissively to provide competence for Shh to induce nkx2.1b forebrain expression (Kobayashi et al., 2002), yet suggest an instructive role in inducing isl1-positive cells in the ventral telencephalon independently of Hh signaling.
foxg1a expression is transiently regulated by Six3 during early segmentation
Foxg1 function is required during early segmentation to promote ventral telencephalic development, and its loss of function results in telencephalic phenotypes highly reminiscent of six3b;six7- or Hh signaling-deficient embryos (Xuan et al., 1995; Martynoga et al., 2005; Danesin et al., 2009). We asked whether foxg1a expression is affected in six3b;six7-deficient embryos when telencephalic DV patterning is established. Indeed, we found that foxg1a expression was not established at the 1-somite stage and remained strongly reduced in six3b;six7-deficient embryos during early segmentation (6-somite stage), compared with control embryos (Fig. 7A,B; data not shown). However, by the 12-somite stage, telencephalic foxg1a expression had largely recovered (Fig. 7C,D). These data demonstrate a biphasic regulation of foxg1a expression where its early but not later expression depend on six3b and six7 function.
Fig. 7.

Interaction between Six3 and Foxg1a in ventral telencephalon development. (A-D) Expression of foxg1a in control (A,C) and six3b;six7-deficient (B,D) embryos at the 6-somite stage (A,B) and 12-somite stage (C,D). (E-H) Expression of six3b (E,F) and six7 (G,H) in uninjected control (UIC) embryos (E,G) and MO2-foxg1a injected embryos (F,H) at the 4-somite stage. Insets in E-H are dorsal views of the same embryo with anterior leftwards. Inset scale bars: 50 μm. (I-P) isl1 (purple) (I-L) or nkx2.1b (purple) (M-P) and six3b (red) expression in UIC embryos (I,M), UIC Tg(hsp70l:Gal4-VP16); Tg(UAS:six3b) embryos misexpressing six3b (J,N), MO2-foxg1a injected embryos (K,O) and MO2-foxg1a injected Tg(hsp70l:Gal4-VP16); Tg(UAS:six3b) embryos misexpressing six3b (L,P) at 24 hpf. Embryos are shown in lateral view with anterior towards the left. Red and green arrowheads indicate dorsal and ventral edges of the telencephalon, respectively. Arrows indicate telencephalic expression domains. Scale bars: 100 μm.
To test whether foxg1a activity also regulates Six3 expression, we analyzed expression of six3b and six7 during early segmentation in embryos injected with MO2-foxg1a. Although foxg1a morphant embryos showed a profound reduction of telencephalic nkx2.1b at 24 hpf (data not shown) (Danesin et al., 2009), expression of six3b and six7 appeared normal in sibling foxg1a morphant embryos at the 4-somite stage (Fig. 7E-H), demonstrating that foxg1a function is not required for six3b and six7 expression. These results place foxg1a function downstream of Six3 in telencephalic DV patterning.
Next, we asked whether foxg1a function was required for the ability of Six3 to promote ventral telencephalic fates by injecting MO2-foxg1a into Tg(hsp70l:Gal4-VP16); Tg(UAS:six3b) embryos and applying heat shock at tailbud stage. Disruption of Foxg1a function did not suppress the expansion of isl1 in six3b-misexpressing embryos (Fig. 7I-L). By contrast, telencephalic expression of nkx2.1b was strongly reduced in six3b-misexpressing embryos injected with MO2-foxg1a (Fig. 7M-P), whereas ectopic nkx2.1b expression was unaffected (40/44 embryos; data not shown). These results demonstrate that in telencephalon, Six3b can promote isl1 but not nkx2.1b expression independently of Foxg1.
