

Abstract
Polyubiquitylation leading to proteasomal degradation is a well-established mechanism for regulating TGF-β signal transduction components such as receptors and Smads. Recently, an equally important role was suggested for monoubiquitylation of both Smad4 and Receptor-associated Smads that regulates their function without protein degradation. Monoubiquitylation of Smads was discovered following the identification of deubiquitylases required for TGF-β signaling, suggesting that continuous cycles of Smad mono- and deubiquitylation are required for proper TGF-β signal transduction. Here we summarize and discuss recent work on Smad mono- and deubiquitylation.
Keywords: BMP, Smad4/Medea, Ectodermin/Tif1-γ/Trim33, Fat facets/Fam/Usp9X, Usp15
Introduction
Members of the TGF–β family of proteins are pleiotropic cell signaling molecules involved in many fundamental biological processes. These include the induction of the embryonic germ layers, patterning the axes of the body plan and maintenance of homeostasis in adult tissues (1). Mirroring these multiple roles, defects in TGF–β signaling are associated with both developmental and adult syndromes such as birth defects, tissue fibrosis and cancer (2). Mechanisms underlying the transduction of TGF–β signals from the cell membrane to the nucleus have been extensively characterized and reviewed elsewhere (3,4). Briefly, secreted TGF–β ligands engage in a complex with two transmembrane kinase receptors leading one member of the receptor complex to phosphorylate C-terminal serine residues of R-Smads (Receptor-associated Smad signal transducers). Phosphorylation enables R-Smads to accumulate in the nucleus where they form a DNA binding complex with their sibling protein Smad4. This Smad heteromeric complex regulates gene expression in conjunction with promoter-specific transcription factors and cofactors. This basic scenario is shared between the TGF–β/Activin/Nodal and Dpp/BMP subfamilies of TGF-β family ligands, although each subfamily employs a different set of R-Smads (Smad2/3 or Smad1/5/8, respectively).
A growing number of studies have shown that numerous mechanisms ensure tight control over the activity of receptors and Smads, thereby regulating cellular responsiveness to TGF–β signals. Some of these strategies influence the primary phosphorylation events of the signaling cascade, such as receptor and R-Smad phosphorylation (5) while others involve a variety of post-translational modifications to pathway components. In the case of Smads these modifications include additional phosphorylation events (6,7), sumoylation (8), parpylation (9), acetylation (10) and ubiquitylation (11,12).
Ubiquitin entered our understanding of TGF–β signaling when the mammalian HECT-domain ubiquitin ligases, Smurf1 and Smurf2, were discovered as negative regulators of the pathway (13,14). Subsequently other molecules with E3 ubiquitin ligase activity were isolated that regulate TGF–β signaling. These include additional HECT-domain family members (e.g., Nedd4L, Wwp1/Tiul1 and Aip4/Itch; 12) and RING-domain proteins such as Arkadia, Highwire and Ectodermin/Tif1–γ/Trim33 (15–17). These molecules, except Arkadia, were isolated as inhibitors of TGF–β signaling thus assigning to ubiquitylation a predominantly negative role in the TGF–β cascade. The role of polyubiquitylation and degradation in TGF–β signaling is well known (12). Here we discuss technical issues related to the analysis of monoubiquitylation and recent data revealing the role of mono- and deubiquitylation in the regulation of Smad activity.
The basics of ubiquitylation
Ubiquitylation is a regulatory mechanism that impinges on a wide variety of processes including cell-cycle checkpoints, DNA damage responses and signal transduction pathways (e.g., 18). Ubiquitylation is the covalent attachment of a ubiquitin polypeptide to a target protein via the sequential action of three enzymes: a ubiquitin activating enzyme (E1), a conjugating enzyme (E2) that carries the activated ubiquitin and transfers it to the target protein and a ubiquitin ligase (E3) that binds both the target and the E2 to promote efficient ubiquitin transfer.
Compared to other post-translational modifications, ubiquitylation is a particularly diverse process (19). Ubiquitin can be linked to a target protein as a monomer on a single lysine (monoubiquitylation), as a monomer to multiple lysines (oligoubiquitylation), or several ubiquitin molecules can be added serially to the same lysine forming a polyubiquitin chain. Furthermore, polyubiquitin chains can assume different geometries according to which of the seven internal lysines of ubiquitin (K48, K63, K29, etc.) is used for polymerization. As a result ubiquitylation can participate in a variety of regulatory mechanisms. For example: 1) as a trigger for proteasomal degradation (K48-linked polyubiquitylation), 2) as a scaffold for protein-protein interactions (K63-linked polyubiquitylation in the NF-κB pathway), 3) or as a tag for vesicle sorting during endocytosis (EGF receptor signaling). In addition, ubiquitylation can be reversed. Cells have at their disposal over 100 different deubiquitylating enzymes that can remove ubiquitin from modified proteins, thus resetting the system or enabling cells to switch from one type of ubiquitylation to another (20).
Monoubiquitylation received particular attention in the last decade when a number of observations in fields as disparate as DNA repair, histone regulation, membrane receptor trafficking and regulation of tumor suppressors (such as PTEN, FOXO and p53) pointed to a key role for monoubiquitylation as a general modulator of protein function, rather than as a dedicated signal for protein destruction (21,22). In this respect, monoubiquitylation is comparable to protein phosphorylation: it can regulate protein activity and subcellular localization, it can form or conceal protein-protein interaction surfaces, it can be used for regulation in a time- and space-dependent manner without the need of regulating total protein levels and it can be rapidly reversed by the activity of deubiquitylases. Moreover, given the size of ubiquitin (76 amino acids), monoubiquitylation could in principle enable interactions based on the newly resulting tridimensional structure of the targeted protein.
Technical challenges in the analysis of ubiquitylation
When studying a regulatory mechanism based on ubiquitylation, there are three key experimental issues to be considered: 1) what is the relevant ubiquitylation pattern of the target - mono-, oligo- or polyubiquitylation; 2) does ubiquitylation regulate degradation of the target or is it regulative; and 3) in the regulative situation - to what extent can the biological function of the ligase (or the deubiquitylase) be explained by regulation of the proposed target?
For the first point (relevant ubiquitylation pattern of the target), it is clear that studying the endogenous ubiquitylation pattern of a protein is challenging. In rare instances, the pattern is so obvious that the mono- or polyubiquitylated isoforms can be readily detected by a simple western blot, as is the case for Hif1α or Fancd2 (23). Generally this is not observed, even for a paradigmatic example of ubiquitin-dependent degradation such as p53. At least in part, this is because polyubiquitylated proteins are rapidly degraded and because many deubiquitylases are thought to be aspecifically activated upon cell lysis. Thus, in most cases it is necessary to immunoprecipitate the protein from cell extracts and identify double positive higher molecular weight bands with both anti-ubiquitin and an antibody to the target protein (or its epitope tag). Even then interpretation of the results may be complicated by the presence of other coprecipitating ubiquitylated proteins and by the fact that available antibodies detecting endogenous ubiquitin are often not very sensitive, such that low levels of ubiquitylation or monoubiquitylation can be easily missed. The use of overexpressed epitope-tagged ubiquitin constructs can solve the last problem, but they introduce an extra variable. Alternatively raising antibodies specific to ubiquitylated proteins would circumvent the need for immunoprecipitation. However this approach is very difficult with only two or three antibodies available. Thus it is often hard to assess, in a quantitative fashion, the degree of ubiquitylation of a protein.
Once the ubiquitylation pattern has been defined, the next question is where this modification is occurring on the target protein (i.e. on which lysine). Bioinformatics studies of evolutionary conservation can provide clues to the identity of targeted lysines based on the idea that evolution will act to conserve important regulatory interactions (24). Alternatively direct mapping of ubiquitylation sites can be addressed either by systematic lysine mutation (25,26) or by mass-spectrometry (27). The first approach can be complicated by the effect of amino acid mutations that are independent from ubiquitylation (e.g. modification of protein structure). The second approach is more direct but is limited to proteolytic peptides that can be detected, so that often there are lysines that cannot be queried. In the end, mutation of the relevant residue(s) should render the protein insensitive, both biochemically and functionally, to ubiquitylation.
