

Abstract
Whereas docking screens have emerged as the most practical way to use protein structure for ligand discovery, an inconsistent track record raises questions about how well docking actually works. In its favor, a growing number of publications report the successful discovery of new ligands, often supported by experimental affinity data and controls for artifacts. Few reports, however, actually test the underlying structural hypotheses that docking makes. To be successful and not just lucky, prospective docking must not only rank a true ligand among the top scoring compounds, it must also correctly orient the ligand so the score it receives is biophysically sound. If the correct binding pose is not predicted, a skeptic might well infer that the discovery was serendipitous. Surveying over 15 years of the docking literature, we were surprised to discover how rarely sufficient evidence is presented to establish whether docking actually worked for the right reasons. The paucity of experimental tests of theoretically predicted poses undermines confidence in a technique that has otherwise become widely accepted. Of course, solving a crystal structure is not always possible, and even when it is, it can be a lot of work, and is not readily accessible to all groups. Even when a structure can be determined, investigators may prefer to gloss over an erroneous structural prediction to better focus on their discovery. Still, the absence of a direct test of theory by experiment is a loss for method developers seeking to understand and improve docking methods. We hope this review will motivate investigators to solve structures and compare them with their predictions whenever possible, to advance the field.
Keywords: virtual screening, ligand discovery
Introduction
Given the atomic resolution structure of a protein, it should be possible to find molecules that bind to it, modulating its activity. This is the premise behind all structure-based ligand design and discovery efforts. Here we consider one aspect of this field, docking large databases of small molecules to discover novel ligands, an approach that has emerged as the most practical way to use protein structure for ligand discovery [1, 2].
Docking is an in silico technique that places a small molecule in the binding site of a protein and estimates its binding affinity. The docking program samples the conformational and orientational degrees of freedom of the small molecule within the constraints of the binding pocket of the protein, often accessing millions of distinct poses. The program uses a scoring function to select the best pose of each molecule and to rank-order a database. Top scoring hits are reviewed by investigators who prioritize molecules for acquisition and testing. Many docking programs have been developed and used successfully [Table (1)] and have been well described elsewhere [3–6], as have the many scoring functions available [7–12].
Table 1.
Predictions from docking screens confirmed by experiment, 2004–2008.
| Target | Docking program | Lead inhibitor IC50, µM |
Year |
|---|---|---|---|
| AChE [48] | ADAM&EVE | 0.6 | 2004 |
| AdoMetDC [49] | Glide | 12 | 2007 |
| AHAS [50] | DOCK 4/AutoDock | 15.2 | 2007 |
| AICAR transformylase [41] | AutoDock | 11.6 | 2004 |
| AICAR transformylase [42] | AutoDock | 0.6 | 2004 |
| Aldose reductase [40] | N/A | 0.53 | 2007 |
| CCP cavity site [27, 51] | DOCK 3.5 | 20 | 2006 |
| CDC25 phosphatase [52] | FRED/Surflex/Lig andFit | 13 | 2008 |
| CDK [53] | 2004 | ||
| CDK2 [54] | QXP | 0.8 | 2006 |
| Chitinase [36] | LIGTOR | 24.8 | 2006 |
| Chk1 kinase [37] | rDock | 13.4 | 2006 |
| Cyclophilin A [55] | LD1.0 | 0.25 | 2006 |
| DNA gyrase [56] | DOCK 5 | 50 | 2007 |
| EphB4 [57] | DAIM-SEED-FFLD | 1.5 | 2008 |
| FFAR1 [58] | Glide | 3.6 | 2008 |
| Histamine H4 [59] | FlexX | 95.8 | 2008 |
| Human Pregnane X [60] | Surflex | 0.049 | 2007 |
| Integrin αvβ3 [61] | DOCK 4 | 33.5 | 2006 |
| Lysozyme cavity site [25] | DOCK 3.5 | 56 | 2006 |
| Lysozyme cavity sites [24] | DOCK 3.5 / MM-GBSA | N/A | 2008 |
| MCH-R1 [62] | ICM | 7.5 | 2008 |
| Pim-1 kinase [63] | Glide | 0.091 | 2008 |
| PNP [23] | GOLD | 18.9 | 2007 |
| PPAR-γ [64] | Glide/IFD | 2.9 | 2008 |
| SARS coronavirus main protease [35] | GOLD | N/A | 2006 |
| Tm0936 [30] | DOCK 3.5 | 44 (Kd) | 2007 |
| TRH-R1/TRH-R2 [65] | FlexX | 0.29 | 2008 |
| β2-adrenergic receptor [66] | DOCK 3 | 0.009 | 2009 |
| β-lactamase [31] | DOCK 3 | 140 | 2008 |
| β-secretase [67] | DAIM-SEED-FFLD | >25 | 2006 |
| β-secretase [68] | DAIM-SEED-FFLD | 7.1 | 2005 |
| HSP90 [43] | rDock | N/A | 2005 |
| SHP2 [69] | DOCK | 100 | 2008 |
| Al-2 quorum sensing [70] | DOCK 5 | 35 | 2008 |
| Anthrax edema factor [71] | HINT/AutoDock | 1.7 | 2008 |
| hPRMT1 [72] | GOLD | 12 | 2008 |
To truly successfully identify binders or ligands among a large set of non-binders or decoys, docking must do two things. It must correctly predict the ligand pose – within some tolerance limit – so that the biophysical interactions captured by docking can be considered meaningful. It must also rank the ligands among the best molecules in the database, typically in the top few hundred, so that they will likely be picked, acquired and tested. Given the large number of sampling and scoring approximations necessary to make large database docking screens computationally tractable, one might wonder how docking could ever work. Yet the approach has been successful numerous times, with over thirty reports in the last four years alone [Table (1)]. Here, we have gathered evidence to assess whether docking fulfils the first requirement: to correctly position a ligand in the binding site in a prospective manner, i.e. when the x-ray structure of the complex is not yet known.
Whereas correct pose prediction is critical to docking, it is rarely tested by crystallography and reported in the literature. There must be many examples available inside companies, but generally these are not published. In the few cases where a crystal structure is reported in the literature, the docking prediction is often only shown if it corresponds fairly closely with experiment. This is a big loss, because the underlying hypothesis of the docking prediction is not tested and the opportunity to learn from failure is lost.
Retrospective studies attest that docking can often recapitulate experimental ligand poses. Typically, crystallographically observed geometries can be reproduced to within 2 Å RMSD in about 70–80% of the cases in such studies [13–16]. Whereas this is an admirable achievement for retrospective evaluations, one still may wonder whether any one ligand is positioned correctly – and therefore ranked highly for the right reasons – in a prospective study — after all, there is at least a 20–30% chance that this is not the case. Retrospective studies are furthermore accompanied by the worry that results may be biased by knowledge of the desired outcome, no matter how fair-minded the authors intended to be. Even if neither a ligand nor a target were part of the data used to parameterize or calibrate the docking method, they may be similar enough to a member of the training set to have been fit, perhaps overfit, by the docking parameterization. Moreover, retrospective calculations may be run and re-run to get the best results. We therefore do not consider retrospective studies to be a sufficiently stringent way to judge whether docking works for the right reasons when it is used to identify novel ligands.
Prospective studies followed up by careful experiment are critical tests which are highly informative and can often be surprising [17, 18]. If a docked ligand scored well and bound experimentally, but had a docked pose substantially different from the experimentally observed pose, it would have to be suspected that the discovery was lucky. Nevertheless, such a result would be immensely informative, because it demonstrates a failure of our model. If we want to understand docking and how to improve it we need to ask hard questions about whether docking successes are real or serendipitous.
Unfortunately, crystallographic determination of ligand poses predicted by docking screens is surprisingly rare. Of the thirty eight papers that have claimed high-throughput docking as the method of discovery of new ligands in the last four years [Table (1)], fewer than 20% reported a crystal structure of the bound ligand, and just six compared predicted with experimental poses. In cases where no crystal structure was determined, we can sometimes still infer that docking has worked for sound reasons, for instance if a structure-activity relationship series is studied. More often than not, however, such information is not available. We do not mean to imply that any of these studies are chance discoveries, but without an experimental structure, we remain unsure.
Why might the identification of an active compound through docking not be considered compelling evidence that docking works for the right reasons? Or, how might docking help find new ligands if it does not predict the actual binding pose? One possibility is that docking is not so much the sophisticated tool that evaluates protein-small molecule interactions we think it is, but rather a crude filter that relies on gross physical properties such as molecular weight, net charge or logP, or a simple evaluator of shape and size compatibility. Another way docking might work for the wrong reasons is database and investigator bias: pure docking results interpreted by experts allow for cherry picking of compounds that past experience might have picked anyway, even without docking. Libraries focused around known actives are heavily biased for success whether docking works or not. While success is always welcome, any of these circumstances would mean that docking is less general, or less useful, or is highly dependent on the investigator.
Given the critical importance of being right for the right reasons, and its significance for future methods development, we have searched the literature for studies in which a ligand pose predicted by docking is tested by crystallography. We consider five features to be essential to demonstrate docking’s ability to discover new ligands: a) use of an unfocused ligand database, and thus not biased towards ligand scaffolds that are already known; b) target structure-based docking only; c) experimental testing including controls for artifacts such as aggregation and covalent binding, as appropriate; d) crystallographic structure determination of the ligand pose; and e) comparison of the crystal structure pose to the docked pose. As mentioned before, we found few cases that fulfill all these criteria.
We begin with a discussion of each complete study, with subsections on model systems, fragment docking, docking for substrates, and direct comparison to high throughput screening (HTS) [Table (2)]. We then take up papers that fall short of our ideal, for instance because only a limited database was screened or the docking prediction is not shown [Table (3)]. Some of these reports offer indirect evidence that docking is working for the right reasons. In cases where a docking prediction is not reported but was doubtlessly made, we are inclined to suspect that the structural prediction was far away from the experimental position; if it were even close, it would have been shown, as that would have strengthened the paper. We conclude with an analysis of what seems to work best, according to the literature, and a plea for investigators to provide more evidence in future reports.