Together with previous studies (Kobayashi et al., 2002; Danesin et al., 2009; Beccari et al., 2012), our data place Foxg1a downstream of both Hh signaling and Six3 in promoting nkx2.1-positive cells in the telencephalon. Given that Six3 can promote isl1-positive cells independent of Foxg1, we asked whether such differential dependence existed also between Hh signaling and Foxg1. Expansion of telencephalic expression of nkx2.1b and dlx2a due to shha misexpression requires foxg1a function (Danesin et al., 2009). We tested whether this is also the case for isl1. Tg(hsp70l:Gal4-VP16); Tg(UAS:shha-NH-EGFP) embryos were injected with MO2-foxg1a, heat shocked at tailbud stage and analyzed at 24 hpf for isl1 expression. Unlike Six3, overactivation of Hh signaling could not restore isl1 expression in foxg1a morphants (supplementary material Fig. S3), further supporting the notion that Foxg1a functions downstream of Hh signaling.
wnt8b expression is upregulated in six3b;six7-deficient embryos during early segmentation
Wnt ligands are expressed in the dorsal forebrain, and Wnt/β-catenin signaling has been shown to promote dorsal telencephalic fates (van de Water et al., 2001; Carl et al., 2007; Danesin et al., 2009). In mouse, excess Wnt/β-catenin signaling is sufficient to expand dorsal telencephalic fates ventrally and reduce ventral fates (Backman et al., 2005), similar to the phenotype observed in six3b;six7-deficient zebrafish embryos. Indeed, we find that overactivation of the Wnt/β-catenin pathway at early segmentation leads to telencephalic phenotypes almost identical to those observed in six3b;six7-deficient embryos (supplementary material Fig. S4). We therefore examined expression of the Wnt/β-catenin target gene axin2 (Kelly et al., 1995; Leung et al., 2002; Carl et al., 2007) in six3b;six7-deficient embryos. Whereas at tailbud stage axin2 expression appeared similar in control and six3b;six7-deficient embryos (supplementary material Fig. S5), at the 8-somite stage it was expanded anteriorly into the telencephalon of six3b;six7-deficient embryos (Fig. 8A,B), suggesting increased Wnt/β-catenin activity in the telencephalon.
Fig. 8.

Six3 represses wnt8b expression in a Foxg1a-independent manner. (A-D) Expression of axin2 (A,B) and wnt8b (C,D) in control (A,C) and six3b;six7-deficient embryos (B,D) at the 8-somite stage. Black arrowhead indicates anterior limit of expression. (E-H) wnt8b (purple) and six3b (red) expression in UIC embryos (E), UIC Tg(hsp70l:Gal4-VP16); Tg(UAS:six3b) embryos misexpressing six3b (F), MO2-foxg1a-injected embryos (G) and MO2-foxg1a-injected Tg(hsp70l:Gal4-VP16); Tg(UAS:six3b) embryos misexpressing six3b (H) at 24 hpf. Embryos are shown in lateral view with anterior towards the left. Red and green arrowheads indicate dorsal and ventral edges of the telencephalon, respectively. Scale bars: 100 μm. (I) Genetic model of Six3 function in zebrafish telencephalon DV patterning. Six3 and Hh signaling function in parallel to promote foxg1a expression, which in turn promotes ventral telencephalon. Six3 and Foxg1a each can repress expression of Wnt/β-catenin ligands such as wnt8b, which can repress ventral telencephalon.
The Wnt/β-catenin ligand wnt8b is expressed in the dorsal forebrain at early segmentation, and its expression is directly repressed by both Six3 and Foxg1a (Carl et al., 2007; Danesin et al., 2009; Liu et al., 2010). We therefore analyzed wnt8b expression in six3b;six7-deficient embryos at the 8-somite stage, and found that expression was also expanded anteriorly, and this anterior expansion was noted as early as the 5-somite stage (Fig. 8C,D; data not shown). As Six3 has been shown to directly repress Wnt8b expression in mouse embryos (Liu et al., 2010), we asked whether Six3b can repress wnt8b expression in zebrafish and whether such repression was dependent on Foxg1a function. To test this, MO2-foxg1a was injected into Tg(hsp70l:Gal4-VP16); Tg(UAS:six3b) embryos and heat shock was applied at tailbud stage. As previously shown, expression of wnt8b is anteriorly expanded owing to disruption of Foxg1a function (Danesin et al., 2009); however, misexpression of six3b repressed wnt8b in both uninjected and foxg1a morphant embryos (Fig. 8E-H). Together, these results support the notion that Six3b can repress wnt8b expression in a Foxg1a-independent manner, and suggest that expanded activity of the Wnt/β-catenin pathway could contribute to the reduced ventral telencephalic fates in six3b;six7-deficient embryos.