For the second point (ubiquitylation regulates degradation of the target or is regulative), this too can be challenging. For a few proteins such as Hif1α, p53 and β-catenin it is self-apparent because inhibition of the proteasome greatly enhances detection of the polyubiquitylated protein and readily stabilizes its steady-state levels. In general it is more difficult to establish which is the relevant process. For example, degradation may be visible only in pulse-chase assays. Alternatively, polyubiquitylation and degradation may be visible only upon overexpression of the E3 ligase. Here, in the absence of supporting loss-of-function evidence E3 overexpression can be misleading as it can mask regulative ubiquitylation. Lastly, polyubiquitylation does not automatically lead to degradation. For example in the NF-κB pathway, K63-linked polyubiquitylation acts as a “scaffold” to enable signal transduction (19). Also, in the TGF-β pathway K63-linked polyubiquitylation promoted by TRAF6 plays a key role in the regulation of non-Smad TGF-β receptor signaling (28,29,30).
For the third point (the biological function of an E3 ligase or a deubiquitylase is dependent upon its target protein), it is essential to identify the appropriate enzyme/target pair. In the ubiquitylation reaction, target specificity is thought to be primarily determined at the level of E3 ligase/target interaction (31). However, E3 specificity is not absolute as one ligase can have multiple targets. This also applies to deubiquitylases where the potential for multiple interactions is even higher as the human genome encodes for over 100 deubiquitylases (23). To determine the functional enzyme/target pair one relies on both biochemical and genetic evidence. For biochemical evidence one should observe a requirement of the E3 ligase for ubiquitylation. For genetic evidence, phenotypes due to loss of the E3 ligase should also depend on the target and phenotypes due to loss of the target should dominate those due to loss of the E3 ligase in double mutants (i.e. the target should be epistatic to the ligase). An example of strong genetic data supporting an enzyme/target pair is that of Mdm2 and p53: mouse knockouts for the p53 ligase Mdm2 die during embryogenesis and are fully rescued by the concomitant knockout of p53 (32).
Mono- and deubiquitylation of Smad4 by Ecto/Tif1-γ and Fam/Usp9X
An early example of monoubiquitylation as a mechanism regulating Smad activity derived from studies of the E3 ligase Ecto/Tif1–γ and the deubiquitylase FAM/Usp9x (17,25). Ecto/Tif1–γ was cloned via a cDNA overexpression screen in Xenopus embryos looking for molecules opposing the differentiation of ectodermal cells. Its subsequent characterization showed that Ecto/Tif1–γ is required to protect cells of the ectoderm, located in the animal region of the embryo, from Nodal/TGF–β signals that induce endoderm and mesoderm in the vegetal and equatorial regions, respectively. Within the ectoderm, Ecto/Tif1–γ restricts BMP signaling, enabling the balanced differentiation of ectodermal cells into epidermal and neural lineages. Accordingly, in human cells Ecto/Tif1–γ restrains both TGF–β and BMP responses.
Ecto/Tif1–γ was shown to bind Smad4, the common transducer of BMP and TGF–β signals and to promote its ubiquitylation. In knock down experiments Ecto/Tif1–γ was required for Smad4 ubiquitylation in Xenopus embryos, for nuclear exclusion of Smad4 in human cells and for the instability of the Smad4 colon tumor allele R100T. Consistent with this, mutation of the RING domain of Ecto/Tif1–γ was sufficient to abolish its Smad4 inhibitory activity. Thus, Ecto/Tif1–γ was proposed to act a Smad4 ubiquitin ligase and Smad4 antagonist (33,34).
The role of Smad4 ubiquitylation as a key regulatory step in TGF–β signaling was later substantiated by the isolation of the deubiquitylase FAM/Usp9x as a required factor for Smad activity. In mammalian cells and Xenopus embryos, FAM/Usp9x sustains both TGF–β and BMP signaling by deubiquitylating Smad4 and by counteracting the inhibitory activity of Ecto/Tif1–γ (25). Smad4 is primarily, though not fully, monoubiquitylated in several cellular systems including Xenopus embryos, depending upon the endogenous levels of FAM/Usp9x and of Ecto/Tif1–γ. This modification was mapped to lysine 519 (K519) of Smad4 that is located close to an interaction interface with R-Smads. Given its position, K519-monoubiquitylated Smad4 is unable to form a complex with phosphorylated Smad2. Thus, the antagonistic activities of Ecto/Tif1–γ and FAM/Usp9x on Smad4, mono- and deubiquitylation respectively, regulate active Smad complex formation and, ultimately, TGF–β responsiveness (Fig. 1A).
Fig. 1. Smad4 regulation by mono- and deubiquitylation.
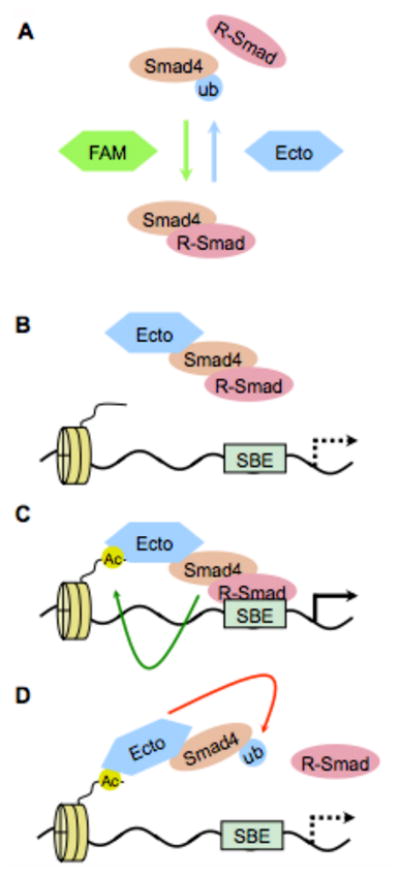
A) Opposing roles of Ecto/Tif1-γ (an E3 ligase adds a monoubiquitin that destabilizes Smad4-R-Smad complexes - blue arrow) and FAM/Usp9x (a deubiquitylase that removes monoubiquitin and allows Smad4-R-Smad complex formation - green arrow) in TGF-β signaling. B-D) Model for activation of Smad4 monoubiquitylation by association with DNA. B) Ecto/Tif1-γ bound to an activated R-Smad-Smad4 complex is recruited to Smad-binding elements (SBE) in promoter DNA. C) The presence of specific histone marks such as acetylation (AC), possibly induced by transcriptionally-active Smads (green arrow), then activates Ecto ubiquitin ligase activity. D) Ecto/Tif1-γ then monoubiquitylates Smad4 (red arrow) causing the dissociation of the Smad complex and the cessation of transcription.
Other studies also observed that Smad4 could be monoubiquitylated (35,36). One report noted that Smad4 is mainly mono- or oligoubiquitylated, mapped this modification to lysine 507 (K507) and proposed it as a positive input for TGF–β signaling (35). This interpretation was primarily based on the lack of activity of Smad4 K507-mutants. However K507 is one of the residues of Smad4 that bind to the phosphorylated R-Smad C-terminal tail, such that its mutation prevents efficient R-Smad/Smad4 interactions (37,38). Thus, there is the possibility of a different rationale for the inactivity of Smad4 K507 mutants. Still, in some assays Smad4 appears linked to two ubiquitins (35,25), with K507 perhaps acting as an alternative or additional monoubiquitylation site to K519. Monoubiquitylation at K507 would also interfere with phospho-R-Smad binding and thus functionally inactivate Smad4. Future studies are necessary to discern how these two ubiquitylation events are regulated in different cellular contexts and whether different ligases are responsible for K507 and K519 ubiquitylation.