Table 2.
Docking reports where a ligand was predicted by virtual screening based on general library screening, and subsequently confirmed by crystallography, and the docking pose is compared to the crystal pose. We explicitly exclude those cases where a member of the series was already known, and/or a library around a limited number of scaffolds was used, since our interest is in gauging the performance of virtual screening for ligand discovery
|
Target |
Docking program | Lead inhibitor IC50, µM |
Screening Database / Size |
Further optimization reported? Best ligand? (IC50, µM) |
|---|---|---|---|---|
| AmpC β-lactamase [19] | DOCK 3.5 | 26 | ACD / 200,000 | Y / 1.0 |
| AmpC β-lactamase [29] | DOCK 3.5 | 700 | ZINC / 137639 | N |
| CCP cavity site [27, 51] | DOCK 3.5 | 20 (Kd) | ACD / 5500 | N |
| CDK2 [32] | LIDEAUS | 0.9 (2.2) | 50,000 | Y / 1.3 – 4 |
| CTX-M β-lactamase [28] | DOCK 3.5 | 21 | ZINC / 67489 | Y / 10 |
| Lysozyme cavity sites [24] | DOCK 3.5 / MM-GBSA | N/A | ACD | N/A |
| Lysozyme M102Q site [25] | DOCK 3.5 | 6.3 mM | ACD / 172,000 | N/A |
| Thymidylate synthase [22] | DOCK 3 | 9.3 | FCD / 55,000 | Y / 2.3 |
| Tm0936 [30] | DOCK 3.5 | 44 (Kd) | KEGG / 4200 | N/A |
| β-lactamase [31] | DOCK 3 | 80 | MLSMR / 70,500 | Y / 8 |
| TGT [17] | LUDI | 8.3 | ACD / 120,000 | Y / 0.3 |
| Thrombin [21] | SANDOCK | 65 | 365,000 | N |
Table 3.
Predictions from docking screens confirmed by crystallography but with insufficient information to assess whether virtual screening really works for the right reasons.
| Target | Docking program | Lead inhibitor IC50, µM |
MWT of lead |
Screening Database / Size |
Further optimization reported? Best ligand? (IC50 µM) |
|---|---|---|---|---|---|
| AICAR transformylase [41] | AutoDock | 7.1 | 492 | NCI/1990 | Y / 1.4 |
| Aldose reductase [39, 40] | FlexX | 2.4 | 276 | 1261 | Y / 0.53 |
| Carbonic anhydrase II [38] | FlexX | 0.0006 | 285 | 90,000 | Y |
| CDK4 [73] | LEGEND/SEEDS | 44 | 390 | custom | Y / 0.042 |
| Chitinase [36] | LIGTOR | 4.8 | 386 | 50,193 | N |
| Chk1 kinase [37] | rDock | 13.4 | 404 | 700,000 | N |
| Factor Xa [74] | MCSS/CAVEAT | 48 | 436 | custom | N |
| Neutrophil Elastase [75] | 632 | N | |||
| Pim-1 kinase [63] | Glide | 0.091 | 327 | 700,000 | N |
| SARS coronavirus main protease [35] | GOLD | ~10 | 459 | 58,855 | Y |
| Trypsin [33] | 100 | 236 | ~50,000 | N | |
| Thymidylate synthase [34] | DOCK 3.5 | 65 | ACD / 153,000 | Y / 1.3 | |
| PNP [23] | GOLD | 18.9 | 300,000 | N |
Complete studies
We found ten reports where structural predictions from docking screens are tested by crystallography, and the docking prediction is shown and compared to the crystal structure. We briefly consider each in turn.
A compelling example that docking can work for the right reasons is the discovery of an inhibitor of AmpC β-lactamase, a clinically important antibiotic resistance target [19]. The Available Chemicals Directory (ACD) containing 200,000 compounds was screened using DOCK 3.5.54, and 56 of the top scoring docking hits were selected for purchase and experimental testing. Three of these had Ki values of 650 µM or better, the best being 26 µM. A structure was determined to 1.94 Å and showed the inhibitor structure in good agreement with the docking prediction [Fig. (1)]. Further optimization of this ligand was reported in a subsequent paper and the best compound in this chemical series to date has a Ki of 1 µM [20]. Since the crystallographic pose agreed closely with the docking prediction, and the series could be progressed, this is an unambiguous success.
Figure 1.
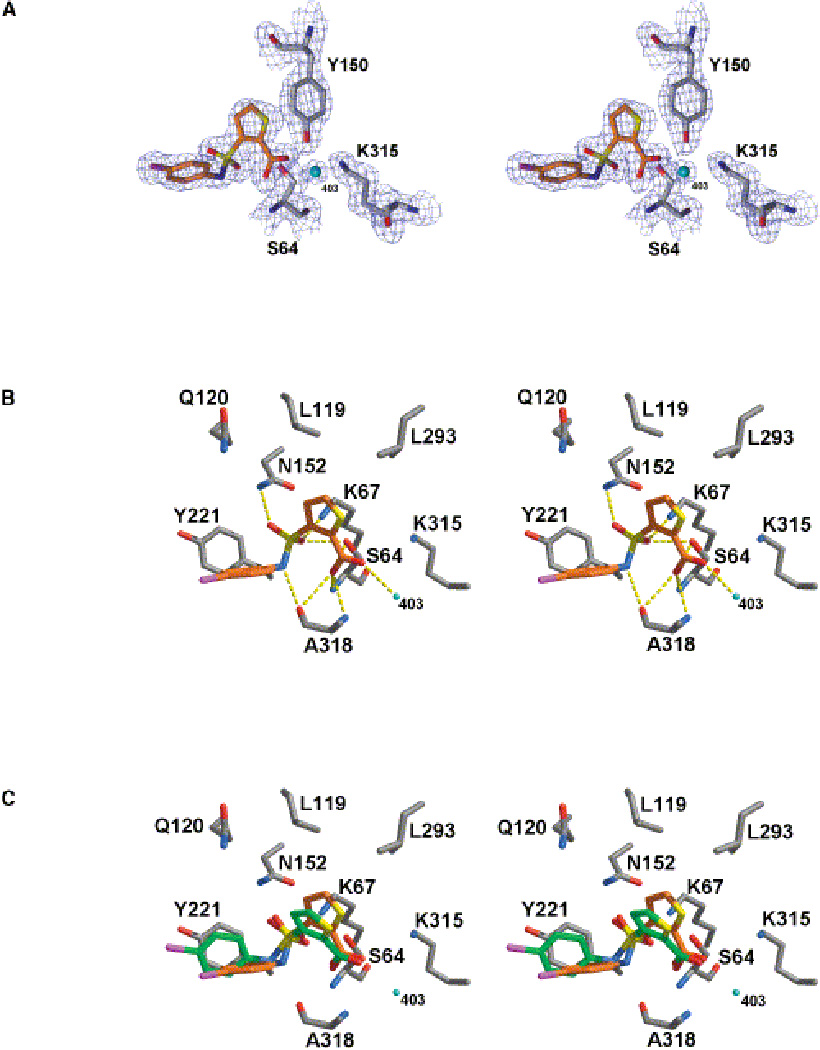
Stereo View of the Active Site Region of the AmpC/1 Complex Determined to 1.94 Å Resolution. (A) The 2Fo-Fc electron density map is shown in blue, contoured at 1.0 σ. This figure was made with SETOR [41]. (B) Interactions observed between AmpC and compound 1 in the crystallographic complex. Cyan spheres represent water molecules. Dashed yellow lines indicate hydrogen bonds. Atoms are colored as in Figure 2, except that the carbon atoms of compound 1 are colored orange.(C) Overlay of the docked and crystallographic conformations of compound 1 in the AmpC site. Carbon atoms of compound 1 in the docked conformation, green; carbon atoms of compound 1 in the crystal structure, orange. (from Ref. [19])
Some docking predictions can be very close to the crystal structure, as in the study against tRNA-guanine transglycosylase (TGT) of S. flexneri, a target for Shigellosis [17]. In a LUDI screen of a 120,000 molecule subset of the ACD, three compounds were discovered, the best of which inhibited the enzyme with a Ki of 8.3 µM. A 1.95 Å x-ray crystal structure of TGT in complex with 4-aminophthalhydrazide showed that the docked structure predicted the crystallographic pose within 0.24 Å RMSD. This small and rigid compound was the first in a series that lead to a molecule with a Ki of 0.3 µM. We consider this study therefore to be a clear success of docking.
An already known ligand re-discovered by docking is less than ideal, because of the potential for bias, but a crystallographic test of a predicted pose is still welcome evidence since so few are available. Using the program SANDOCK, Burkhard et al. screened 365,000 molecules having 8 to 15 non-hydrogen atoms for inhibitors of thrombin [21]. The pose of one hit, p-amino-benzamidine, previously reported to bind at 65 µM, fell within 0.7 Å of the subsequently determined crystal structure [Fig. (2)]. Although an unknown ligand would have been more compelling, it is reassuring that docking could pick this compound out of the database, and get it right for the right reasons.
Figure 2.
