

Abstract
Understanding the causes and consequences of transposable element (TE) activity in the genomic era requires sophisticated bioinformatics approaches to accurately identify individual insertion sites. Next-generation sequencing technology now makes it possible to rapidly identify new TE insertions using resequencing data, opening up new possibilities to study the nature of TE-induced mutation and the target site preferences of different TE families. While the identification of new TE insertion sites is seemingly a simple task, the mechanisms of transposition present unique challenges for the annotation of de novo transposable element insertions mapped to a reference genome. Here I discuss these challenges and propose a framework for the annotation of de novo TE insertions that accommodates known mechanisms of TE insertion and established coordinate systems for genome annotation.
Keywords: coordinate systems, genome bioinformatics, next generation sequencing, target site duplications, transposable elements
Overview
Next-generation sequencing (NGS) offers unparalleled opportunities to study the causes and consequences of transposable element (TE) activity across an ever-widening range of host species. Consequently, a large number of computational methods have recently been developed to identify both artificially and naturally induced TE insertions using NGS data.1-10 These methods use diverse approaches, but share the fundamental aim of assessing whether a particular TE insertion is present in a given individual, or pool of, resequenced genome(s).
TE insertions discovered in resequenced genomes can either be known (i.e., insertions present in the reference genome) or novel (i.e., de novo insertions not present in the reference genome). Since known TE insertions occupy an identifiable span in the reference genome, representing them as a range of coordinate is no different than any other annotated genomic feature, such as genes or regulatory elements. However, de novo TE insertions are by definition not present in the genome sequence and their representation, while conceptually simple, is technically not straightforward. Here I discuss the challenges relating to the reference-based annotation of de novo TE insertions. I then propose a solution for representing de novo TE insertions that accommodates known mechanisms of TE insertion and established coordinate systems for genome annotation.
Base vs. Interbase Genome Coordinate Systems
Before considering issues relating to the reference-based annotation of de novo TE annotations, it is necessary to introduce the two major coordinate systems for genome annotation. The so-called “base” coordinate system anchors genomic feature to nucleotide positions in the genome. In contrast, the “interbase” (also known as “zero-based” or “space-based”11) coordinate system anchors genomic feature to the spaces between nucleotide positions in the genome. While they may seem trivially different, these two alternate representations have important implications for the mapping of de novo TE insertions relative to a reference genome, and often cause confusion in the genomics community. For example, The UCSC genome bioinformatics team provides an answer to a “frequently asked question” (http://genome.ucsc.edu/FAQ/FAQtracks.html#tracks1) about this issue since this site uses base coordinate system (which they refer to as “one-based, fully-closed”) in the UCSC genome browser display but interbase coordinate system (referred to as “zero-based, half-open”) in their analysis tools and file formats.
Base coordinate systems are in many ways more intuitive biologically, since features encoded by specific nucleotides in the genome are mapped to corresponding regions of the reference sequence. As such, most genome annotation portals (e.g., NCBI or Ensembl), bioinformatics software (e.g., BLAST) and annotation file formats (e.g., GFF) use the base coordinate system. Interbase coordinate systems, despite being biologically non-intuitive, have a number of features that make them more computationally attractive, and thus are used by a growing number of genome bioinformatics systems, such as the UCSC Genome Browser (http://genome.ucsc.edu/FAQ/FAQtracks.html#tracks1), Chado (http://gmod.org/wiki/Introduction_to_Chado#Interbase_Coordinates), and DAS2 (http://biodas.org/documents/das2/das2_get.html#segment_ranges).
To see why many genome informatics systems use the interbase coordinate system, it is first necessary to see how base and interbase coordinates are represented numerically for an annotation that is present in the reference genome (such as a known TE insertion that is present in the reference sequence). Let's assume that we have an annotated feature spanning the nucleotides GGGCCC in a hypothetical reference genome shown in Figure 1A. Under the base coordinate system, this feature would be represented as a pair of coordinates: start = 3 and end = 8. Under the interbase coordinate system, the coordinates are instead: start = 2 and end = 8. The numerical difference between the two coordinate systems lies in terms of how the start coordinate is represented and how the coordinate range is interpreted.