In parallel to these studies, a bioinformatics analysis of lysine conservation in Smad proteins identified Smad4 K507 as a lysine that is universally conserved at the homologous position in all Smads from nematodes, flies and mice. A subgroup analysis of mouse Smad4 and its closest relatives (fly Medea and nematode Sma-4) then identified K519 as a Smad4 specific lysine. Based on the fact that K507 was known to be monoubiquitylated (35) it was proposed that K519 would be as well (24). This prediction was subsequently validated (25).
One unsolved question related to Smad4 monoubiquitylation is where this takes place within the cell. Two observations compound the uncertainty: 1) although Smad4 monoubiquitylation is incompatible with Smad4/R-Smad interactions Ecto/Tif1 γ was found in a trimeric complex with Smad4 and phospho-Smad2, and 2) TGF-β stimulation promoting Smad4/R-Smad complex formation enhanced Smad4 monoubiquitylation (25). Recently, a possible solution to this question was provided by the discovery of a link between Ecto/Tif1-γ and the transcriptional engagement of Smads (39). To inhibit TGF–β signaling Ecto/Tif1–γ requires not only its RING domain but also its C-terminal PHD–Bromo domain. This domain-enables Ecto/Tif1–γ to bind to histones in a manner dependent upon acetylation, and this in turn activates Ecto/Tif1–γ ubiquitin ligase activity for Smad4 (39). A positive effect of the p300 histone acetyltransferase on Smad4 monoubiquitylation was observed, thus reinforcing the notion that Smad4 ubiquitylation can be regulated by chromatin (36). This mechanism would allow Smad transcriptional complexes to self-limit their own activity (Fig. 1B-D). When active Smads bind a promoter in response to TGF–β stimulation they carry with them Ecto/Tif1–γ. Once bound to chromatin Ecto/Tif1–γ can be locally activated (possibly by histone modifications induced by Smads such as acetylation), to ubiquitylate Smad4 and destabilize the Smad complex (39). The role of the PHD-Bromo domain of Ecto/Tif1–γ as a chromatin-interaction module was recently observed in another study, where a distinct histone-code for PHD–Bromo domain interaction was observed 40. Future studies will be required to address the possibility that cell type specific histone-codes are responsible for distinct activities of Ecto/Tif1–γ (40,41).
RING and PHD–Bromo domains, promoting ubiquitylation and histone reading respectively, are found in all Tif1 family members. Tif1-α/Trim24 and KAP1/Tif1-β/Trim28 also act at the chromatin level via their PHD–Bromo domain (42.43) and have the potential to promote ubiquitylation through their RING-domain (44,45). This suggests that a general role of Tif1 proteins is connecting epigenetic information to the regulation of transcription factors and that their E3 ligase activity is integral to this role (46). The finding that Ecto/Tif1–γ is regulated by chromatin opens new questions about how Smads themselves interact with chromatin such as: what are the histone modifications promoted by Smads and what are the epigenetic contexts in which Ecto/Tif1–γ regulation of Smads is permitted or prohibited?
Regulation of Ecto/Tif1–γ at the chromatin level also provides an explanation for the observation that Smad4 in most cells is not completely ubiquitylated: monoubiquitylation occurs on the fraction of the protein that is transcriptionally active. In this model Ecto/Tif1–γ primarily acts at the promoter by creating an equilibrium between Smad complexes containing Ecto/Tif1–γ that are readily inactivated and pure Smad complexes that successfully engage in transcription.
According to this model, there would always be a fraction of the pool of Smad4 in a cell that is inactive. This is consistent with observations that Smad4 availability is context dependent. On the one hand, cells have at their disposal a wide amount of Smad4 such that in vitro only very efficient Smad4 knockdown can unveil the requirement of Smad4 for TGF–β responses (47). On the other hand, evidence suggests that the amount of Smad4 available for signaling is limited in vivo, such that Smad4 heterozygosity is sufficient to unveil its tumor-suppressive functions and in some cases BMP-induced and TGF–β-induced Smads compete for a seemingly limited pool of Smad4 (48,49). Thus, perhaps some factors entrap Smad4 in an inactive pool and the remaining free Smad4 is regulated by monoubiquitylation. Candidates for Smad4 entrapping factors are Sno-Ski family proteins. These proteins require Smad4 interaction for their TGF-β inhibitory functions (50) and they can regulate Smad4 monoubiquitylation (36). Collectively, these biochemical studies point to the modulation of Smad4 activity as a key step in the cellular regulation of TGF–β signal transduction, acting in parallel to the regulation of R-Smad phosphorylation. Overall, it appears that TGF–β stimulation sets the maximum possible R-Smad activity in a cell but the response will be precisely determined by Smad4 availability.
Genetic evidence in support of Smad4 regulation by Ecto/Tif1–γ and FAM/Usp9X
The regulation of Smad4 by Ecto/Tif1–γ was further documented in vivo by studies of early mouse embryos. Analysis of Ecto/Tif1–γ homozygous knockout mice (Ecto−/−) showed that lack of an intracellular Smad antagonist caused embryonic defects that were comparable to deregulation of Nodal, the primary TGF–β ligand in the early embryo (41). Ecto−/− embryos die at the time of gastrulation and show phenotypes caused by unrestrained Nodal effects on extraembryonic tissues. In particular, the induction of the anterior visceral endoderm (AVE) by Nodal occurs in a much broader domain than normal, as shown by the increased expression of the Smad targets Lefty1 and Cerl1 (Fig. 2). Consistent with the biochemical characterization of Ecto/Tif1–γ as a Smad4 antagonist, for the AVE phenotype Smad4 is epistatic to Ecto/Tif1–γ: double Ecto−/−; Smad4−/− embryos are equal to Smad4−/− embryos.
Fig. 2. Ecto/Tif1–γ in early mouse development.
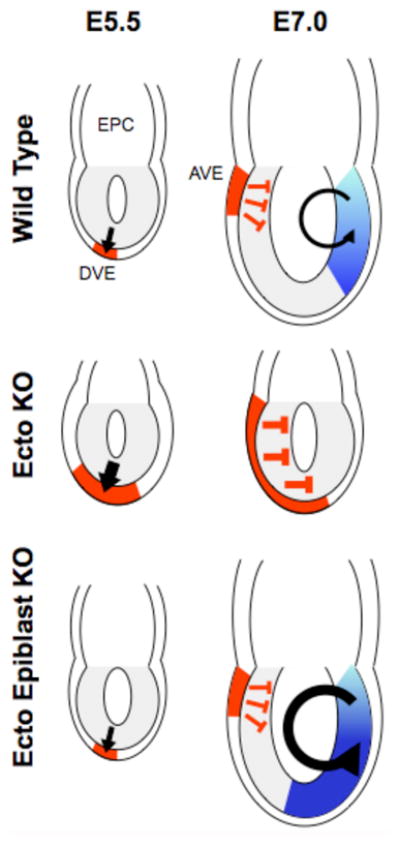
Schematic of mouse embryos with the epiblast (progenitor of the embryo proper) in gray adjacent to extraembryonic cells of the ectoplacental cone (EPC). The thin layer surrounding the epiblast and the EPC is the primitive endoderm. Top row is wild type. At embryonic day 5.5 (E5.5 - one day past uterine implantation) Nodal signaling (black arrow) induces differentiation of the Distal Visceral Endoderm (DVE - in red). At E7.0, one-half day past the initiation of gastrulation, Nodal (black arrow) induces differentiation within the epiblast of mesodermal tissues (shades of blue). Rotation of the DVE toward the anterior forms the Anterior Visceral Endoderm (AVE – in red) that secretes the Nodal antagonists Lefty1 and Cerberus-like (red T-bars). These proteins limit the activity of Nodal within the epiblast. Middle row is Ecto −/−. At stage E5.5. the lack of Ecto/Tif1–γ causes an expansion of the DVE and subsequently the AVE (larger red area). The latter leads to the absence of mesoderm due to production of excess Lefty1 and Cerberus-like (larger red T-bars). Bottom row is Ecto−/− only in epiblast cells. At stage E5.5 these embryos are indistinguishable from wild type. At stage E7.0, AVE expression of Lefty1 and Cerberus-like is wild type but enhanced Nodal signaling (larger black arrow) within the epiblast leads to expansion of mesoderm (larger blue area).