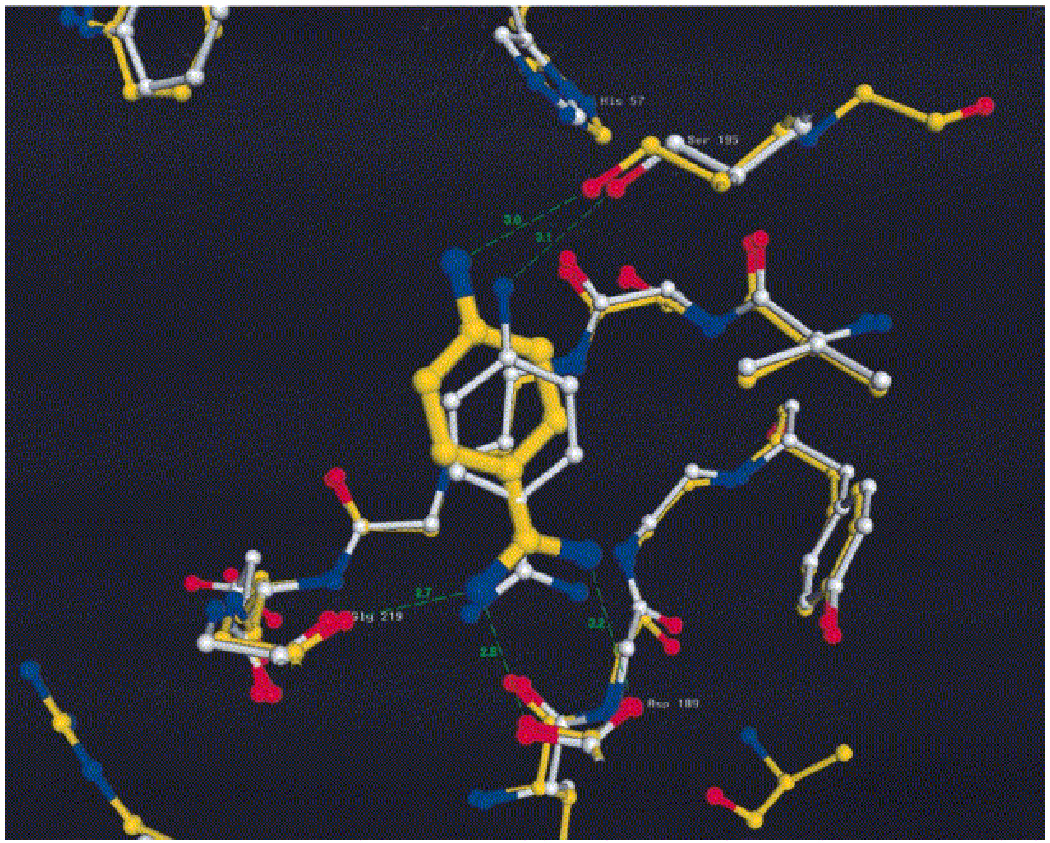
Overlay of the docked and the refined NBEN structures. Yellow: the refined NBEN structure as in Figure 10. White: protein atoms of the docked structure. The fit is based on the least-squares fit of all Cα atoms of the model and X-ray thrombin molecules in the vicinity of the ligand. Note the differences of the two structures of the active site residues His57 and Ser195. The r.m.s. difference between the atoms of the two ligands is 0.726 Å. (from Ref. [21])
These three successes show that docking can be used to find hits against real drug targets, with poses that are biophysically sensible, and that the docking model can be used to guide optimization of these hits. The first report of ligand discovery by docking that included a crystal structure of the predicted ligand was the discovery of phenolphthalein derivatives as inhibitors of thymidylate synthase (TS) [22]. This screening used the Fine Chemicals Database (FCD, the predecessor of the ACD), which contained 55,000 molecules, and the DOCK 2 program for docking. Among the top scoring hits, many of which were known ligands, were three novel compounds. These were tested experimentally and shown to inhibit the enzyme. One of the novel binders was crystallized and appeared in two configurations that differed from the DOCK-favored geometry, which was attributable to a counterion that appeared [Fig. (3a)]. The structure of these complexes suggested another binding region, which was then targeted by docking. This led to the discovery of a series of phenolphthalein analogs that inhibited TS in the 1 to 30 µM range. A crystal structure of phenolphthalein with TS shows that it binds in the target site in a configuration that resembles the one suggested by DOCK [Fig. (3b)], but the pose is sufficiently different that we feel it is hard to argue that docking worked for the right reasons in this case.
Figure 3.
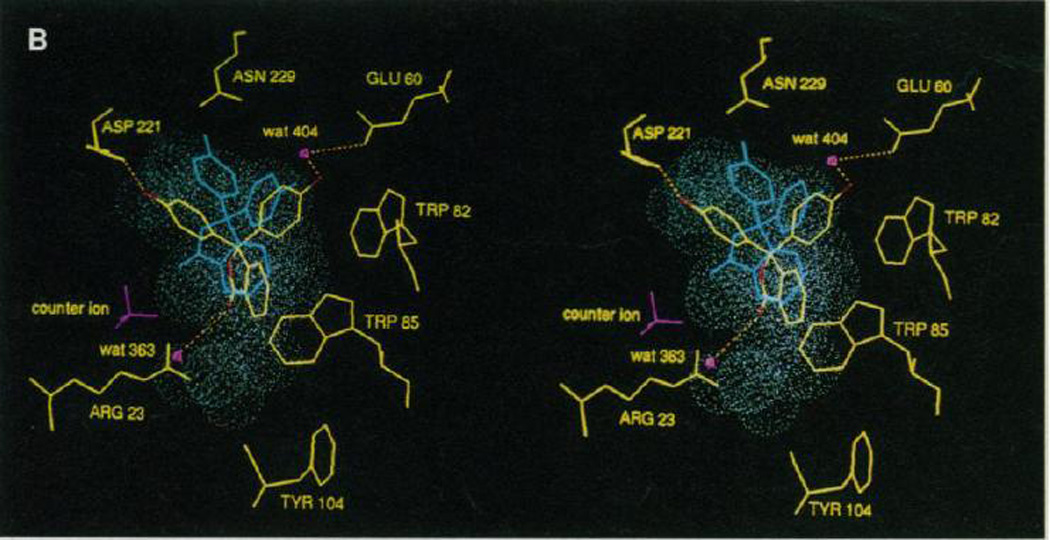
Predicted and experimental ligand orientations for the hydrophobic L99A cavity. The carbon atoms of the crystallographic pose, the DOCK predicted pose, the AMBERDOCK predicted pose, and the PLOP predicted pose are colored gray, yellow, cyan, and magenta, respectively. The Fo-Fc omit electron density maps (green mesh) are contoured at 2.5–3.0 σ (a) β-chlorophenetole (1), (b) 4-(methylthio)nitrobenzene (2), (c) 2,6-difluorobenzylbromide (4), (d) 2-ethoxyphenol (5), and (e) 3-methylbenzylazide (6) bound to L99A. Rendered with the program PyMOL. (from Ref. [24])
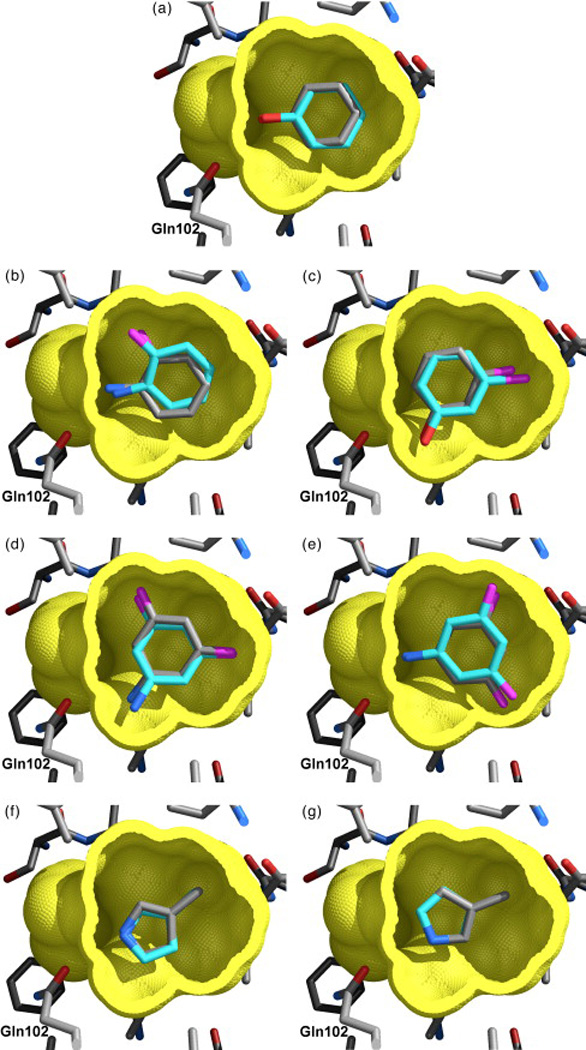
A paper by Pereira et al. has an interesting twist beyond the identification of a binder [23]. They were searching for inhibitors of purine nucleoside phosphorylase (PNP) using the program GOLD. In a screen of a library of 300,000 molecules, they identified two compounds with reasonable affinity, with the most potent compound inhibiting at 18.9 µM. When they solved the crystal structure, the computed binding mode of the compound – a purine analog – overlapped with the x-ray pose in the 6-ring, but was rotated by 60° [Fig. (4)]. Interestingly, they then tried to improve the docked pose, and found that they succeeded in this endeavor when they protonated a glutamate residue and flipped the side chain of a glutamine in the binding site. This study thus provides further evidence that most docking programs are indeed very sensitive to small changes in atom positions and hydrogen bond arrangements. We consider this study to be a partial docking success.
Figure 4.

Comparison of the geometries of binding predicted using the AMSOL charges and the experimentally observed modes for five ligands of L99A/M102Q: phenol (a), 2-fluoroaniline (b), 3-chlorophenol (c), 3,5-difluoroaniline ((d) and (e)), and 3-methylpyrrole ((f) and (g)). Carbon atoms are colored cyan in the predicted structures, and gray in the crystallographic structures, nitrogen atoms in blue, oxygen atoms in red, halogen atoms are magenta. The cavity's molecular surface (yellow) has been cut away to show the ligands. Gln102 is labeled. Because the receptor structure shows little change upon binding of the ligands, only the apo-receptor structure, used in docking, is shown. (from Ref. [25]
These two studies show that docking sometimes makes predictions that are correct, even though the pose seems wrong enough that it cannot capture the key elements of biophysical recognition.