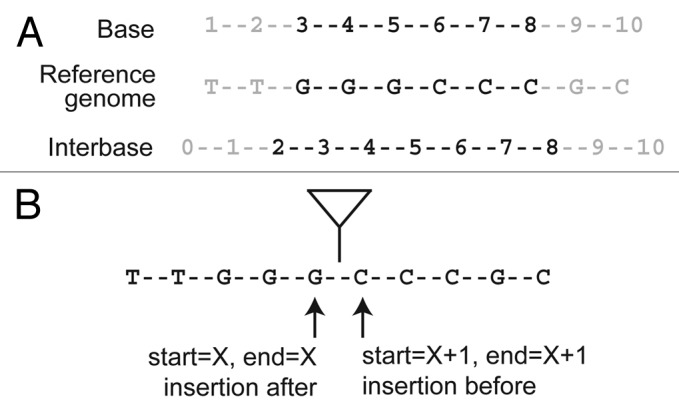
Figure 1. Genome coordinate systems and the annotation of TE insertions. The location of an arbitrary genomic feature encoded by the sequence GGGCCC is represented differently in base and interbase coordinate systems (A). Since de novo TE insertions occur between bases in the reference genome, they are more naturally represented by interbase coordinate systems. On the widely-used base coordinate system, mapping a de novo TE insertion requires the invocation of arbitrary rules (either before or after the insertion site) (B). These arbitrary rules can lead to ambiguity in the mapping and interpretation of de novo TE insertions.
As noted above, there are several advantages for using the interbase coordinate system including: (1) the ability to represent features that occur between nucleotides (like a splice site or de novo TE insertion), (2) simpler arithmetic for computing the length of features (e.g., the length of a coordinate span is end-start, rather than end-start+1 as it is for base coordinates), (3) simpler arithmetic for calculating range overlaps, and (4) more rational conversion of coordinates from the positive to the negative strand (For further discussion, see http://genomewiki.ucsc.edu/index.php/Coordinate_Transforms).
A Proposal for Annotating De Novo TE Insertions on Base Coordinate Systems
So why is the choice of coordinate system important for the annotation of de novo TE insertions mapped to a reference sequence? The short answer is that de novo TE insertions are not a part of the reference sequence and occur between nucleotides in the reference coordinate system. Therefore it is intrinsically difficult to accurately represent the location of a de novo TE insertion on base coordinates. Nevertheless, one-base coordinate systems dominate most of genome bioinformatics systems and are an established framework that one has to work within. So how then should we annotate de novo TE insertions on base coordinates? Answering this question leads to several unanticipated considerations, and why I believe that a standard must be established in the field of TE genomics if we wish to create easily interpretable annotations of de novo TE insertions identified using NGS technologies. Moreover, solving this problem is particularly crucial for applications where we wish to map TE insertions with nucleotide-level precision, such as extracting information about the exact nature of a TE-induced mutation or detailed understanding of the target site preferences of a TE family.12
To begin, let’s consider a TE that inserts between positions X and X + 1 in a genome. Under a base coordinate system, if we wish to map a TE insertion to single base resolution, we quickly encounter our first problem. Do we annotate both the start and stop coordinates at position X, or both coordinates at position X + 1 (Fig. 1B)? If we chose to annotate the insertion at position X, then we need to invoke a rule that the TE inserts after nucleotide X to interpret this annotation correctly. Conversely, if we chose to annotate the insertion at position X + 1, then we need to invoke a rule that the TE inserts before nucleotide X to interpret this annotation correctly.
So, should we instead annotate the TE as a two base span starting at X and ending at X + 1, with the interpretation that the insertion occurs between the start and end positions? This too is an unsatisfactory solution since at face value it incorrectly implies that the TE insertion spans two base pairs in the genome or that it is imprecisely mapped.
In addition to the fact that TEs insert between bases in the reference genome and therefore present an intrinsic challenge to base coordinate systems, a second problem concerning the annotation of de novo insertions arises from the joint effects of (1) the presence of target site duplications (TSDs) and (2) the sequence information used to map TE insertions to a reference genome. First, most TEs create staggered cuts in the genomic DNA that are filled on TE integration leading to short TSDs at the ends of TE insertion. TSDs, however short they may be, represent duplication of sequence that is present as a single copy in the pre-insertion sequence represented by the reference genome. Second, methods used to map de novo TE insertions to precise coordinates in the genome use sequence information in the junction region between a TE and its unique flanking sequence (such methods are sometimes referred to as “split-read” methods). These TE-flank junction sequences can be obtained from either the 5′ or 3′ end of the TE insertion (Fig. 2). Because the TSD is present on both ends of the TE insertion but only occurs once in the reference genome, it turns out that where a de novo TE insertion is annotated depends on whether one uses the TE-flank sequence from the 5′ or 3′ end and the orientation of the TE insertion in the genome.