In mouse embryos, the AVE plays a key role in setting the anterior-posterior axis: Lefty1 and Cerl1 encode for secreted Nodal antagonists that diffuse into the anterior epiblast abutting the AVE and prevent Nodal from inducing mesoderm differentiation. In Ecto−/− AVE, overproduction of Lefty1 and Cerl causes, as a secondary event, a nearly complete inhibition of Nodal ligands that are active in the epiblast. As a consequence Ecto−/− embryos do not form mesoderm. Thus, in Ecto−/− embryos, a precocious excess of Smad signaling in AVE cells results in lack of Nodal/TGF–β signaling to epiblast cells at later time-points. This causal relationship was supported by the observation that genetic reduction of Nodal in Ecto−/− embryos led to normal AVE induction and the rescue of mesoderm differentiation.
Ecto−/− embryos also have phenotypes that are the opposite of those caused by the loss of R-Smads in AVE cells: lack of AVE induction or Lefty1/Cerl expression and unrestrained mesoderm induction in the epiblast (51). Further analyses showed that lack of mesoderm in Ecto−/− embryos is specifically due to a direct effect of Ecto/Tif1–γ in AVE cells. Tissue-specific inactivation of Ecto/Tif1–γ only in epiblast cells showed a different phenotype - normal posterior mesoderm induction with expansion of anterior primitive streak derivatives. This phenotype is consistent with enhanced Nodal signaling (whose maximal dose is required to induce anterior primitive streak) and is the opposite of phenotypes seen with epiblast-specific inactivation of Smad4 (52). Together, these data indicate that negative regulation of Smad4 is essential for the correct interpretation of TGF–β signaling in embryonic tissues. Further, in the AVE of Ecto−/− embryos Smad target genes were dramatically enhanced, but without overt changes in the expression of Nodal or in the intensity of Smad2 phosphorylation. In other words, this is an example of cellular interpretation of a TGF–β morphogen signal that depends on variation in the cellular perception of the signal, rather than on its extracellular concentration.
Inactivation of Ecto/Tif1–γ in mouse adult tissues indicates that this protein has additional functions. For example, tissue-specific inactivation of Ecto/Tif1–γ in the pancreas and liver unveiled a tumor suppressive function (53,54) that was unexpected from the deletion of a TGF–β antagonist. In zebrafish, Ecto/Tif1–γ was identified as the affected gene in the moonshine mutant that displays defective red-cell progenitor survival (55). For this phenotype, it was subsequently proposed that Ecto/Tif1–γ serves as a positive R-Smad cofactor in a Smad4-independent TGF–β pathway (56). However, this is at odds with data from mice with inactivation of Ecto/Tif1–γ in hematopoietic stem cells. These mice initially display normal circulating red-cell parameters but developed a progressive myeloproliferative syndrome leading to leukemic conditions and unbalanced hematopoietic stem cell differentiation within the bone marrow (57,58). A recent re-examination of zebrafish moonshine mutants uncovered defects in the myeloid lineage that are more similar to those observed in mice (59), suggesting a conserved function of Ecto/Tif1–γ in the vertebrate hematopoietic system. Moreover, at least for the erythroid phenotype, a zebrafish genetic screen identified the PolII-associated factors pTEF–β and FACT as required downstream of Ecto/Tif1–γ. This suggested that Ecto/Tif1–γ acts by promoting transcriptional elongation at promoters of erythroid differentiation genes (60), although the specific transcription factors recruiting Ecto/Tif1–γ to chromatin and any requirement of its RING and PHD-Bromo domains remain unknown. Collectively, the data indicate that Ecto/Tif1–γ likely regulates transcription factors other than Smads. Thus it is important to dissect to what extent regulation of TGF-β/BMP-dependent or other factors influence the various Ecto/Tif1–γ mutant phenotypes.
Regarding the genetic validation of FAM/Usp9x as a Smad4 deubiquitylase, it is important to note that Drosophila Fat facets (homolog of FAM/Usp9x) is a well known regulator of cell fate (61). During eye development Fat facets regulates ubiquitylation of the endocytic adaptor Liquid facets/Epsin influencing the Receptor Tyrosine Kinase/Ras and Notch pathways (62,63). Subsequent studies identified a requirement for fat facets during development of larval neuro-muscular junctions and found that this was accomplished by opposing the activity of the E3 ligase Highwire (64), a negative regulator of Gbb/BMP signaling (16). Building upon this initial connection between Fat Facets and BMP signaling, experiments exploiting Drosophila wing development showed that overexpression of Fat facets produced vein overgrowth phenotypes that are typically caused by ectopic Dpp/BMP signaling (25). Most recently, a closer examination of embryos generated by transheterozygous combinations of several fat facets mutant alleles uncovered a previously unsuspected requirement for fat facets in Dpp/BMP signaling during dorsal-ventral patterning (65). We observed that a subset of fat facets mutants resemble dpp mutants and that introduction of a fat facets mutation enhances the phenotype of dpp mutants. These new results suggest that regulation of Smad4, Medea in flies, by ubiquitylation and deubiquitylation is an evolutionarily conserved mechanism by which cells interpret TGF–β morphogen gradients.
Interestingly, Ecto/Tif1–γ is not cleanly conserved in flies. The closest fly protein, Bonus, is equally related to the triplicated mammalian Tif1 proteins by sequence but appears functionally most similar to Tif1-α/Trim24 (66). The lack of conservation implies that another Medea ubiquitin ligase exists opposite Fat facets to modulate Dpp/BMP signaling during embryonic dorsal-ventral axis formation.
Monoubiquitylation of R-Smads and their deubiquitylation by Usp15
Recently, the ubiquitylation pattern of R-Smads was revisited. This study found that a previously unappreciated aspect of their regulation occurs by oligoubiquitylation. Utilizing a scheme similar to the one that lead to FAM/Usp9x, the deubiquitylase Usp15 was identified as an important factor for R-Smad activity (26). Usp15 is required in multiple cellular contexts for Smad transcription and for TGF–β/BMP-induced responses including cell migration and germ layer patterning in Xenopus embryos. Consistent with this, a role for Usp15 in dorsal-ventral patterning (and by extension BMP signaling) was recently proposed in zebrafish (67).
Mechanistically, Usp15 interacts with R-Smads of the TGF–β (i.e. Smad2/3) and BMP (i.e. Smad1/5/8) branches and opposes their ubiquitylation. In the absence of Smurf overexpression R-Smads are primarily monoubiquitylated on two lysines that are essential for DNA recognition (K33 and K81), such that monoubiquitylation of R-Smads was sufficient to prevent DNA binding. It was thus proposed that Usp15 primarily functions by opposing R-Smad monoubiquitylation and by that this sustains their DNA binding activity. This finding is supported by the observation that in absence of Usp15, R-Smads were less efficiently recruited to target promoters in ChIP assays. In contrast, upstream events in TGF-β signaling such as subcellular Smad2/3 localization and formation of endogenous R-Smad/Smad4 complexes were unaffected by the loss of Usp15. Overall, it appears that monoubiquitylation of R-Smads, in the absence of degradation and independent of overt effects from phosphorylation-dephosphorylation, is sufficient to control R-Smad activity (Fig. 3A).
Fig. 3. Ubiquitylation and regulation of R-Smads.