Model systems
Protein-ligand interactions in real drugs sites are complex, so a pragmatic approach to investigate the strengths and weaknesses of docking is to look at less complex sites, where failures can be attributed to particular errors in the method. One of the simplest model sites is the L99A mutant of T4 lysozyme, a synthetic, hydrophobic, and completely buried site. In a re-scoring study, Graves and colleagues picked 33 compounds that scored well by MM-GBSA and had not ranked well in an initial DOCK 3.5 screen of the ACD [24]. When tested, 23 of these 33 had measurable affinities. X-ray structures were determined for 21 of these 23 molecules. In many but not all cases, the docking pose generated by MM-PBSA more closely resembled the crystallographic result [Fig. (5)]. Docking predicted many poses correctly, unmistakable evidence that docking is working for the right reasons with very small molecules against such a simple site.
Figure 5.

Crystal structures of selected ligands from Table 2 bound to CCP W191G. Part 3 of the original figure only. Left column: |Fo|-|Fc| omit map for the refined complexes, except for (a), (c), (k), (m), and (y) where the map of the unrefined complex is shown, contoured at 2.5 σ (green) with the ligand left out of the calculation, but shown in the Figure for clarity. Right column: Superposition of the highest ranking dock pose (green carbon atoms) with the crystallographically determined binding mode (yellow carbon atoms). Hydrogen bonds are drawn as broken lines. (a) and (b) 14; (c) and (d) 15; (e) and (f) 16; (g) and (h) 17; (i) and (j) 18; (k) and (l) 21, the |Fo|-|Fc| map, contoured at 10 σ (red) is also shown; (m) and (n) 22; (o) and(p) 24; (q) and (r) 25, the |Fo|-|Fc| map, contoured at 9 σ (red) is also shown; (s) and (t) 28; (u) and (v) 33, the |Fo|-|Fc| map, contoured at 14 σ (red) is also shown; (w) and (x) 34; (y) and (z) 35; (aa) and (bb) 36. (from Ref. [27])
A more complex site is the L99A/M102Q double mutant of T4 lysozyme, which is more polar than L99A. Wei and colleagues reported the discovery of ligands selective for L99A/M102Q by screening against the 172,000 compound ACD database using DOCK 3.5 [25]. Seven compounds that scored well against the double mutant, but not against the L99A single mutant, were purchased and tested for binding. Five of these were determined by x-ray crystallography. Docking predictions corresponded to the crystallographic results within 0.4 Å RMSD [Fig. (6)], and were so detailed as to include predictions of double occupancy as seen in some crystal structures. Again, docking clearly works very well on small polar molecules against this simple site.
Figure 6.
Superposition of the crystal structure of Tm0936 in complex with SIH (red) and the docking predicted structure of the high-energy intermediate of SAH (carbons in green). Enzyme carbons are coloured light blue, SAH and enzyme oxygen atoms are coloured red, nitrogens blue and sulphurs orange. The purple sphere represents the divalent metal ion. An Fo-Fc omit electron density map for SIH is shown, contoured at 4.1 σ. The structure was determined at 2.1 Å resolution. (from Ref. [30])
The charged, wet site of the synthetic model site of the cytochrome C peroxidase W191G mutant affords a slightly more complex site, as it allows for a water molecule to be present. Brenk and co-workers reported the discovery of new binders by docking a library of 5300 compounds from the ACD using DOCK 3.5 [26, 27]. They picked 16 high scoring compounds not previously known to bind. Fifteen of these showed experimentally measurable binding affinity, ranging from 20 to 60 µM. Crystal structures were determined for ten of these and the observed geometries corresponded closely to those predicted by docking [Fig. (7)]. This is more evidence that docking is clearly working most of the time against simple sites with very small ligands.
Figure 7.

Docking discovers two novel β-lactamase inhibitors. (A) Structures of the two new inhibitors. (B) Initial unbiased Fo-Fc density contoured at 3 σ from the 1.8 Å structure with compound 3 modeled. Full crystallographic statistics are given in Table S3, Supporting Information. (C) Hydrogen bonding pattern of 3 with AmpC. (D) Overlay of the crystal structure (white carbons) overlaid to the initial pose proposed by DOCK (magenta carbons). The RMSD of the heavy atoms between the two poses was 0.9 Å. (from Ref. [31]
Docking small molecules to these simple model sites works reliably, but what about real drug sites? Whereas the complexity of a real drug site cannot be simplified, the ligands docked to it can be, using fragments, often defined as molecules up to about 250 Daltons. Chen and Shoichet identified fragment-sized ligands for CTX-M β-lactamase, a clinically important antibiotic resistance enzyme [28]. They docked 67,000 fragments from of the ZINC database, using DOCK 3.5. The best inhibitor found has a Ki of 21 µM and corresponds closely to the crystal structure.
Another important antibiotic resistance enzyme, AmpC β lactamase has been targeted by Teotico et al. in a screen of 137,639 fragments of the ZINC database [29]. The study produced a very high hit rate of 46% and Ki values that ranged from 700 µM to 9.2 mM. Upon crystallization of eight fragments, they found RMSDs ranging from 0.2–2.6 Å for six of the inhibitors, while the remaining two were in very different poses due to substantial changes in the protein conformation.
These results indicate quite strongly that docking predicts the poses of fragment-sized molecules against not only simple model sites, but against real drug sites, with a few notable exceptions.
Docking for substrate discovery
Since enzymes stabilize the transition state and not the ground state of a molecule being turned over, docking for substrate discovery can benefit from representing the ligand in a high energy intermediate form to better capture the key molecular recognition event. Hermann and colleagues used this strategy to discover the substrate and thereby the function of Tm0936, an enzyme of then unknown function from T. maritima [30]. They docked 4200 metabolites from the KEGG database that had undergone the first assumed step of nucleophilic attack by hydroxyl to form either a tetrahedral or trigonal-bipyramidal high energy intermediate. On the basis of docking, five top scoring compounds, among them S-adenosylmethionine, were selected for experimental testing. Of these five, three had substantial activity as substrates (better than 105·M−1·s−1). A crystal structure of the product resulting from the deamination of S-adenosylhomocysteine, S-inosylhomocysteine in complex with Tm0936 was determined, and agreed very closely with the docking prediction. From these confirmed predictions, the function of the enzyme as a highly active deaminase was inferred [Fig. (8)].
Figure 8.

Superposition of experimental (yellow) and low-energy DOCK consensus configuration I (cyan) positions of PTH in the target site [graphics made with MIDAS+ (31)]. Atoms corresponding to the SB orientations are represented as white, van der Waals spheres. Phenolphthalein binary complex crystals were grown in 100 mM KHPO4, 0.1 mM EDTA, 1 mM DTT, and 0.5% dimethyl sulfoxide at pH 7.4. The well contained buffer without (NH4)2SO4. The interaction energies of the low-energy consensus configurations from families one and two are −11.2 and −3.2 kcal.mol, respectively. Energies were calculated with Chemgrid (23) with the use of the AMBER potential (32). The maximum van der Waals energy for any atom was set to 10 kcal/mol with a 10 Å distance cutoff in a 28 Å box centered on the SB atoms. Grid spacing was 0.33 Å and an ε=4R distance-dependent dielectric was used. (from Ref. [22])
Comparison with HTS
Of interest especially for industry is the direct comparison of docking and high throughput screening. It offers the possibility to follow up on each hit from either method and – ideally – improve both methods. Babaoglu and colleagues screened the 70,563 compound “Molecular Libraries Small Molecule Repository” (MLMSR) database against AmpC β lactamase looking for non-covalent inhibitors of this target involved in antibiotic resistance [31]. They used DOCK 3.5 to prioritize the library, and picked sixteen compounds for purchase and testing. Of these, two were modest inhibitors, the best having a Ki of 80 µM. A crystal structure was determined and showed that the non-H atoms of the docking prediction were within 0.9 Å RMSD of the crystallographic pose [Fig. (9)]. Subsequent medicinal chemistry afforded an inhibitor with a Ki of 8 µM. Interestingly, high throughput screening of the same compounds at a maximum concentration of 30 µM failed to identify any non-covalent, validated hits.
Figure 9.

The base binding site of bovine PNP. Comparison of the crystal structure of the complex (yellow) and that predicted by VS (green). In (A) the VS was performed assuming E201 to be non-protonated and the side chain of N243 to have the same orientation as that observed in 1A9P. In (B) E201 was treated as protonated and the side chain of N243 was flipped with respect to its orientation in 1A9P. In the former case the docked compound is orientated differently to that observed in the crystal structure while in the latter the superposition is markedly improved. (from Ref. [23])
These ten papers are the only ones we could find that tested a pose prediction from docking screens by crystallography and compared the prediction with the crystal structure. It is noteworthy that most of these successes are on very small ligands, with very few internal degrees of freedom. After 15 years of docking, we are amazed by how few cases were reported, and how few real “lead like” or “drug like” molecules are represented. It has to be emphasized again that we do not consider only studies that were clear docking successes as valuable reports that will advance the field; quite the contrary, those studies that also report the failures are the ones that will allow changes to the docking engines.
We now turn to cases that are still instructive without being completely ideal case studies according to the criteria we set forth in the Introduction [Table (3)]. We do not claim that this list is complete, many more studies might fulfill the relaxed criteria, but we would like to highlight some aspects we consider important.