DISCUSSION
Zebrafish Six3-related genes as a tool for dissecting the function of Six3 in forebrain development
We have taken advantage of the functional redundancies between three Six3-related genes in the zebrafish genome to dissect the roles of Six3 in telencephalic development (Seo et al., 1998a; Seo et al., 1998b). The homeodomains of zebrafish Six3-related genes can bind the same DNA sequence (Suh et al., 2010), and misexpression of six3a, six3b, six7 or human SIX3 in zebrafish embryos leads to the same early phenotypes (i.e. dorsalization, increased head and eye size) (D.C., L.S.-K. and A.I., unpublished) (Domene et al., 2008; Geng et al., 2008). These data strongly suggest that zebrafish Six3-related genes could act redundantly during development and in conserved fashion with mammalian orthologs. Indeed, we have previously shown that whereas the loss of six3b or six7 function alone did not result in observable phenotypes, their combined loss of function resulted in microphthalmia or anophthalmia and brain laterality defects (Inbal et al., 2007). In the current study, we identified abnormal telencephalic DV patterning as a consequence of combined loss of six3b and six7 function. Both eye malformations and telencephalic patterning defects are consistent with phenotypes observed when Six3 function is perturbed in other vertebrates and in cases of HPE (Carl et al., 2002; Lagutin et al., 2003; Ando et al., 2005; Geng et al., 2008).
As Six3 regulates many processes during early development, three redundant genes in zebrafish afford generation of discrete hypomorphic phenotypes through combinatorial loss of function. For example, loss of Six3 function in mouse results in lack of both forebrain and eyes (Lagutin et al., 2003), whereas the loss of eyes in six3b;six7-deficient embryos is uncoupled from lack of forebrain (Inbal et al., 2007). Similarly in medaka fish, differential tissue sensitivities are observed in embryos deficient in Six3.1 or Six3.2 (Carl et al., 2002; Beccari et al., 2012). However, certain phenotypes related to loss of Six3 function have not yet been described in zebrafish, such as midline deficiencies seen in HPE, which are not observed in six3b;six7-deficient embryos (D.C., L.S.-K. and A.I., unpublished). To obtain a more comprehensive understanding of the roles of Six3, it will be important to also analyze loss of six3a function alone and in combination with six3b and/or six7. Indeed, such functional redundancies of three Nodal-related genes facilitated dissection of their roles in mesendoderm induction and patterning, and left-right axis specification (Schier, 2009). Overall, zebrafish provide a powerful system with which to study the specific roles of Six3 in early CNS development.
Parallel functions of Six3 and Hh signaling converge on Foxg1a
Several observations suggest Six3 and Hh signaling cooperate in promoting ventral telencephalic fates. First, reduction of Six3 function or Hh signaling each result in reduction of ventral and expansion of dorsal telencephalic fates (Fig. 1; Fig. 6K,N) (Chiang et al., 1996; Rallu et al., 2002; Danesin et al., 2009). Conversely, gain of Six3 function or excess Hh signaling each result in an expansion of ventral telencephalic fates at the expense of dorsal ones (Fig. 4C,G; Fig. 6E,H) (Kohtz et al., 1998; Rohr et al., 2001; Rallu et al., 2002). Second, both Six3 and Shh function during early segmentation stages to promote ventral telencephalic fates (Fig. 4) (Kohtz et al., 1998; Danesin et al., 2009). Interestingly, our results show nkx2.1b-positive cells require Six3 function slightly earlier or longer than isl1-positive cells, which suggests that the most ventromedial telencephalic fates may be specified earlier than more dorsally located ventral fates. This is consistent with nkx2.1b being expressed earlier than isl1 in the ventral telencephalon (Rohr et al., 2001), and also with data in rat showing Shh first induces Nkx2.1-positive and later Islet-1-positive ventral telencephalic progenitors (Kohtz et al., 1998). Third, our data show that global misexpression of six3b activates ectopic nkx2.1b expression only near a source of Shh, similar to what has been previously described in chick embryos (Kobayashi et al., 2002). We interpret this result to mean that Six3 provides competence for cells to respond to Shh by expressing nkx2.1b. Fourth, exacerbated deficiency of telencephalic isl1 expression in six3bvu87/vu87; smob641/b641 compound mutants demonstrates a strong genetic interaction between Six3 and Hh signaling in the formation of these ventral telencephalic progenitors.