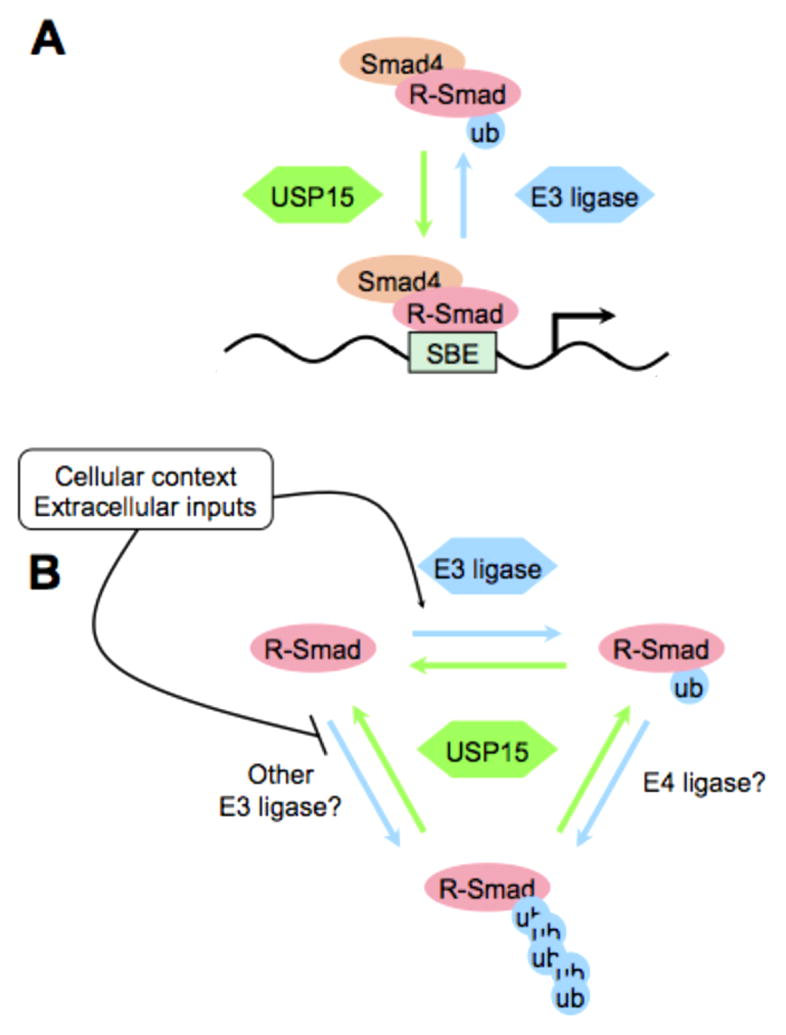
A) Monoubiquitylation of R-Smads inhibits their association with a Smad binding element (SBE) in a target gene promoter (blue arrow). This inhibition is reversed by the deubiquitylation of R-Smads by Usp15 (green arrow) that stimulates Smad complex transcriptional activity. B) Model for potential relationship between mono- and polyubiquitylation of R-Smads. Polyubiquitylation might occur either by elongating an existing monoubiquitin via an E4 ligase or stimulated directly by an E3 ligase distinct from the one inducing monoubiquitylation (blue arrows - ubiquitin addition). Usp15 may oppose all of these reactions (green arrows - deubiquitylation). Extracellular signals or the cell-intrinsic context may regulate each of these three processes positively or negatively.
In addition, Usp15 differentially influences the transcriptional activity of Smad2/3 complexes as shown by the fact that it has only minor effects on promoters when other transcription factors provide the central DNA binding platform (such as FoxH1 on the Mix.2 reporter). Previously differences in Smad transcriptional activity were assigned to preferential regulation of some promoters by Smad3 (able to bind DNA directly) and others by Smad2 (unable to directly bind to DNA). This led to the idea that Smad2 and Smad3 may play non-redundant roles in TGF-β signal transduction. This hypothesis is however inconsistent with genetic studies in mice, where replacement of Smad2 with Smad3 fully rescued phenotypes due to Smad2 inactivation and did not cause any defects in otherwise wild type mice (68). Nevertheless, the potential for direct versus indirect R-Smad recruitment to, or influence on, promoter DNA remains a possibility. From this perspective, R-Smad monoubiquitylation may represent a means of inhibiting R-Smad activity on selected promoters while leaving them active on others. Thus, R-Smad monoubiquitylation may function as a switch between different TGF-β induced gene-expression programs.
In the future, it will be interesting to learn how monoubiquitylation of R-Smads is regulated, and if this regulation represents a new point of crosstalk between TGF–β signaling and other pathways. Some reports suggest the possibility that DNA damage might be one of these. Usp15 has been identified as an ATM phosphorylation target in cells upon irradiation (69), raising the possibility that DNA damage might activate Usp15 activity to foster Smad activity and induce cell-cycle arrest.
HECT-domain proteins are strong candidates for E3 ligases stimulating R-Smad monoubiquitylation. Usp15 is both necessary and sufficient to oppose R-Smad polyubiquitylation as well as degradation induced by Smurf overexpression. Further, a mixture of recombinant Smurf2/Nedd4 can ubiquitylate Smad3 in vitro and promote its detachment from DNA. This effect mirrors depletion of Usp15 (inhibits R-Smad binding to chromatin) and correlates with decreased DNA binding activity of purified monoubiquitylated Smad3. Lastly, Smad3 monoubiquitylation appears dependent on endogenous Smurf2 in mouse MEFs (70).
These recent data suggest that R-Smad monoubiquitylation is a physiologically relevant mechanism of regulation. However, to date R-Smad ubiquitylation was studied in the context of polyubiquitylation and degradation (13,14). There are several possibilities for the relationship between poly- and monoubiquitylation for R-Smad proteins. First, polyubiquitylation may be a parallel input to monoubiquitylation, occurring at different lysines and Usp15 may oppose both modifications. Second, R-Smad polyubiquitylation may represent a secondary event taking place in particular contexts requiring total R-Smad degradation. In this scenario, perhaps linker phosphorylation or other inputs influence the residence of Smurfs on R-Smads thus determining the balance between monoubiquitylation (occurring quickly) and polyubiquitylation (requiring more stable ligase-target interaction). Third, Smurfs could work in some systems as E4 ligases (ubiquitin chain extenders), acting downstream of an as yet unidentified E3 ligase responsible for R-Smad monoubiquitylation. The homolog of Usp15 in yeast, Ubp12, was identified as a resident subunit of the CSN/COP9 signalosome, an important regulator of cullin-RING ubiquitin ligase complexes (71). The availability of cullin-specific inhibitors (72) enables testing of cullin ligases in opposition to Usp15 in R-Smad monoubiquitylation (Fig. 3B).
Addressing these issues will require better characterization of the genetic requirements of HECT ligases in general and Usp15 in particular and then testing for possible interactions. Regarding Usp15, the phenotype of knockout mice has not yet been reported and the extent of overlap with the similar deubiquitylase Usp4 (73) is unknown. Regarding Smurfs, genetic requirements in Dpp/BMP signaling are well known in Drosophila (74,75) but are missing in mammals. With regard to mammalian Smurfs, the redundancy between Smurf1 and Smurf2 and their roles in JNK signaling and planar cell polarity (76,77) represent complicating issues in assessing the function of the protein in vivo. In addition, the effect of other HECT-domain family members on TGF–β signaling may provide further redundancy even though single mutants for HECT ligases in mice and Drosophila do not phenocopy TGF–β mα (78). Another approach to understanding the role of HECT ligases in vivo is the mutation of the R-Smad linker phosphorylation sites that, according to current models (79,80,81,82), should render them resistant to HECT-mediated polyubiquitylation. However in the mouse, mutation of the linker phosphorylation sites of Smad1 do not generate phenotypes due to enhanced BMP signaling and this mutation in primordial germ cells mimics Smad1 loss-of-function (83).
Finally, a cautionary note since our understanding of the effects of HECT ligases has recently shifted from total R-Smad degradation to regulation of the activity of phospho-R-Smads (79,80). Since it has been proposed that Smurfs and other HECT proteins act not only on R-Smads but also on TGF–β receptors (84,85), this leads to difficulty in concluding whether these protein influence R-Smad activity directly or indirectly via the receptors. The importance of ubiquitylation (and thus potentially of HECT ligases) for receptor regulation has been independently confirmed by the role of the deubiquitylase Uch37 in protecting TGF–β receptors from degradation (86,87). Uch37/Uch15 knockout mice die shortly after gastrulation and it will be interesting to determine if this phenotype relates to TGF–β signaling (88).