Less than complete studies
In an article by Wu et al., the discovery of a novel family of inhibitors of cyclin-dependent kinase (CDK) was reported [32]. The software used was LIDAEUS, a further development of its predecessors SANDOCK and PROBIS. With it, a library of 50,000 commercially available drug-like molecules was screened and in the first round, the top-ranking 120 molecules were assayed at a fixed concentration of 5 µM, yielding 17 actives. In a second round, the parameters of the LIDAEUS program were optimized and the entire database was processed again. The top-ranking 28 molecules were tested in a biochemical assay, and 8 showed at least 30% activity at 30 µM. The authors then focused on a trio of molecules with a common 2-amino-4-heteroarylpyrimidine scaffold, and the most potent compound out of this set bound with an IC50 of 2.2 µM. Structure-based design yielded a fourth compound with an improved IC50 of 0.9 µM. All four compounds were crystallized. Regarding the quality of the computational poses, the authors remark that the x-ray pose of compound 1 “shows a significant twist of 35° about the bond linking the two aromatic rings”, whereas the model was planar [Fig. (10)]. They then attempt to refine the docked pose by allowing the docking program to dock the twisted conformation (LIDAEUS rotates single bonds only in order to optimize hydrogen bonds) and the best scoring pose has an RMSD of 1.60 Å. We consider this study to be a partial docking success.
Figure 10.
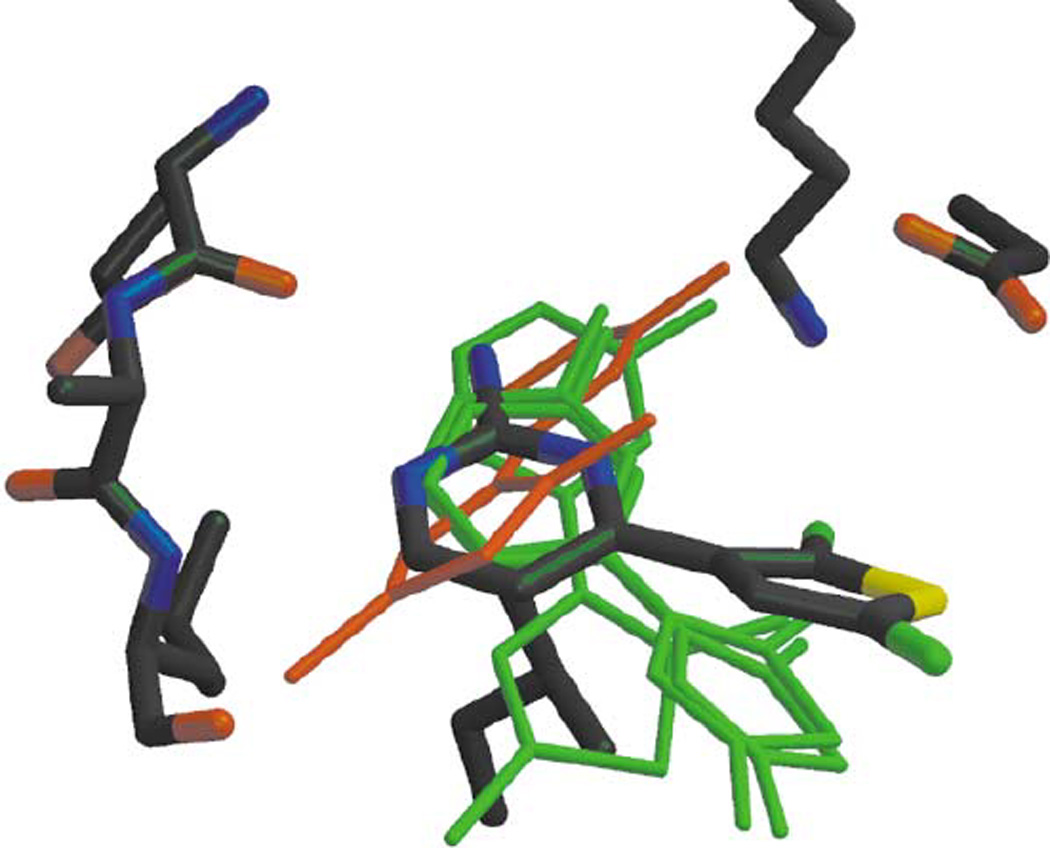
Overlay of the top four positions identified by the program LIDAEUS, in Green, compared with the experimentally determined X-ray structure of CYC1. The ligand in red is the fourth-best fit. It has an unacceptable energyscore (and large RMSD fit on the experimental structure). (from Ref. [32])
The paper by Kurinov and Harrison is among the first applications of docking that was followed by x-ray crystallography [33]. It does not use an unbiased library, however. In the search for inhibitors of trypsin, they selected only compounds that contain an amine and a hydrophobic moiety, such as benzene or cyclohexane, which left them with 50 compounds. These, they docked with an in-house docking program and the best compound had a Ki of 100 µM. All of the inhibitors that were tested and co-crystallized were docked within 2 Å of the x-ray pose. Interestingly, the authors also reported a good correlation between docking score and measured binding affinity.
An important test of a docking prediction in the absence of a crystal structure is how well it can be used to guide ligand optimization. An interesting case is reported by Tondi and colleagues who screened the ACD against the folate site of thymidylate synthase [34]. They initially picked five of the top scoring docking hits for testing, two of which had high micromolar affinity. Guided by two possible docking poses, they purchased and tested analogs, one of which has 65 µM affinity. Having corroborated the docking model of binding, 33 derivatives were made and tested, the best of which had a Ki of 1.3 µM. Thus the docking model was not only probably right, but also useful, even without a crystal structure.
Lu et al. picked a very attractive target for their docking campaign: they tried to find novel binders of the SARS coronavirus main protease [35]. Using the program GOLD, they screened a library of 58,855 compounds, which yielded two hits binding at approximately 10 µM. In the search for more potent inhibitors, they tested 21 analogs, thereby improving the potency to 0.3 µM. Unfortunately, although they solved the crystal structure, they did not compare the binding mode to the docked pose. It thus has to be left to speculation whether or not it shows reasonable overlap.
In a study by Schüttelkopf et al., development of a promising scaffold by means of molecular docking is reported [36]. It started from three 1,3-dimethylxanthine that had been identified as inhibitors of A. fumigates chitinase B1 by the same lab. Then, the authors filtered all 3-methylxanthines from a large library, which left them with 50193 molecules for the docking calculations. In these, they employed the program LIGTOR, which is not a canonical docking program, but instead evaluates possible side chain conformations of a small molecule. However, they identified a novel chitinase inhibitor binding with 2.8 µM affinity and reasonable docking accuracy, as judged by the images showing the two poses.
Foloppe et al. identified inhibitors of Chk1 kinase in a library of 700,000 molecules using the program rDock [37]. They obtained nine hits from the docking screen and one by analogy to one of the hits, and the most potent of these compounds inhibited with an IC50 of 13.4 µM. For four of these compounds, they were able to obtain crystal structures with reasonable resolution (2.20–2.60 Å). Unfortunately, they do not provide RMSDs with respect to the crystal structures, but comment that docking modes similar in orientation and key interactions with the ATP binding site were among the top 12 hits for all four compounds, and in two cases even were the top-ranking solutions. They mention, however, that for the other two cases it would have been difficult to pick the correct solution from among other reasonable solutions offered by the docking program. Especially for the one compound which interacts with the hinge region by means of a nitro group, they commented that “it would have been exceedingly difficult to identify the experimental binding mode from the computational suggestions, maybe because of a subjective reluctance to accept the interaction between a nitro group and the kinase motif”. Moreover, they comment on the complete failure of docking for correctly prioritizing stereoisomers, tautomers and hydrogen bond patterns for these two compounds.
Another kinase is the subject of the investigation by Pierce et al., in which they target the protooncogene Pim-1 kinase. In this docking campaign, they went out from a library of 700,000 molecules and docked them into the ATP binding site of Pim-1 kinase with Glide 3.5. This calculation was not completely unbiased as a hydrogen bond with a residue in the binding site was enforced. Upon testing of 96 selected compounds, four showed Kis below 10 µM in a range from 0.091 to 4.5 µM. Two of these compounds could be crystallized together with the protein and the heavy-atom RMSDs were 1.74 and 0.87 Å, respectively. The authors mention that the rather high RMSD for the first structure stems from the movement of a protein sidechain which renders the formation of a hydrogen bond with the compound impossible. Upon correction of the side chain conformation in the docking calculation, the RMSD drops to 0.96 Å.
Docking is used only as part of a pipeline in an investigation by Grüneberg and colleagues [38], but the authors extensively compare the docking poses with the x-ray structures and draw valuable conclusions, which makes it a highly relevant addition to this review. The target is Human Carbonic Anhydrase II and a library of 90,000 molecules is first screened with UNITY, then the remaining 3314 are filtered through FlexS before being docked with FlexX. The authors note that the “partial hydration of the pocket proved to play an important role”, and that they were able to identify four conserved water molecules from superpositions of several apo-and complex-structures. After docking, 13 hits were tested experimentally, three were shown to be subnanomolar, one nanomolar, and seven micromolar binders. Two of the hits were investigated crystallographically and the authors find that the “binding mode generated by FleX and ranked as highest using DrugScore is in good agreement with the experimental structure.” They caution, however, that FlexX' own scoring function would have ranked the correct solutions at ranks 51 and 61, respectively. Moreover, they note that the correlation between the affinities and the values predicted by either scoring function is disappointing.
Similar to the just mentioned study, the same group has published two papers about a virtual screening campaign on aldose reductase. The first one describes the computational identification of a set of six compounds out of a pre-filtered database of 1261 molecules, the most potent compound of which had an IC50 of 2.4 µM [39]. In the second paper, they follow up on these hits with x-ray crystallography and manage to obtain crystals for the two most potent compounds [40]. While the authors do not give RMSDs, they describe differences qualitatively. The docking was not completely unbiased as the carboxylate warhead of the candidate molecules was forced to adopt a geometry that had previously been observed for a different binder. Even so, the authors note several differences which would have made a correct prediction very hard. First, an essential hydrogen bond is made impossible by a backbone flip in the protein. Second, one of the compounds picks up a water molecule upon binding, whereas the other does not. This divergent behavior could only have been captured employing a flexible water in the docking calculations, which few docking programs do.