In this study, we did not find evidence for Six3 regulating Hh signaling or vice versa. A previous report in zebrafish showed that loss of Hh signaling affects six3b expression by midsegmentation (Sanek et al., 2009); however, we observed no significant changes in six3b or six7 expression due to loss of Hh signaling during early segmentation when Six3 regulates DV telencephalon patterning.
As our results suggest that Six3 and Hh signaling function largely in parallel to specify ventral telencephalic fates, we examined the possibility that Foxg1, which is also required to promote ventral telencephalic cell fates, may link Six3 and Hh signaling. Loss of Foxg1 gene function results in a dorsalized telencephalon almost identical to that observed in six3b;six7-deficient or Hh signaling-deficient embryos, and Foxg1 functions at similar developmental stages (Xuan et al., 1995; Martynoga et al., 2005; Danesin et al., 2009). Our findings show that induction and early maintenance of foxg1a expression is affected in six3b;six7-deficient zebrafish embryos during early segmentation when ventral telencephalon is specified. These data, together with our observation that six3b and six7 expression is not affected by loss of foxg1a function, place Foxg1 downstream of Six3 in telencephalon DV patterning. Consistent with this conclusion, medaka Six3.2 has been shown to bind highly conserved non-coding elements in the Foxg1 regulatory region in vitro (Beccari et al., 2012), and Six3 misexpression in chick embryos could activate ectopic Foxg1 expression near the mid-hindbrain boundary (Kobayashi et al., 2002). Expression of foxg1a during early segmentation stages is also transiently dependent on Hh signaling, and misexpression of foxg1a could restore expression of some ventral telencephalic markers in embryos that lack Hh signaling (Danesin et al., 2009). Consistent with the notion of Foxg1a acting downstream of Hh signaling, misexpression of shha is insufficient to promote ventral telencephalon cell fates in foxg1a-deficient embryos (supplementary material Fig. S3) (Danesin et al., 2009). Therefore, we propose that foxg1a is a common downstream effector of Six3 and Hh signaling in the process of telencephalon patterning during early segmentation (Fig. 8I).
Hh signaling- and Foxg1a-independent function of Six3
Our data support the notion that Six3 and Hh signaling cooperate to establish expression of foxg1a during early segmentation, which is required to promote expression of nkx2.1b in the ventral telencephalon. This is a strict cooperation between Six3 and Hh signaling such that increased activation of one pathway cannot compensate for loss of the other, nor can they compensate for the loss of foxg1a function. Surprisingly, Six3 can promote isl1-positive cells independently of Hh signaling and foxg1a. Although this could be interpreted that Six3 functions downstream of Hh and Foxg1a, given that six3b and six7 expression is not affected by lack of Hh signaling or Foxg1a function during the developmental time window when DV patterning of the telencephalon is established, we favor the interpretation that Six3 acts in parallel to Hh and Foxg1a to specify this cell type.
Both Six3 and Foxg1 have been shown to directly repress expression of Wnt8b (Danesin et al., 2009; Liu et al., 2010), and Six3 has also been shown to directly repress Wnt1 (Lagutin et al., 2003). Wnt/β-catenin activity can promote dorsal and repress ventral telencephalic fates (supplementary material Fig. S4) (van de Water et al., 2001; Backman et al., 2005). We demonstrate here that the Wnt/β-catenin pathway, and specifically wnt8b expression, is upregulated in telencephalon of six3b;six7-deficient embryos. Given that Six3 and Foxg1a can each repress wnt8b expression, regulation of the Wnt/β-catenin pathway by Six3 may be responsible for the Foxg1a- and Hh signaling-independent function of Six3 in promoting telencephalic isl1 (Fig. 8I). As foxg1a misexpression is also sufficient to rescue isl1 expression in embryos lacking Hh signaling (Danesin et al., 2009), it will be interesting to test whether this can also be attributed to Foxg1a repression of Wnt ligands. As several Wnt ligands are present near the developing telencephalon (Ciani and Salinas, 2005; Carl et al., 2007), reduction of wnt8b function alone may be insufficient to suppress the six3b;six7-deficient phenotype in ventral telencephalon, as is the case for foxg1a morphant embryos (Danesin et al., 2009). The role of Six3 and Foxg1a in regulation of other regionally expressed Wnt ligands remains to be tested, and may provide additional insight into the mechanisms of DV patterning in telencephalon.