Cycles of mono- and deubiquitylation are at the core of Smad regulation
Collectively, these recent studies indicate a key role for monoubiquitylation in the regulation of Smad complexes by targeting protein-protein interaction surfaces or by inhibiting their DNA binding activity. Monoubiquitylation of Smads can occur “in solution” before nuclear Smad4 and phospho-R-Smads form a complex on DNA or while Smad complexes are engaged in transcription. Thus monoubiquitylation may provide a system for actively disrupting Smad complexes and switching off transcription. This hypothesis, together with the active role played by deubiquitylases in TGF–β signaling, suggests that Smad monoubiquitylation is subject to on/off cycles that control the perception of TGF–β signals in several contexts.
This model begs the question - why are Smads engaged in these cycles? Mechanisms keeping Smad complexes dynamically unstable were originally proposed to explain the TGF–β pathways ability to induce different genetic programs based on the intensity of the signal - for example in a morphogen gradient. For this to happen, two conditions need to be met. First, the number of receptor complexes activated at the plasma membrane must be kept proportional to the extracellular concentration of the ligand; this is likely ensured by the existence of multiple feedback mechanisms that regulate receptor endocytosis, their persistence in early endosomes, and the ratio of post-endocytosis recycling versus degradation. Indeed, the TGF-β pathway antagonists Smad6/7 and Smurfs are early targets of Smad transcriptional activation representing a potential feedback mechanism to shut down R-Smad activation after the initial response and allow the system to determine if the signal is sustained or has ceased. Second, nuclear Smad complexes must not be too stable over time, so that R-Smads constantly get dephosphorylated, shuttle back to the cytoplasm and are available for another round of phosphorylation should the signal persist. These two mechanisms allow Smad activity to be kept proportional to TGF–β signal concentrations that change not only in space (between neighboring cells) but also in time.
What is the molecular nature of the nuclear mechanism that continuously dissociates Smad complexes? R-Smad degradation as well as dephosphorylation have been proposed to fulfil this role, although it is still not clear at which step in the pathway these events take place. Dephosphorylation of R-Smads, for example, was shown to occur independently of R-Smad binding to Smad4 and thus independently of R-Smad binding to DNA (89). Similarly, phosphoR-Smad degradation might occur only under particular circumstances. Thus, R-Smad monoubiquitylation represents a more likely mechanism for the disassembly of Smad transcriptional complexes, allowing them to maintain a dynamic rate of nucleo/cytoplasmic shuttling and enabling them to repeatedly monitor ligand/receptor activity.
Future Directions
Additional genetic analyses that interrogate existing biochemical models are required to extend the observations reported here and thus expand our understanding of their biological relevance. More biochemical studies are needed to analyze the interplay between the different classes of Smad post-translational modification (primarily ubiquitylation and phosphorylation but also sumoylation and others) and the order in which they occur, so that more physiologically precise models can be generated for future genetic testing. For the biochemical experiments, it is becoming more and more clear that they must consider Smad regulation as a dynamic process that responds even to subtle variations in TGF–β ligand concentration (89,90).
In summary, studies to date have shown that the regulation of Smad signal transducers by mono- and deubiquitylation is a highly conserved mechanism governing the amplitude of TGF-β signal transduction and thus TGF-β responsiveness in many cell types.
Acknowledgments
We apologize to colleagues not cited due to space limitations. Ubiquitylation studies by SJN were supported in part by NIH HG002516. SD is supported by AIRC (Italian Association for Cancer Research) and MIUR (Italian Ministry of Universities). MI is a Marie Curie FP-7 IIF fellow and recipient of Toyobo Biotechnology and Uehara Memorial Foundation grants.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Moustakas A, Heldin CH. The regulation of TGF–β signal transduction. Development. 2009;136(22):3699–714. doi: 10.1242/dev.030338. [DOI] [PubMed] [Google Scholar]
- 2.Pardali E, Goumans MJ, ten Dijke P. Signaling by members of the TGF–β family in vascular morphogenesis and disease. Trends Cell Biol. 2010;20(9):556–67. doi: 10.1016/j.tcb.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19(23):2783–810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 4.Schmierer B, Hill CS. TGF–β-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. 2007;8(12):970–82. doi: 10.1038/nrm2297. [DOI] [PubMed] [Google Scholar]
- 5.Lin X, et al. PPM1A functions as a Smad phosphatase to terminate TGF–β signaling. Cell. 2006;125(5):915–28. doi: 10.1016/j.cell.2006.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kretzschmar M, Doody J, Massague J. Opposing BMP and EGF signaling pathways converge on the TGF-beta family mediator Smad1. Nature. 1997;389(6651):618–22. doi: 10.1038/39348. [DOI] [PubMed] [Google Scholar]
- 7.Pera EM, et al. Integration of IGF, FGF, and anti-BMP signals via Smad1 phosphorylation in neural induction. Genes Dev. 2003;17(24):3023–8. doi: 10.1101/gad.1153603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miles WO, et al. Medea SUMOylation restricts the signaling range of the Dpp morphogen in the Drosophila embryo. Genes Dev. 2008;22(18):2578–90. doi: 10.1101/gad.494808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lonn P, et al. PARP-1 attenuates Smad-mediated transcription. Mol Cell. 2011;40(4):521–32. doi: 10.1016/j.molcel.2010.10.029. [DOI] [PubMed] [Google Scholar]
- 10.Grönroos E, et al. Control of Smad7 stability by competition between acetylation and ubiquitylation. Mol Cell. 2002;10(3):483–493. doi: 10.1016/s1097-2765(02)00639-1. [DOI] [PubMed] [Google Scholar]
- 11.Izzi L, Attisano L. Ubiquitin-dependent regulation of TGF–β signaling in cancer. Neoplasia. 2006;8(8):677–88. doi: 10.1593/neo.06472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inoue Y, Imamura T. Regulation of TGF–β family signaling by E3 ubiquitin ligases. Cancer Sci. 2008;99(11):2107–12. doi: 10.1111/j.1349-7006.2008.00925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu H, et al. A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature. 1999;400(6745):687–93. doi: 10.1038/23293. [DOI] [PubMed] [Google Scholar]
- 14.Lin X, Liang M, Feng XH. Smurf2 is a ubiquitin E3 ligase mediating degradation of Smad2 in TGF–β signaling. J Biol Chem. 2000;275(47):36818–22. doi: 10.1074/jbc.C000580200. [DOI] [PubMed] [Google Scholar]
- 15.Episkopou V, et al. Induction of the mammalian node requires Arkadia function in the extraembryonic lineages. Nature. 2001;410(6830):825–30. doi: 10.1038/35071095. [DOI] [PubMed] [Google Scholar]
- 16.McCabe BD, et al. Highwire regulates presynaptic BMP signaling essential for synaptic growth. Neuron. 2004;41(6):891–905. doi: 10.1016/s0896-6273(04)00073-x. [DOI] [PubMed] [Google Scholar]
- 17.Dupont S, et al. Germ-layer specification and control of cell growth by Ectodermin, a Smad4 ubiquitin ligase. Cell. 2005;121(1):87–99. doi: 10.1016/j.cell.2005.01.033. [DOI] [PubMed] [Google Scholar]
- 18.Ulrich HD, Walden H. Ubiquitin signaling in DNA replication and repair. Nat Rev Mol Cell Biol. 2010;11(7):479–89. doi: 10.1038/nrm2921. [DOI] [PubMed] [Google Scholar]