Xu and co-workers report the discovery of a novel non-folate inhibitor of AICAR transformylase, a target for anti-neoplastic chemotherapy in the purine biosynthesis pathway [41, 42]. They used AutoDock 3 to screen the NCI diversity set containing 1990 compounds. Docking identified 44 compounds that differed substantially from known antifolates. Of these 16 that were soluble were tested, and 8 had IC50s in the µM range. The best compound had a Ki of 7.1 µM. A 1.80 Å crystal structure was determined, but unfortunately, the comparison with the predicted geometry was not shown. Afterwards, the ACD was searched for analogues of these actives and ten compounds were ranked by docking, purchased, and tested. The best of these had an IC50 of 1.4 µM, an 8-fold improvement.
Barril et al. report the identification of a novel class of inhibitors of Hsp90, an attractive target in oncology [43]. Their original library consisted of 700,000 compounds and was docked to the binding site of the N-terminal domain of Hsp90 using the program rDock. Out of five compounds belonging to the same family of 1-(2-phenol)-2-naphthols, three were active and two had low micromolar activity. The investigators then searched for modifications of the basic scaffold and came up with a compound that had an IC50 of 0.7 µM and could be crystallized in complex with the protein. In their Fig. 4, the authors show the overlay of the pose of the best compound with the pose of the most potent compound coming out of docking. They remark that the common scaffold overlaps very well at an RMSD of 0.6 Å. However, the variable part adopts significantly different positions, which they attribute to a difference in the protein region close to that moiety.
This concludes the report of the studies that we found to fulfill all or most of our criteria for an unbiased assessment of docking. Interestingly, many studies that failed to obtain the correct pose in docking were able to rescue it retrospectively, i.e., some mostly simple tweaks can considerably improve the result. While this is encouraging for experienced researchers, it currently lets “blind” docking seem out of reach.
Conclusions and Outlook
When they work, do docking screens really discover ligands for the right reasons? For simple model systems with very small ligands, docking appears to work amazingly well. For fragment-sized molecules in drug-like sites, we also found evidence that docking works, at least sometimes, for the right reasons. But for the most popular use of docking, using “drug-like” molecules against real drug sites, there are very few complete studies available on which to base an opinion. At the same time, many reports of docking successes without crystal structure continue to appear, now at a rate well over a dozen per year [Table 1]. With this level of success, should dockers simply declare victory and go home? There are at least two reasons to hold out for more data. First, docking remains challenging, even for experts, and urgently needs improvement. Experimental tests of docking predictions can expose methodological weaknesses and serve to focus efforts to improve the calculation. Second, retrospective studies, the current gold standard for the field, are beset by worries of hard-to-quantify bias that only prospective studies can overcome.
Why does docking sometimes fail to predict the biologically significant pose? We can only speculate based on the literature surveyed in this report, but the reasons seem to have changed little over 15 years: structural water molecules, flexible side chains, unexpected loop flips, side chain protonation states, and other varieties of receptor flexibility are all too common, and remain challenging to treat computationally. For the ligand, representation of protonation, tautomerism and stereoisomerism is often crucial, but it is unclear whether exhaustive enumeration or careful generation and curation of selected isomers is more successful. All these concerns, and many others, are why structure determination to corroborate or refute predicted poses remains important — because it provides an honest test of the sampling and scoring algorithms as well as the entire protocol used to represent, sample and score protein-small molecule interactions.
If we cannot be sure that docking works for the right reasons, why does it work at all? We have already suggested some ways in which docking might enrich true binders even when it fails to predict the correct binding geometry. Mostly, these are cases in which docking functions as a filter to eliminate non-binders based on gross physico-chemical properties, such as molecular weight, overall charge and size, for instance. Discovering new ligands is good regardless of how it is done, but we miss the opportunity to improve the method if we do not understand it. Along these lines, it has been shown that it is a lot harder to identify known ligands among a set of property-matched decoys, i.e., in cases where getting the pose and energy right really matters [44]. Another factor that helps docking is the bias inherent in many of today’s libraries: depending on the target, libraries will contain a large number of molecules that were specifically made for that target or target family [45]. In these cases, docking becomes less of a search for a needle in a haystack than a search for hay in a haystack.
Encouragingly, there are several lessons we learned from this review. First and foremost, there seems to be no best method, since a variety of programs have been successful on a variety of targets [Tables 1–3]. On the other hand, we can say something about the targets for which docking has worked: one group are the systems with small binding sites and/or small ligands. This is not surprising: smaller sites allow fewer degrees of freedom and are less complex in the Hann sense [46]. The second group is formed by the cases where the binding sites have strong orienting constraints, for instance by being long and narrow, or having a charged residue that imposes a preferred direction. Again, these systems are easier, since gross physical properties eliminate many candidates. And thirdly, in all the cases where investigators used specific knowledge about the target, they were able to obtain the correct poses and sometimes rescue incorrect predictions. In the hands of an experienced investigator, docking therefore is a powerful technique. All of these points can be considered common knowledge of the field, but to the best of our knowledge this is the first review that bases them on the combination of the available literature.
How does this translate to practical rules for people new to the field or for researchers tackling a new system? First and foremost, small is beautiful for docking. The available evidence shows that small “fragmentlike” ligands work well. There is very little structural experimental evidence for the discovery of bigger ligands with docking. This fits with the industry-wide trend towards the fragment based approach [47]. Otherwise, the papers reviewed here support a recommendation that is short, but not easy: get to know the system by trying to find data that will help you make the right decisions in terms of protonations, conformations and library composition. If ligands are known, evaluate the docking model for its ability to enrich actives from among property matched decoys. If a ligand bound crystal structure is available, docking should recapitulate the structure well enough to capture the main biophysical interactions. In the absence of a crystal structure to corroborate a prediction, one way to evaluate experimentally confirmed docking hits is to ask if they can be progressed based on the structural prediction. Although this does not mean the prediction must be right, it is a reassuring sign. Another form of indirect evidence is structure activity data relating to the docking prediction.
Does docking work for the right reasons? For simple model systems, the answer is Yes, and often. For real drug sites with fragment-sized ligands, the answer is also an encouraging “probably”. For big (> 350 Daltons) ligands, however, there is currently very little evidence. We hope investigators will come forward with crystallographic confirmation of docking predictions for higher molecular weight compounds to shed more light on this important problem. Until that time, it is still too early to say.
Acknowledgements
This work is supported by NIH GM071896 (to Brian K. Shoichet and JJI). PK acknowledges the support of the Swiss National Science Foundation, grant PBZHA-118815.
References
- 1.Leach AR, Shoichet BK, Peishoff CE. Prediction of protein-ligand interactions. Docking and scoring: successes and gaps. J. Med. Chem. 2006;49:5851–5855. doi: 10.1021/jm060999m. [DOI] [PubMed] [Google Scholar]
- 2.Shoichet BK. Virtual screening of chemical libraries. Nature. 2004;432:862–865. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 4.Perola E, Walters WP, Charifson PS. A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins. 2004;56:235–249. doi: 10.1002/prot.20088. [DOI] [PubMed] [Google Scholar]
- 5.Brooijmans N, Kuntz ID. Molecular recognition and docking algorithms. Annu. Rev. Biophys. Biomol. Struct. 2003;32:335–373. doi: 10.1146/annurev.biophys.32.110601.142532. [DOI] [PubMed] [Google Scholar]