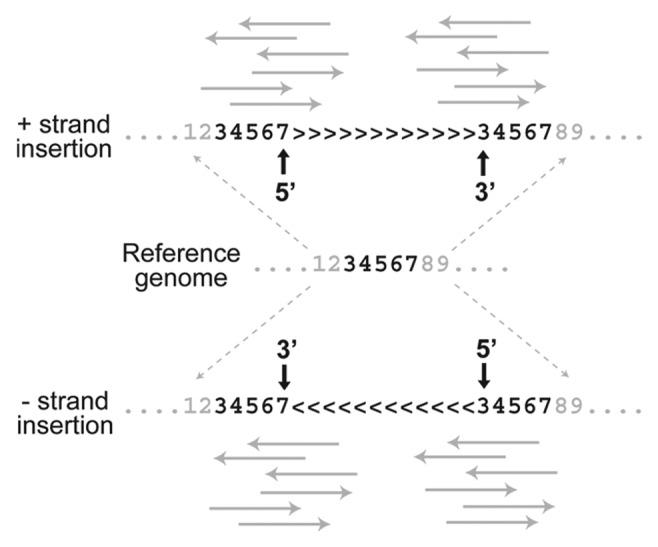
Figure 2. TSDs create ambiguity in the annotation of de novo TE insertion sites. Unique DNA in the reference genome (e.g., positions 3–7 for a 5 bp TSD) is duplicated on insertion of a TE for both insertions on the positive strand (> > > ) and negative strand (< < < ). When NGS reads (solid gray arrows) that span the TE-flanking region junction are used to map de novo TE insertions on the positive strand, the placement of the insertion relative the TSD differs for reads from the 5′ (after TSD) and 3′ (before TSD) ends of the TE. Differential annotation of TE insertion sites is also observed for negative strand insertions, but placement relative to the TSD is reversed relative to positive strand insertions. These TSD-induced effects can lead to ambiguity in the mapping and interpretation of de novo TE insertions.
An example of how these effects together create problems for mapping TE insertions is shown in Figure 2. In this case, imagine that at TE creates a five base pair TSD on insertion, represented once in the reference genome but in two places in the genome with the TE insertion. For an insertion on the positive strand (> > > ), a TE-flank sequence from the 5′ end is annotated to occur at the 3′ end of the TSD. In contrast, an insertion mapped using information from the 3′ TE-flank sequence is placed at the 5′ end of the TSD. On the other hand, for an insertion on the negative strand (< < < ), the opposite effect occurs. Regardless of orientation, TE-flank junction sequences from the 5′ or 3′ end map the TE insertion to different locations in the genome, which is highly undesirable and could lead to differences in interpretation among researchers.
In fact, depending on (1) the orientation of the TE insertion and (2) which end of the TE is mapped to the genome, a given target site can lead to a total of four potential mappings. As a consequence, both the one- and two-base coordinate representations suggested above to map insertion sites are flawed, since even with consistent rules about mapping from either the 5′ or 3′ end, TEs that insert into the same target site but occur on different strands would be annotated at two different locations. This is precisely the case for the annotation of artificial P-element insertions into the D. melanogaster genome (which have the same reference-based mapping problems as TE insertions discovered using NGS), and why we previously observed an unexpected excess of insertions spaced exactly eight base pairs apart (the length of the TSD for the P-element13) in the genome annotation on opposite strands for this TE.14
As an solution to the problem of mapping de novo TE insertions on base coordinate systems, I propose that we abandon the idea of annotating the insertion site and instead annotate the genomic sequence that is duplicated to give rise to the TSD (the “pre-TSD” sequence). Specifically, I suggest annotating the start and end of the pre-TSD sequence as the feature span and labeling the orientation of the TE in the strand field. This formulation works because the pre-TSD actually does exist in the genome and therefore can be naturally annotated on base coordinate systems. Moreover, this solution bypasses having to chose an arbitrary rule about where to locate the TE relative to the TSD, as is required under the one-base/two-base annotation framework (see for example ref. 15). Furthermore, it represents insertions into the same target site, but which occur on different strands, at the same location in the genome. Finally, under this framework one can use both 5′ and 3′ TE-flanking sequence information jointly to map de novo TE insertion sites. In fact, the overlap on genome coordinates from sequences supporting the 5′ and 3′ TE-flanking regions defines the pre-TSD.12 This solution is flexible enough to accommodate most mechanisms of TE integration, since it requires no prior information about TSD length for a given TE family, and it also works for TE families that generate variable length TSDs, since the pre-TSD is annotated on a per insertion basis.