Supplementary Material
Acknowledgements
We thank Bruce Appel, Corinne Houart, Randall Moon, Bruce Riley, Gilbert Weidinger and Steve Wilson for sending reagents and fish, and members of the Solnica-Krezel and Inbal laboratories for helpful comments and suggestions. We also thank Heidi Beck, Amanda Bradshaw, Tina Ho and Erik Sanders for excellent zebrafish care and technical support.
Footnotes
Funding
This work was supported by National Institutes of Health – National Institute of Neurological Disorders and Stroke Grant [R01 NS52386 to L.S.-K.] and by The Israel Science Foundation [791/09 to A.I.]. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.076018/-/DC1
References
- Akimenko M. A., Ekker M., Wegner J., Lin W., Westerfield M. (1994). Combinatorial expression of three zebrafish genes related to distal-less: part of a homeobox gene code for the head. J. Neurosci. 14, 3475-3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando H., Kobayashi M., Tsubokawa T., Uyemura K., Furuta T., Okamoto H. (2005). Lhx2 mediates the activity of Six3 in zebrafish forebrain growth. Dev. Biol. 287, 456-468 [DOI] [PubMed] [Google Scholar]
- Appolloni I., Calzolari F., Corte G., Perris R., Malatesta P. (2008). Six3 controls the neural progenitor status in the murine CNS. Cereb. Cortex 18, 553-562 [DOI] [PubMed] [Google Scholar]
- Backman M., Machon O., Mygland L., van den Bout C. J., Zhong W., Taketo M. M., Krauss S. (2005). Effects of canonical Wnt signaling on dorso-ventral specification of the mouse telencephalon. Dev. Biol. 279, 155-168 [DOI] [PubMed] [Google Scholar]
- Beccari L., Conte I., Cisneros E., Bovolenta P. (2012). Sox2-mediated differential activation of Six3.2 contributes to forebrain patterning. Development 139, 151-164 [DOI] [PubMed] [Google Scholar]
- Berghmans S., Murphey R. D., Wienholds E., Neuberg D., Kutok J. L., Fletcher C. D., Morris J. P., Liu T. X., Schulte-Merker S., Kanki J. P., et al. (2005). tp53 mutant zebrafish develop malignant peripheral nerve sheath tumors. Proc. Natl. Acad. Sci. USA 102, 407-412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bovolenta P., Mallamaci A., Puelles L., Boncinelli E. (1998). Expression pattern of cSix3, a member of the Six/sine oculis family of transcription factors. Mech. Dev. 70, 201-203 [DOI] [PubMed] [Google Scholar]
- Carl M., Loosli F., Wittbrodt J. (2002). Six3 inactivation reveals its essential role for the formation and patterning of the vertebrate eye. Development 129, 4057-4063 [DOI] [PubMed] [Google Scholar]
- Carl M., Bianco I. H., Bajoghli B., Aghaallaei N., Czerny T., Wilson S. W. (2007). Wnt/Axin1/beta-catenin signaling regulates asymmetric nodal activation, elaboration, and concordance of CNS asymmetries. Neuron 55, 393-405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C., Litingtung Y., Lee E., Young K. E., Corden J. L., Westphal H., Beachy P. A. (1996). Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 383, 407-413 [DOI] [PubMed] [Google Scholar]
- Ciani L., Salinas P. C. (2005). WNTs in the vertebrate nervous system: from patterning to neuronal connectivity. Nat. Rev. Neurosci. 6, 351-362 [DOI] [PubMed] [Google Scholar]
- Concordet J. P., Lewis K. E., Moore J. W., Goodrich L. V., Johnson R. L., Scott M. P., Ingham P. W. (1996). Spatial regulation of a zebrafish patched homologue reflects the roles of sonic hedgehog and protein kinase A in neural tube and somite patterning. Development 122, 2835-2846 [DOI] [PubMed] [Google Scholar]
- Danesin C., Peres J. N., Johansson M., Snowden V., Cording A., Papalopulu N., Houart C. (2009). Integration of telencephalic Wnt and hedgehog signaling center activities by Foxg1. Dev. Cell 16, 576-587 [DOI] [PubMed] [Google Scholar]