- 19.Grabbe C, Husnjak K, Dikic I. The spatial and temporal organization of ubiquitin networks. Nat Rev Mol Cell Biol. 2011;12(5):295–307. doi: 10.1038/nrm3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10(8):550–63. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- 21.Sigismund S, Polo S, Di Fiore PP. Signaling through monoubiquitylation. Curr Top Microbiol Immunol. 2004;286:149–85. doi: 10.1007/978-3-540-69494-6_6. [DOI] [PubMed] [Google Scholar]
- 22.Salmena L, Pandolfi PP. Changing venues for tumor suppression: balancing destruction and localization by monoubiquitylation. Nat Rev Cancer. 2007;7:409–13. doi: 10.1038/nrc2145. [DOI] [PubMed] [Google Scholar]
- 23.Nijman SM, et al. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell. 2005;17(3):331–9. doi: 10.1016/j.molcel.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 24.Konikoff C, Wisotzkey R, Newfeld SJ. Lysine conservation and context in TGF-β and Wnt signaling suggests new targets and general themes for post-translational modification. J Mol Evol. 2008;67:323–333. doi: 10.1007/s00239-008-9159-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dupont S, et al. FAM/USP9x, a deubiquitinating enzyme essential for TGF–β signaling, controls Smad4 monoubiquitylation. Cell. 2009;136(1):123–35. doi: 10.1016/j.cell.2008.10.051. [DOI] [PubMed] [Google Scholar]
- 26.Inui M, et al. USP15 is a deubiquitylating enzyme for receptor-activated SMADs. Nat Cell Biol. 2011;13(11):1368–75. doi: 10.1038/ncb2346. [DOI] [PubMed] [Google Scholar]
- 27.Trotman LC, et al. Ubiquitylation regulates PTEN nuclear import and tumor suppression. Cell. 2007;128(1):141–56. doi: 10.1016/j.cell.2006.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sorrentino, et al. The type I TGF-β receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat Cell Biol. 2008;10(10):1199–1207. doi: 10.1038/ncb1780. [DOI] [PubMed] [Google Scholar]
- 29.Yamashita, et al. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-β. Mol Cell. 2008;31(6):918–924. doi: 10.1016/j.molcel.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mu, et al. TRAF6 ubiquitinates TGF-β type I receptor to promote its cleavage and nuclear translocation in cancer. Nat Commun. 2011;2:330. doi: 10.1038/ncomms1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lipkowitz S, Weissman AM. RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis. Nat Rev Cancer. 2011;11(9):629–43. doi: 10.1038/nrc3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Rozieres S, et al. The loss of mdm2 induces p53-mediated apoptosis. Oncogene. 2000;19(13):1691–7. doi: 10.1038/sj.onc.1203468. [DOI] [PubMed] [Google Scholar]
- 33.Levy L, et al. Arkadia activates Smad3/Smad4-dependent transcription by triggering signal-induced SnoN degradation. Mol Cell Biol. 2007;27(17):6068–83. doi: 10.1128/MCB.00664-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hesling C, et al. Antagonistic regulation of EMT by TIF1-γ and Smad4 in mammary epithelial cells. EMBO Rep. 2011;12(7):665–72. doi: 10.1038/embor.2011.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moren A, et al. Differential ubiquitylation defines the functional status of the tumor suppressor Smad4. J Biol Chem. 2003;278(35):33571–82. doi: 10.1074/jbc.M300159200. [DOI] [PubMed] [Google Scholar]
- 36.Wang B, Suzuki H, Kato M. Roles of monoubiquitinated Smad4 in formation of Smad transcriptional complexes. Biochem Biophys Res Comm. 2008;376(2):288–92. doi: 10.1016/j.bbrc.2008.08.143. [DOI] [PubMed] [Google Scholar]
- 37.Chacko BM, et al. The L3 loop and C-terminal phosphorylation jointly define Smad protein trimerization. Nat Struct Biol. 2001;8(3):248–53. doi: 10.1038/84995. [DOI] [PubMed] [Google Scholar]
- 38.Wu JW, et al. Crystal structure of phosphorylated Smad2: recognition of phosphoserine by the MH2 domain and insights on Smad function in TGF–β signaling. Mol Cell. 2001;8(6):1277–89. doi: 10.1016/s1097-2765(01)00421-x. [DOI] [PubMed] [Google Scholar]
- 39.Agricola E, et al. Recruitment of TIF1-γ to chromatin via its PHD finger-bromodomain activates its ubiquitin ligase and transcriptional repressor activities. Mol Cell. 2011;43(1):85–96. doi: 10.1016/j.molcel.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 40.Xi Q, et al. A Poised Chromatin Platform for TGF–β Access to Master Regulators. Cell. 2011;147(7):1511–24. doi: 10.1016/j.cell.2011.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morsut L, et al. Negative control of Smad activity by Ectodermin/Tif1-γ patterns the mammalian embryo. Development. 2010;137(15):2571–8. doi: 10.1242/dev.053801. [DOI] [PubMed] [Google Scholar]
- 42.Schultz D, Friedman J, Rauscher FJ., 3rd Targeting HDACs via KRAB-zinc finger proteins: the PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2alpha subunit of NuRD. Genes Dev. 2001;15(4):428–43. doi: 10.1101/gad.869501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsai WW, et al. TRIM24 links a non-canonical histone signature to breast cancer. Nature. 2010;468(7326):927–32. doi: 10.1038/nature09542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang C, et al. MDM2 interaction with nuclear corepressor KAP1 contributes to p53 inactivation. EMBO J. 2005;24(18):3279–90. doi: 10.1038/sj.emboj.7600791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Allton K, et al. Trim24 targets endogenous p53 for degradation. Proc Natl Acad Sci U S A. 2009;106(28):11612–6. doi: 10.1073/pnas.0813177106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herquel B, Ouararhni K, Davidson I. TRIM cofactors couple chromatin modifications to transcriptional regulation, signaling and tumor suppression. Transcription. 2011;2(5):231–6. doi: 10.4161/trns.2.5.17725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gomis RR, et al. A FoxO-Smad synexpression group in human keratinocytes. Proc Natl Acad Sci U S A. 2006;103(34):12747–52. doi: 10.1073/pnas.0605333103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Candia AF, et al. Cellular interpretation of multiple TGF–β signals: intracellular antagonism between activin/BVg1 and BMP-2/4 signaling mediated by Smads. Development. 1997;124(22):4467–80. doi: 10.1242/dev.124.22.4467. [DOI] [PubMed] [Google Scholar]
- 49.Takaesu NT, et al. dSno facilitates Baboon signaling in the Drosophila brain by switching the affinity of Medea away from Mad and toward dSmad2. Genetics. 2006;174:1299–1313. doi: 10.1534/genetics.106.064956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu JW, et al. Structural mechanism of Smad4 recognition by nuclear oncoprotein Ski: insights on Ski-mediated repression of TGF–β signaling. Cell. 2002;111(3):357–67. doi: 10.1016/s0092-8674(02)01006-1. [DOI] [PubMed] [Google Scholar]
- 51.Waldrip WR, et al. Smad2 signaling in extraembryonic tissues determines anterior-posterior polarity of the early mouse embryo. Cell. 1998;92(6):797–808. doi: 10.1016/s0092-8674(00)81407-5. [DOI] [PubMed] [Google Scholar]
- 52.Chu GC, et al. Differential requirements for Smad4 in TGF–β-dependent patterning of the early mouse embryo. Development. 2004;131(15):3501–12. doi: 10.1242/dev.01248. [DOI] [PubMed] [Google Scholar]
- 53.Vincent DF, et al. Inactivation of Tif1–γ cooperates with Kras to induce cystic tumors of the pancreas. PLoS Genet. 2009;5(7):e1000575. doi: 10.1371/journal.pgen.1000575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Herquel B, et al. Transcription cofactors TRIM24, TRIM28, and TRIM33 associate to form regulatory complexes that suppress murine hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2011;108(20):8212–7. doi: 10.1073/pnas.1101544108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ransom DG, et al. The zebrafish moonshine gene encodes Tif1–γ, an essential regulator of hematopoiesis. PLoS Biol. 2004;2(8):E237. doi: 10.1371/journal.pbio.0020237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.He W, et al. Hematopoiesis controlled by distinct Tif1–γ and Smad4 branches of the TGFbeta pathway. Cell. 2006;125(5):929–41. doi: 10.1016/j.cell.2006.03.045. [DOI] [PubMed] [Google Scholar]