Exceptions and Concluding Remarks
One problem left open by this solution is that posed by exceptional TE families that do not create a TSD, which do exist.16 However, since these families by definition do not generate a TSD, several of the key problems with the one-base/two-base representations discussed above do not apply. Thus either of these strategies could suffice and would be in principle compatible with the pre-TSD annotation scheme advocated here. I suggest using the one-base representation, with insertions mapped consistently to the X position regardless of strand. Finally, the framework proposed here should be seen not as the ultimate solution to the problem of representing de novo TE insertions, but as a step toward establishing a standard for studies that harness the power of NGS technology to answer fundamental questions about the role of TEs in functional and evolutionary genomics. By raising the issues relating to the seemingly simple task of mapping TEs to a reference genome here, it is hoped that further consideration of this matter will lead to the adoption of a general solution that allows for the annotation of TE insertions in a concerted and uniform manner in the field.
Acknowledgments
The author thanks Raquel Linheiro and Alexandru Al. Ecovoiu for helpful discussion on the ideas presented here.
Footnotes
Previously published online: www.landesbioscience.com/journals/mge/article/19479
References
- 1.Sackton TB, Kulathinal RJ, Bergman CM, Quinlan AR, Dopman EB, Carneiro M, et al. Population genomic inferences from sparse high-throughput sequencing of two populations of Drosophila melanogaster. Genome Biol Evol. 2009;1:449–65. doi: 10.1093/gbe/evp048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–71. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ewing AD, Kazazian HH., Jr High-throughput sequencing reveals extensive variation in human-specific L1 content in individual human genomes. Genome Res. 2010;20:1262–70. doi: 10.1101/gr.106419.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quinlan AR, Clark RA, Sokolova S, Leibowitz ML, Zhang Y, Hurles ME, et al. Genome-wide mapping and assembly of structural variant breakpoints in the mouse genome. Genome Res. 2010;20:623–35. doi: 10.1101/gr.102970.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iskow RC, McCabe MT, Mills RE, Torene S, Pittard WS, Neuwald AF, et al. Natural mutagenesis of human genomes by endogenous retrotransposons. Cell. 2010;141:1253–61. doi: 10.1016/j.cell.2010.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Witherspoon DJ, Xing J, Zhang Y, Watkins WS, Batzer MA, Jorde LB. Mobile element scanning (ME-Scan) by targeted high-throughput sequencing. BMC Genomics. 2010;11:410. doi: 10.1186/1471-2164-11-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fiston-Lavier AS, Carrigan M, Petrov DA, González J. T-lex: a program for fast and accurate assessment of transposable element presence using next-generation sequencing data. Nucleic Acids Res. 2011;39:e36. doi: 10.1093/nar/gkq1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ewing AD, Kazazian HH., Jr. Whole-genome resequencing allows detection of many rare LINE-1 insertion alleles in humans. Genome Res. 2011;21:985–90. doi: 10.1101/gr.114777.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mei L, Ding X, Tsang SY, Pun FW, Ng SK, Yang J, et al. AluScan: a method for genome-wide scanning of sequence and structure variations in the human genome. BMC Genomics. 2011;12:564. doi: 10.1186/1471-2164-12-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hormozdiari F, Alkan C, Ventura M, Hajirasouliha I, Malig M, Hach F, et al. Alu repeat discovery and characterization within human genomes. Genome Res. 2011;21:840–9. doi: 10.1101/gr.115956.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li K, Stockwell TB. VariantClassifier: A hierarchical variant classifier for annotated genomes. BMC research notes. 2010;3:191. doi: 10.1186/1756-0500-3-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Linheiro RS, Bergman CM. Whole genome resequencing reveals natural target site preferences of transposable elements in Drosophila melanogaster. PLoS ONE. 2012;7:e30008. doi: 10.1371/journal.pone.0030008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O’Hare K, Rubin GM. Structures of P transposable elements and their sites of insertion and excision in the Drosophila melanogaster genome. Cell. 1983;34:25–35. doi: 10.1016/0092-8674(83)90133-2. [DOI] [PubMed] [Google Scholar]
- 14.Linheiro RS, Bergman CM. Testing the palindromic target site model for DNA transposon insertion using the Drosophila melanogaster P-element. Nucleic Acids Res. 2008;36:6199–208. doi: 10.1093/nar/gkn563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stewart C, Kural D, Strömberg MP, Walker JA, Konkel MK, Stütz AM, et al. 1000 Genomes Project A comprehensive map of mobile element insertion polymorphisms in humans. PLoS Genet. 2011;7:e1002236. doi: 10.1371/journal.pgen.1002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bedzyk LA, Shoemaker NB, Young KE, Salyers AA. Insertion and excision of Bacteroides conjugative chromosomal elements. J Bacteriol. 1992;174:166–72. doi: 10.1128/jb.174.1.166-172.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]