- Davidson A. E., Balciunas D., Mohn D., Shaffer J., Hermanson S., Sivasubbu S., Cliff M. P., Hackett P. B., Ekker S. C. (2003). Efficient gene delivery and gene expression in zebrafish using the Sleeping Beauty transposon. Dev. Biol. 263, 191-202 [DOI] [PubMed] [Google Scholar]
- Del Bene F., Tessmar-Raible K., Wittbrodt J. (2004). Direct interaction of geminin and Six3 in eye development. Nature 427, 745-749 [DOI] [PubMed] [Google Scholar]
- Domene S., Roessler E., El-Jaick K. B., Snir M., Brown J. L., Velez J. I., Bale S., Lacbawan F., Muenke M., Feldman B. (2008). Mutations in the human SIX3 gene in holoprosencephaly are loss of function. Hum. Mol. Genet. 17, 3919-3928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubourg C., Bendavid C., Pasquier L., Henry C., Odent S., David V. (2007). Holoprosencephaly. Orphanet. J. Rare Dis. 2, 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekker S. C., Ungar A. R., Greenstein P., von Kessler D. P., Porter J. A., Moon R. T., Beachy P. A. (1995). Patterning activities of vertebrate hedgehog proteins in the developing eye and brain. Curr. Biol. 5, 944-955 [DOI] [PubMed] [Google Scholar]
- Geng X., Speirs C., Lagutin O., Inbal A., Liu W., Solnica-Krezel L., Jeong Y., Epstein D. J., Oliver G. (2008). Haploinsufficiency of Six3 fails to activate Sonic hedgehog expression in the ventral forebrain and causes holoprosencephaly. Dev. Cell 15, 236-247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George R. A., Heringa J. (2002). An analysis of protein domain linkers: their classification and role in protein folding. Protein Eng. 15, 871-879 [DOI] [PubMed] [Google Scholar]
- Gestri G., Carl M., Appolloni I., Wilson S. W., Barsacchi G., Andreazzoli M. (2005). Six3 functions in anterior neural plate specification by promoting cell proliferation and inhibiting Bmp4 expression. Development 132, 2401-2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinblat Y., Gamse J., Patel M., Sive H. (1998). Determination of the zebrafish forebrain: induction and patterning. Development 125, 4403-4416 [DOI] [PubMed] [Google Scholar]
- Hebert J. M., Fishell G. (2008). The genetics of early telencephalon patterning: some assembly required. Nat. Rev. Neurosci. 9, 678-685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inbal A., Topczewski J., Solnica-Krezel L. (2006). Targeted gene expression in the zebrafish prechordal plate. Genesis 44, 584-588 [DOI] [PubMed] [Google Scholar]
- Inbal A., Kim S. H., Shin J., Solnica-Krezel L. (2007). Six3 represses nodal activity to establish early brain asymmetry in zebrafish. Neuron 55, 407-415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A., Takahashi M., Hatta K., Hotta Y., Okamoto H. (1994). Developmental regulation of islet-1 mRNA expression during neuronal differentiation in embryonic zebrafish. Dev. Dyn. 199, 1-11 [DOI] [PubMed] [Google Scholar]
- Jeong Y., Leskow F. C., El-Jaick K., Roessler E., Muenke M., Yocum A., Dubourg C., Li X., Geng X., Oliver G., et al. (2008). Regulation of a remote Shh forebrain enhancer by the Six3 homeoprotein. Nat. Genet. 40, 1348-1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly G. M., Greenstein P., Erezyilmaz D. F., Moon R. T. (1995). Zebrafish wnt8 and wnt8b share a common activity but are involved in distinct developmental pathways. Development 121, 1787-1799 [DOI] [PubMed] [Google Scholar]
- Kimmel C. B., Ballard W. W., Kimmel S. R., Ullmann B., Schilling T. F. (1995). Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253-310 [DOI] [PubMed] [Google Scholar]
- Kobayashi M., Toyama R., Takeda H., Dawid I. B., Kawakami K. (1998). Overexpression of the forebrain-specific homeobox gene six3 induces rostral forebrain enlargement in zebrafish. Development 125, 2973-2982 [DOI] [PubMed] [Google Scholar]