- 6.Kellenberger E, Rodrigo J, Muller P, Rognan D. Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins. 2004;57:225–242. doi: 10.1002/prot.20149. [DOI] [PubMed] [Google Scholar]
- 7.Gohlke H, Klebe G. Statistical potentials and scoring functions applied to protein-ligand binding. Curr. Opin. Struct. Biol. 2001;11:231–235. doi: 10.1016/s0959-440x(00)00195-0. [DOI] [PubMed] [Google Scholar]
- 8.Halperin I, Ma B, Wolfson H, Nussinov R. Principles of Docking: An Overriew of Search Algorithms and a Guide to Scoring Functions. Proteins. 2002;47:409–443. doi: 10.1002/prot.10115. [DOI] [PubMed] [Google Scholar]
- 9.Stahl M, Rarey M. Detailed analysis of scoring functions for virtual screening. J. Med. Chem. 2001;44:1035–1042. doi: 10.1021/jm0003992. [DOI] [PubMed] [Google Scholar]
- 10.Ferrara P, Gohlke H, Price DJ, Klebe G, Brooks CL., III Assessing scoring functions for protein-ligand interactions. J. Med. Chem. 2004;47:3032–3047. doi: 10.1021/jm030489h. [DOI] [PubMed] [Google Scholar]
- 11.Wang R, Lu Y, Fang X, Wang S. An extensive test of 14 scoring functions using the PDBbind refined set of 800 protein-ligand complexes. J. Chem. Inf. Comput. Sci. 2004;44:2114–2125. doi: 10.1021/ci049733j. [DOI] [PubMed] [Google Scholar]
- 12.Velec HF, Gohlke H, Klebe G. DrugScore(CSD)-knowledge-based scoring function derived from small molecule crystal data with superior recognition rate of near-native ligand poses and better affinity prediction. J. Med. Chem. 2005;48:6296–6303. doi: 10.1021/jm050436v. [DOI] [PubMed] [Google Scholar]
- 13.Moustakas DT, Lang PT, Pegg S, Pettersen E, Kuntz ID, Brooijmans N, Rizzo RC. Development and validation of a modular, extensible docking program: DOCK 5. J. Comput.-Aided Mol. Des. 2006;20:601–619. doi: 10.1007/s10822-006-9060-4. [DOI] [PubMed] [Google Scholar]
- 14.Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS. A critical assessment of docking programs and scoring functions. J. Med. Chem. 2006;49:5912–5931. doi: 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]
- 15.Hartshorn MJ, Verdonk ML, Chessari G, Brewerton SC, Mooij WT, Mortenson PN, Murray CW. Diverse, high-quality test set for the validation of protein-ligand docking performance. J. Med. Chem. 2007;50:726–741. doi: 10.1021/jm061277y. [DOI] [PubMed] [Google Scholar]
- 16.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved protein-ligand docking using GOLD. Proteins. 2003;52:609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 17.Gradler U, Gerber HD, Goodenough-Lashua DM, Garcia GA, Ficner R, Reuter K, Stubbs MT, Klebe G. A new target for shigellosis: rational design and crystallographic studies of inhibitors of tRNA-guanine transglycosylase. J. Mol. Biol. 2001;306:455–467. doi: 10.1006/jmbi.2000.4256. [DOI] [PubMed] [Google Scholar]
- 18.Horn JR, Shoichet BK. Allosteric inhibition through core disruption. J. Mol. Biol. 2004;336:1283–1291. doi: 10.1016/j.jmb.2003.12.068. [DOI] [PubMed] [Google Scholar]
- 19.Powers RA, Morandi F, Shoichet BK. Structure-based discovery of a novel, noncovalent inhibitor of AmpC β-lactamase. Structure. 2002;10:1013–1023. doi: 10.1016/s0969-2126(02)00799-2. [DOI] [PubMed] [Google Scholar]
- 20.Tondi D, Morandi F, Bonnet R, Costi MP, Shoichet BK. Structure-based optimization of a non-beta-lactam lead results in inhibitors that do not up-regulate β-lactamase expression in cell culture. J. Am. Chem. Soc. 2005;127:4632–4639. doi: 10.1021/ja042984o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burkhard P, Taylor P, Walkinshaw MD. An example of a protein ligand found by database mining: description of the docking method and its verification by a 2.3 A X-ray structure of a thrombin-ligand complex. J. Mol. Biol. 1998;277:449–466. doi: 10.1006/jmbi.1997.1608. [DOI] [PubMed] [Google Scholar]
- 22.Shoichet BK, Stroud RM, Santi DV, Kuntz ID, Perry KM. Structure-based discovery of inhibitors of thymidylate synthase. Science. 1993;259:1445–1450. doi: 10.1126/science.8451640. [DOI] [PubMed] [Google Scholar]
- 23.Pereira HM, Berdini V, Cleasby A, Garratt RC. Crystal structure of calf spleen purine nucleoside phosphorylase complexed to a novel purine analogue. FEBS Lett. 2007;581:5082–5086. doi: 10.1016/j.febslet.2007.09.051. [DOI] [PubMed] [Google Scholar]
- 24.Graves AP, Shivakumar DM, Boyce SE, Jacobson MP, Case DA, Shoichet BK. Rescoring docking hit lists for model cavity sites: predictions and experimental testing. J. Mol. Biol. 2008;377:914–934. doi: 10.1016/j.jmb.2008.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wei BQ, Baase WA, Weaver LH, Matthews BW, Shoichet BK. A model binding site for testing scoring functions in molecular docking. J. Mol. Biol. 2002;322:339–355. doi: 10.1016/s0022-2836(02)00777-5. [DOI] [PubMed] [Google Scholar]
- 26.Graves AP, Brenk R, Shoichet BK. Decoys for docking. J. Med. Chem. 2005;48:3714–3728. doi: 10.1021/jm0491187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brenk R, Vetter SW, Boyce SE, Goodin DB, Shoichet BK. Probing molecular docking in a charged model binding site. J. Mol. Biol. 2006;357:1449–1470. doi: 10.1016/j.jmb.2006.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Y, Shoichet BK. Molecular docking and ligand specificity in fragment-based inhibitor discovery. Nat. Chem. Biol. 2009 doi: 10.1038/nchembio.155. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Teotico D, Shoichet BK. Docking for fragment inhibitors of AmpC β-lactamase: hit-rates, structures and chemical space. Proc. Natl. Acad. Sci. U.S.A. 2009 doi: 10.1073/pnas.0813029106. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hermann JC, Marti-Arbona R, Fedorov AA, Fedorov E, Almo SC, Shoichet BK, Raushel FM. Structure-based activity prediction for an enzyme of unknown function. Nature. 2007;448 doi: 10.1038/nature05981. 775-U2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Babaoglu K, Simeonov A, Lrwin JJ, Nelson ME, Feng B, Thomas CJ, Cancian L, Costi MP, Maltby DA, Jadhav A, Inglese J, Austin CP, Shoichet BK. Comprehensive mechanistic analysis of hits from high-throughput and docking screens against β-lactamase. J. Med. Chem. 2008;51:2502–2511. doi: 10.1021/jm701500e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu SY, McNae I, Kontopidis G, McClue SJ, McInnes C, Stewart KJ, Wang S, Zheleva DI, Marriage H, Lane DP, Taylor P, Fischer PM, Walkinshaw MD. Discovery of a novel family of CDK inhibitors with the program LIDAEUS: structural basis for ligand-induced disordering of the activation loop. Structure. 2003;11:399–410. doi: 10.1016/s0969-2126(03)00060-1. [DOI] [PubMed] [Google Scholar]
- 33.Kurinov IV, Harrison RW. Prediction of new serine proteinase inhibitors. Nat. Struct. Biol. 1994;1:735–743. doi: 10.1038/nsb1094-735. [DOI] [PubMed] [Google Scholar]
- 34.Tondi D, Slomczynska U, Costi MP, Watterson DM, Ghelli S, Shoichet BK. Structure-based discovery and in-parallel optimization of novel competitive inhibitors of thymidylate synthase. Chem. Biol. 1999;6:319–331. doi: 10.1016/S1074-5521(99)80077-5. [DOI] [PubMed] [Google Scholar]
- 35.Lu IL, Mahindroo N, Liang PH, Peng YH, Kuo CJ, Tsai KC, Hsieh HP, Chao YS, Wu SY. Structure-based drug design and structural biology study of novel nonpeptide inhibitors of severe acute respiratory syndrome coronavirus main protease. J. Med. Chem. 2006;49:5154–5161. doi: 10.1021/jm060207o. [DOI] [PubMed] [Google Scholar]
- 36.Schuttelkopf AW, Andersen OA, Rao FV, Allwood M, Lloyd C, Eggleston IM, van Aalten DM. Screening-based discovery and structural dissection of a novel family 18 chitinase inhibitor. J. Biol. Chem. 2006;281:27278–27285. doi: 10.1074/jbc.M604048200. [DOI] [PubMed] [Google Scholar]
- 37.Foloppe N, Fisher LM, Howes R, Potter A, Robertson AGS, Surgenor AE. Identification of chemically diverse Chk1 inhibitors by receptor-based virtual screening. Bioorganic & Medicinal Chemistry. 2006;14:4792–4802. doi: 10.1016/j.bmc.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 38.Gruneberg S, Stubbs MT, Klebe G. Successful virtual screening for novel inhibitors of human carbonic anhydrase: strategy and experimental confirmation. J. Med. Chem. 2002;45:3588–3602. doi: 10.1021/jm011112j. [DOI] [PubMed] [Google Scholar]
- 39.Krämer O, Hazemann I, Podjarny AD, Klebe G. Virtual screening for inhibitors of human aldose reductase. Proteins: Structure Function and Bioinformatics. 2004;55:814–823. doi: 10.1002/prot.20057. [DOI] [PubMed] [Google Scholar]
- 40.Steuber H, Heine A, Klebe G. Structural and thermodynamic study on aldose reductase: nitro-substituted inhibitors with strong enthalpic binding contribution. J. Mol. Biol. 2007;368:618–638. doi: 10.1016/j.jmb.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 41.Xu L, Li CL, Olson AJ, Wilson IA. Crystal structure of avian aminoimidazole-4-carboxamide ribonucleotide transformylase in complex with a novel non-folate inhibitor identified by virtual ligand screening. J. Biol. Chem. 2004;279:50555–50565. doi: 10.1074/jbc.M406801200. [DOI] [PubMed] [Google Scholar]
- 42.Li CL, Xu L, Wolan DW, Wilson IA, Olson AJ. Virtual screening of human 5-aminoimidazole-4-carboxamide ribonucleotide transformylase against the NCI diversity set by use of AutoDock to identify novel nonfolate inhibitors. J. Med. Chem. 2004;47:6681–6690. doi: 10.1021/jm049504o. [DOI] [PubMed] [Google Scholar]