- 57.Aucagne R, et al. Tif1-γ is a tumor suppressor in mouse and human chronic myelomonocytic leukemia. J Clin Invest. 2011;121(6):2361–70. doi: 10.1172/JCI45213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kusy S, et al. Adult hematopoiesis is regulated by Tif1-γ, a repressor of TAL1 and PU.1 transcriptional activity. Cell Stem Cell. 2011;8(4):412–25. doi: 10.1016/j.stem.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 59.Monteiro R, Pouget C, Patient R. The gata1/pu.1 lineage paradigm varies between blood populations and is modulated by Tif1-γ. EMBO J. 2011;30(6):1093–103. doi: 10.1038/emboj.2011.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bai X, et al. Tif1–γ controls erythroid cell fate by regulating transcription elongation. Cell. 2010;142(1):133–43. doi: 10.1016/j.cell.2010.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huang Y, Baker RT, Fischer-Vize JA. Control of cell fate by a deubiquitinating enzyme encoded by the fat facets gene. Science. 1995;270(5243):1828–31. doi: 10.1126/science.270.5243.1828. [DOI] [PubMed] [Google Scholar]
- 62.Chen X, Zhang B, Fischer JA. A specific protein substrate for a deubiquitinating enzyme: Liquid facets is the substrate of Fat facets. Genes Dev. 2002;16(3):289–94. doi: 10.1101/gad.961502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Overstreet E, Fitch E, Fischer JA. Fat facets and Liquid facets promote Delta endocytosis and signaling. Development. 2004;131(21):5355–66. doi: 10.1242/dev.01434. [DOI] [PubMed] [Google Scholar]
- 64.DiAntonio A, et al. Ubiquitylation-dependent mechanisms regulate synaptic growth and function. Nature. 2001;412(6845):449–52. doi: 10.1038/35086595. [DOI] [PubMed] [Google Scholar]
- 65.Stinchfield MJ, et al. Fat facets deubiquitylation of Medea modulates interpretation of the Dpp dorsal-ventral morphogen gradient. 2012. manuscript in revision. [DOI] [PubMed] [Google Scholar]
- 66.Beckstead R, et al. Bonus, Drosophila homolog of Tif1 proteins, interacts with nuclear receptors and inhibit betaFTZ-F1-dependent transcription. Mol Cell. 2001;7:753–65. doi: 10.1016/s1097-2765(01)00220-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tse WK, et al. Genome-wide loss-of-function analysis of deubiquitylating enzymes for zebrafish development. BMC Genomics. 2009;10:637. doi: 10.1186/1471-2164-10-637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dunn NR, et al. Mice exclusively expressing the short isoform of Smad2 develop normally and are viable and fertile. Genes Dev. 2005;19(1):152–63. doi: 10.1101/gad.1243205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mu JJ, et al. A proteomic analysis of (ATM)/(ATR) substrates identifies the ubiquitin-proteasome system as a regulator for DNA damage checkpoints. J Biol Chem. 2007;282(24):17330–4. doi: 10.1074/jbc.C700079200. [DOI] [PubMed] [Google Scholar]
- 70.Tang LY, et al. Ablation of Smurf2 reveals an inhibition in TGF–β signaling through multiple mono-ubiquitylation of Smad3. EMBO J. 2011;30(23):4777–89. doi: 10.1038/emboj.2011.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhou C, et al. Fission yeast COP9/signalosome suppresses cullin activity through recruitment of the deubiquitylating enzyme Ubp12p. Mol Cell. 2003;11:927–938. doi: 10.1016/s1097-2765(03)00136-9. [DOI] [PubMed] [Google Scholar]
- 72.Soucy TA, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458(7239):732–6. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 73.Angelats C, et al. Isolation and characterization of the mouse ubiquitin-specific protease Usp15. Mamm Genome. 2003;14(1):31–46. doi: 10.1007/s00335-002-3035-0. [DOI] [PubMed] [Google Scholar]
- 74.Podos SD, et al. The dSmurf ubiquitin-protein ligase restricts BMP signaling spatially and temporally during Drosophila embryogenesis. Dev Cell. 2001;1(4):567–78. doi: 10.1016/s1534-5807(01)00057-0. [DOI] [PubMed] [Google Scholar]
- 75.Xia L, et al. The Fused/Smurf complex controls the fate of Drosophila germline stem cells by generating a gradient BMP response. Cell. 2010;143(6):978–90. doi: 10.1016/j.cell.2010.11.022. [DOI] [PubMed] [Google Scholar]
- 76.Yamashita M, et al. Ubiquitin ligase Smurf1 controls osteoblast activity and bone homeostasis by targeting MEKK2 for degradation. Cell. 2005;121(1):101–13. doi: 10.1016/j.cell.2005.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Narimatsu M, et al. Regulation of planar cell polarity by Smurf ubiquitin ligases. Cell. 2009;137(2):295–307. doi: 10.1016/j.cell.2009.02.025. [DOI] [PubMed] [Google Scholar]
- 78.Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol. 2009;10(6):398–409. doi: 10.1038/nrm2690. [DOI] [PubMed] [Google Scholar]
- 79.Fuentealba LC, et al. Integrating patterning signals: Wnt/GSK3-β regulates the duration of the BMP/Smad1 signal. Cell. 2007;131(5):980–93. doi: 10.1016/j.cell.2007.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sapkota G, et al. Balancing BMP signaling through integrated inputs into the Smad1 linker. Mol Cell. 2007;25(3):441–54. doi: 10.1016/j.molcel.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 81.Eivers E, et al. Phosphorylation of Mad controls competition between Wingless and BMP signaling. Sci Signal. 2011;4(194):ra68. doi: 10.1126/scisignal.2002034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Quijano JC, et al. Wg signaling via Zw3 and Mad restricts self-renewal of sensory organ precursor cells in Drosophila. Genetics. 2011;189:809–824. doi: 10.1534/genetics.111.133801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Aubin J, Davy A, Soriano P. In vivo convergence of BMP and MAPK signaling pathways: impact of differential Smad1 phosphorylation on development and homeostasis. Genes Dev. 2004;18(12):1482–94. doi: 10.1101/gad.1202604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kavsak P, et al. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF–β receptor for degradation. Mol Cell. 2000;6(6):1365–75. doi: 10.1016/s1097-2765(00)00134-9. [DOI] [PubMed] [Google Scholar]
- 85.Ebisawa T, et al. Smurf1 interacts with TGF–β type I receptor through Smad7 and induces receptor degradation. J Biol Chem. 2001;276(16):12477–80. doi: 10.1074/jbc.C100008200. [DOI] [PubMed] [Google Scholar]
- 86.Wicks SJ, et al. The deubiquitinating enzyme Uch37 interacts with Smads and regulates TGF–β signaling. Oncogene. 2005;24(54):8080–4. doi: 10.1038/sj.onc.1208944. [DOI] [PubMed] [Google Scholar]
- 87.Al-Shami A, et al. Regulators of the proteasome pathway, Uch37 and Rpn13, play distinct roles in mouse development. PLoS One. 2010;5(10):e13654. doi: 10.1371/journal.pone.0013654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cutts AJ, et al. Early phase TGF–β receptor signalling dynamics stabilised by the deubiquitinase Uch37 promotes cell migratory responses. Int J Biochem Cell Biol. 2011;43(4):604–12. doi: 10.1016/j.biocel.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 89.Schmierer B, et al. Mathematical models identify Smad nucleocytoplasmic shuttling as a dynamic signal-interpreting system. Proc Natl Acad Sci USA. 2008;105:6608–13. doi: 10.1073/pnas.0710134105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zi Z, et al. Quantitative analysis of transient and sustained TGF–β signaling dynamics. Mol Systems Biol. 2011;7:492. doi: 10.1038/msb.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]