- Kobayashi D., Kobayashi M., Matsumoto K., Ogura T., Nakafuku M., Shimamura K. (2002). Early subdivisions in the neural plate define distinct competence for inductive signals. Development 129, 83-93 [DOI] [PubMed] [Google Scholar]
- Kohtz J. D., Baker D. P., Corte G., Fishell G. (1998). Regionalization within the mammalian telencephalon is mediated by changes in responsiveness to Sonic Hedgehog. Development 125, 5079-5089 [DOI] [PubMed] [Google Scholar]
- Lacbawan F., Solomon B. D., Roessler E., El-Jaick K., Domene S., Velez J. I., Zhou N., Hadley D., Balog J. Z., Long R., et al. (2009). Clinical spectrum of SIX3-associated mutations in holoprosencephaly: correlation between genotype, phenotype and function. J. Med. Genet. 46, 389-398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagutin O. V., Zhu C. C., Kobayashi D., Topczewski J., Shimamura K., Puelles L., Russell H. R., McKinnon P. J., Solnica-Krezel L., Oliver G. (2003). Six3 repression of Wnt signaling in the anterior neuroectoderm is essential for vertebrate forebrain development. Genes Dev. 17, 368-379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavado A., Lagutin O. V., Oliver G. (2008). Six3 inactivation causes progressive caudalization and aberrant patterning of the mammalian diencephalon. Development 135, 441-450 [DOI] [PubMed] [Google Scholar]
- Leung J. Y., Kolligs F. T., Wu R., Zhai Y., Kuick R., Hanash S., Cho K. R., Fearon E. R. (2002). Activation of AXIN2 expression by beta-catenin-T cell factor. A feedback repressor pathway regulating Wnt signaling. J. Biol. Chem. 277, 21657-21665 [DOI] [PubMed] [Google Scholar]
- Liu W., Lagutin O., Swindell E., Jamrich M., Oliver G. (2010). Neuroretina specification in mouse embryos requires Six3-mediated suppression of Wnt8b in the anterior neural plate. J. Clin. Invest. 120, 3568-3577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loosli F., Koster R. W., Carl M., Krone A., Wittbrodt J. (1998). Six3, a medaka homologue of the Drosophila homeobox gene sine oculis is expressed in the anterior embryonic shield and the developing eye. Mech. Dev. 74, 159-164 [DOI] [PubMed] [Google Scholar]
- Martynoga B., Morrison H., Price D. J., Mason J. O. (2005). Foxg1 is required for specification of ventral telencephalon and region-specific regulation of dorsal telencephalic precursor proliferation and apoptosis. Dev. Biol. 283, 113-127 [DOI] [PubMed] [Google Scholar]
- Mione M., Shanmugalingam S., Kimelman D., Griffin K. (2001). Overlapping expression of zebrafish T-brain-1 and eomesodermin during forebrain development. Mech. Dev. 100, 93-97 [DOI] [PubMed] [Google Scholar]
- Monuki E. S. (2007). The morphogen signaling network in forebrain development and holoprosencephaly. J. Neuropathol. Exp. Neurol. 66, 566-575 [DOI] [PubMed] [Google Scholar]
- Morita T., Nitta H., Kiyama Y., Mori H., Mishina M. (1995). Differential expression of two zebrafish emx homeoprotein mRNAs in the developing brain. Neurosci. Lett. 198, 131-134 [DOI] [PubMed] [Google Scholar]
- Oliver G., Mailhos A., Wehr R., Copeland N. G., Jenkins N. A., Gruss P. (1995). Six3, a murine homologue of the sine oculis gene, demarcates the most anterior border of the developing neural plate and is expressed during eye development. Development 121, 4045-4055 [DOI] [PubMed] [Google Scholar]
- Rallu M., Machold R., Gaiano N., Corbin J. G., McMahon A. P., Fishell G. (2002). Dorsoventral patterning is established in the telencephalon of mutants lacking both Gli3 and Hedgehog signaling. Development 129, 4963-4974 [DOI] [PubMed] [Google Scholar]
- Rohr K. B., Barth K. A., Varga Z. M., Wilson S. W. (2001). The nodal pathway acts upstream of hedgehog signaling to specify ventral telencephalic identity. Neuron 29, 341-351 [DOI] [PubMed] [Google Scholar]