- 43.Barril X, Brough P, Drysdale M, Hubbard RE, Massey A, Surgenor A, Wright L. Structure-based discovery of a new class of Hsp90 inhibitors. Bioorg. Med. Chem. Lett. 2005;15:5187–5191. doi: 10.1016/j.bmcl.2005.08.092. [DOI] [PubMed] [Google Scholar]
- 44.Huang N, Shoichet BK, Irwin JJ. Benchmarking sets for molecular docking. J. Med. Chem. 2006;49:6789–6801. doi: 10.1021/jm0608356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hert J, Keiser MJ, Irwin JJ, Oprea TI, Shoichet BK. Quantifying the relationships among drug classes. J. Chem. Inf. Model. 2008;48:755–765. doi: 10.1021/ci8000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hann MM, Leach AR, Harper G. Molecular complexity and its impact on the probability of finding leads for drug discovery. J. Chem. Inf. Comput. Sci. 2001;41:856–864. doi: 10.1021/ci000403i. [DOI] [PubMed] [Google Scholar]
- 47.Jhoti H. Fragment-based drug discovery using rational design. Ernst Schering Found. Symp. Proc. 2007:169–185. doi: 10.1007/2789_2007_064. [DOI] [PubMed] [Google Scholar]
- 48.Mizutani MY, Itai A. Efficient method for high-throughput virtual screening based on flexible docking: Discovery of novel acetylcholinesterase inhibitors. J. Med. Chem. 2004;47:4818–4828. doi: 10.1021/jm030605g. [DOI] [PubMed] [Google Scholar]
- 49.Brooks WH, McCloskey DE, Daniel KG, Ealick SE, Secrist JA, Waud WR, Pegg AE, Guida WC. In silico chemical library screening and experimental validation of a novel 9-aminoacridine based lead-inhibitor of human S-adenosylmethionine decarboxylase. J. Chem. Inf. Model. 2007;47:1897–1905. doi: 10.1021/ci700005t. [DOI] [PubMed] [Google Scholar]
- 50.Wang JG, Xiao YJ, Li YH, Ma Y, Li ZM. Identification of some novel AHAS inhibitors via molecular docking and virtual screening approach. Bioorganic & Medicinal Chemistry. 2007;15:374–380. doi: 10.1016/j.bmc.2006.09.050. [DOI] [PubMed] [Google Scholar]
- 51.Rosenfeld RJ, Goodsell DS, Musah RA, Morris GM, Goodin DB, Olson AJ. Automated docking of ligands to an artificial active site: augmenting crystallographic analysis with computer modeling. J. Comput.-Aided Mol. Des. 2003;17:525–536. doi: 10.1023/b:jcam.0000004604.87558.02. [DOI] [PubMed] [Google Scholar]
- 52.Montes M, Braud E, Miteva MA, Goddard ML, Mondesert O, Kolb S, Brun MP, Ducommun B, Garbay C, Villoutreix BO. Receptor-based virtual ligand screening for the identification of novel CDC25 phosphatase inhibitors. J. Chem. Inf. Model. 2008;48:157–165. doi: 10.1021/ci700313e. [DOI] [PubMed] [Google Scholar]
- 53.Wang S, Meades C, Wood G, Osnowski A, Anderson S, Yuill R, Thomas M, Mezna M, Jackson W, Midgley C, Griffiths G, Fleming I, Green S, McNae I, Wu SY, McInnes C, Zheleva D, Walkinshaw MD, Fischer PM. 2-Anilino-4-(thiazol-5-yl)pyrimidine CDK inhibitors: synthesis, SAR analysis, X-ray crystallography, and biological activity. J. Med. Chem. 2004;47:1662–1675. doi: 10.1021/jm0309957. [DOI] [PubMed] [Google Scholar]
- 54.Vulpetti A, Casale E, Roletto F, Amici R, Villa M, Pevarello P. Structure-based drug design to the discovery of new 2-aminothiazole CDK2 inhibitors. J. Mol. Graph. 2006;24:341–348. doi: 10.1016/j.jmgm.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 55.Li J, Zhang J, Chen J, Luo XM, Zhu WL, Shen JH, Liu H, Shen X, Jiang HL. Strategy for discovering chemical inhibitors of human cyclophilin A: Focused library design, virtual screening, chemical synthesis and bioassay. J. Comb. Chem. 2006;8:326–337. doi: 10.1021/cc0501561. [DOI] [PubMed] [Google Scholar]
- 56.Ostrov DA, Prada JAH, Corsino PE, Finton KA, Le N, Rowe TC. Discovery of novel DNA gyrase inhibitors by high-throughput virtual screening. Antimicrob. Agents Chemother. 2007;51:3688–3698. doi: 10.1128/AAC.00392-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kolb P, Berset Kipouros C, Huang D, Caflisch A. Structure-based tailoring of compound libraries for high-throughput screening: Discovery of novel EphB4 inhibitors. Proteins. 2008 doi: 10.1002/prot.22028. [DOI] [PubMed] [Google Scholar]
- 58.Tikhonova IG, Sum CS, Neumann S, Engel S, Raaka BM, Costanzi S, Gershengorn MC. Discovery of novel Agonists and antagonists of the free fatty acid receptor 1 (FFAR1) using virtual screening. J. Med. Chem. 2008;51:625–633. doi: 10.1021/jm7012425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kiss R, Kiss B, Konczol A, Szalai F, Jelinek I, Laszlo V, Noszal B, Falus A, Keseru GM. Discovery of Novel Human Histamine H4 Receptor Ligands by Large-Scale Structure-Based Virtual Screening. J. Med. Chem. 2008 doi: 10.1021/jm7014777. [DOI] [PubMed] [Google Scholar]
- 60.Lemaire G, Benod C, Nahoum V, Pillon A, Boussioux AM, Guichou JFO, Subra G, Pascussi JM, Bourguet W, Chavanieu A, Balaguer P. Discovery of a highly active ligand of human pregnane X receptor: a case study from pharmacophore Modeling and virtual screening to "in vivo" biological activity. Mol. Pharmacol. 2007;72:572–581. doi: 10.1124/mol.106.033415. [DOI] [PubMed] [Google Scholar]
- 61.Zhou Y, Peng H, Ji Q, Qi J, Zhu ZP, Yang CZ. Discovery of small molecule inhibitors of integrin alpha v beta 3 through structure-based virtual screening. Bioorg. Med. Chem. Lett. 2006;16:5878–5882. doi: 10.1016/j.bmcl.2006.08.061. [DOI] [PubMed] [Google Scholar]
- 62.Cavasotto CN, Orry AJW, Murgolo NJ, Czarniecki MF, Kocsi SA, Hawes BE, O'Neill KA, Hine H, Burton MS, Voigt JH, Abagyan RA, Bayne ML, Monsma FJ. Discovery of novel chemotypes to a G-protein-coupled receptor through ligand-steered homology modeling and structure-based virtual screening. J. Med. Chem. 2008;51:581–588. doi: 10.1021/jm070759m. [DOI] [PubMed] [Google Scholar]
- 63.Pierce AC, Jacobs M, Stuver-Moody C. Docking study yields four novel inhibitors of the protooncogene Pim-1 kinase. J. Med. Chem. 2008;51:1972–1975. doi: 10.1021/jm701248t. [DOI] [PubMed] [Google Scholar]
- 64.Salam NK, Huang THW, Kota BP, Kim MS, Li YH, Hibbs DE. Novel PPAR-gamma agonists identified from a natural product library: A virtual screening, induced-fit docking and biological assay study. Chem. Biol. Drug. Des. 2008;71:57–70. doi: 10.1111/j.1747-0285.2007.00606.x. [DOI] [PubMed] [Google Scholar]
- 65.Engel S, Skoumbourdis AP, Childress J, Neumann S, Deschamps JR, Thomas CJ, Colson AO, Costanzi S, Gershengorn MC. A virtual screen for diverse ligands: Discovery of selective G protein-coupled receptor antagonists. J. Am. Chem. Soc. 2008;130:5115–5123. doi: 10.1021/ja077620l. [DOI] [PubMed] [Google Scholar]
- 66.Kolb P, Rosenbaum DM, Irwin JJ, Fung J, Kobilka BK, Shoichet BK. Structure-based discovery of β2-adrenergic receptor ligands. Proc. Natl. Acad. Sci. U.S.A. 2009;106:6843–6848. doi: 10.1073/pnas.0812657106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang DZ, Luthi U, Kolb P, Cecchini M, Barberis A, Caflisch A. In silico discovery of β-secretase inhibitors. J. Am. Chem. Soc. 2006;128:5436–5443. doi: 10.1021/ja0573108. [DOI] [PubMed] [Google Scholar]
- 68.Huang DZ, Luthi U, Kolb P, Edler K, Cecchini M, Audetat S, Barberis A, Caflisch A. Discovery of cell-permeable non-peptide inhibitors of β-secretase by high-throughput docking and continuum electrostatics calculations. J. Med. Chem. 2005;48:5108–5111. doi: 10.1021/jm050499d. [DOI] [PubMed] [Google Scholar]
- 69.Yu WM, Guvench O, Mackerell AD, Jr, Qu CK. Identification of Small Molecular Weight Inhibitors of Src Homology 2 Domain-Containing Tyrosine Phosphatase 2 (SHP-2) via in Silico Database Screening Combined with Experimental Assay. J. Med. Chem. 2008 doi: 10.1021/jm800229d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li M, Ni N, Chou HT, Lu CD, Tai PC, Wang B. Structure-based discovery and experimental verification of novel AI-2 quorum sensing inhibitors against Vibrio harveyi. ChemMedChem. 2008;3:1242–1249. doi: 10.1002/cmdc.200800076. [DOI] [PubMed] [Google Scholar]
- 71.Chen D, Misra M, Sower L, Peterson JW, Kellogg GE, Schein CH. Novel inhibitors of anthrax edema factor. Bioorg. Med. Chem. 2008;16:7225–7233. doi: 10.1016/j.bmc.2008.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Heinke R, Spannhoff A, Meier R, Trojer P, Bauer I, Jung M, Sippl W. Virtual Screening and Biological Characterization of Novel Histone Arginine Methyltransferase PRMT1 Inhibitors. ChemMedChem. 2008;4:69–77. doi: 10.1002/cmdc.200800301. [DOI] [PubMed] [Google Scholar]
- 73.Honma T, Hayashi K, Aoyama T, Hashimoto N, Machida T, Fukasawa K, Iwama T, Ikeura C, Ikuta M, Suzuki-Takahashi I, Iwasawa Y, Hayama T, Nishimura S, Morishima H. Structure-based generation of a new class of potent Cdk4 inhibitors: new de novo design strategy and library design. J. Med. Chem. 2001;44:4615–4627. doi: 10.1021/jm0103256. [DOI] [PubMed] [Google Scholar]
- 74.Takano Y, Koizumi M, Takarada R, Kamimura MT, Czerminski R, Koike T. Computer-aided design of a factor Xa inhibitor by using MCSS functionality maps and a CAVEAT linker search. J. Mol. Graph. 2003;22:105–114. doi: 10.1016/s1093-3263(03)00140-2. [DOI] [PubMed] [Google Scholar]
- 75.Cregge RJ, Durham SL, Farr RA, Gallion SL, Hare CM, Hoffman RV, Janusz MJ, Kim HO, Koehl JR, Mehdi S, Metz WA, Peet NP, Pelton JT, Schreuder HA, Sunder S, Tardif C. Inhibition of human neutrophil elastase. 4. Design, synthesis, X-ray crystallographic analysis, and structure-activity relationships for a series of P2-modified, orally active peptidyl pentafluoroethyl ketones. J. Med. Chem. 1998;41:2461–2480. doi: 10.1021/jm970812e. [DOI] [PubMed] [Google Scholar]