- Sanek N. A., Taylor A. A., Nyholm M. K., Grinblat Y. (2009). Zebrafish zic2a patterns the forebrain through modulation of Hedgehog-activated gene expression. Development 136, 3791-3800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schier A. F. (2009). Nodal morphogens. Cold Spring Harb. Perspect. Biol. 1, a003459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo H. C., Drivenes Ellingsen S., Fjose A. (1998a). Expression of two zebrafish homologues of the murine Six3 gene demarcates the initial eye primordia. Mech. Dev. 73, 45-57 [DOI] [PubMed] [Google Scholar]
- Seo H. C., Drivenes O., Ellingsen S., Fjose A. (1998b). Transient expression of a novel Six3-related zebrafish gene during gastrulation and eye formation. Gene 216, 39-46 [DOI] [PubMed] [Google Scholar]
- Shin J., Poling J., Park H. C., Appel B. (2007). Notch signaling regulates neural precursor allocation and binary neuronal fate decisions in zebrafish. Development 134, 1911-1920 [DOI] [PubMed] [Google Scholar]
- Suh C. S., Ellingsen S., Austbo L., Zhao X. F., Seo H. C., Fjose A. (2010). Autoregulatory binding sites in the zebrafish six3a promoter region define a new recognition sequence for Six3 proteins. FEBS J. 277, 1761-1775 [DOI] [PubMed] [Google Scholar]
- Toresson H., Martinez-Barbera J. P., Bardsley A., Caubit X., Krauss S. (1998). Conservation of BF-1 expression in amphioxus and zebrafish suggests evolutionary ancestry of anterior cell types that contribute to the vertebrate telencephalon. Dev. Genes Evol. 208, 431-439 [DOI] [PubMed] [Google Scholar]
- van de Water S., van de Wetering M., Joore J., Esseling J., Bink R., Clevers H., Zivkovic D. (2001). Ectopic Wnt signal determines the eyeless phenotype of zebrafish masterblind mutant. Development 128, 3877-3888 [DOI] [PubMed] [Google Scholar]
- Varga Z. M., Amores A., Lewis K. E., Yan Y. L., Postlethwait J. H., Eisen J. S., Westerfield M. (2001). Zebrafish smoothened functions in ventral neural tube specification and axon tract formation. Development 128, 3497-3509 [DOI] [PubMed] [Google Scholar]
- Verduzco D., Amatruda J. F. (2011). Analysis of cell proliferation, senescence, and cell death in zebrafish embryos. Methods Cell Biol. 101, 19-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis D. E., Roessler E., Hehr U., Nanni L., Wiltshire T., Richieri-Costa A., Gillessen-Kaesbach G., Zackai E. H., Rommens J., Muenke M. (1999). Mutations in the homeodomain of the human SIX3 gene cause holoprosencephaly. Nat. Genet. 22, 196-198 [DOI] [PubMed] [Google Scholar]
- Weidinger G., Thorpe C. J., Wuennenberg-Stapleton K., Ngai J., Moon R. T. (2005). The Sp1-related transcription factors sp5 and sp5-like act downstream of Wnt/beta-catenin signaling in mesoderm and neuroectoderm patterning. Curr. Biol. 15, 489-500 [DOI] [PubMed] [Google Scholar]
- Westerfield M. (1993). The Zebrafish Book: a guide for the laboratory use of zebrafish (Brachydanio rerio). Eugene, OR: M. Westerfield; [Google Scholar]
- Wilson S. W., Rubenstein J. L. (2000). Induction and dorsoventral patterning of the telencephalon. Neuron 28, 641-651 [DOI] [PubMed] [Google Scholar]
- Woo K., Fraser S. E. (1995). Order and coherence in the fate map of the zebrafish nervous system. Development 121, 2595-2609 [DOI] [PubMed] [Google Scholar]
- Xuan S., Baptista C. A., Balas G., Tao W., Soares V. C., Lai E. (1995). Winged helix transcription factor BF-1 is essential for the development of the cerebral hemispheres. Neuron 14, 1141-1152 [DOI] [PubMed] [Google Scholar]
- Zhou X., Hollemann T., Pieler T., Gruss P. (2000). Cloning and expression of xSix3, the Xenopus homologue of murine Six3. Mech. Dev. 91, 327-330 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}