

Abstract
Pseudogout and the associated calcium pyrophosphate dihydrate (CPPD)- crystal-related arthropathies are common conditions that present particular management problems in clinical practice as they often affect older patients with multiple medical comorbidities. The epidemiology, metabolic and endocrine disease associations, and routine investigations used in the diagnostic workup are briefly reviewed. Current treatment approaches that are mainly directed at relieving the symptoms of joint inflammation are outlined. Unlike gout, there are no agents available that have been shown to decrease crystal load in CPPD-related joint disease. Recent novel insights into the pathogenesis of crystal-induced joint inflammation and subsequent joint degeneration are also discussed. The potential of colchicine as a prophylactic agent in managing recurrent attacks and the likely mechanisms of its effects on the NACHT, LRR and PYD domains-containing protein 3 (NALP-3) inflammasome of the innate immune system are highlighted. The use of agents that directly target the inflammasome, in particular drugs which inhibit the interleukin 1 pathway, in the treatment of severe, refractory pseudogout is also discussed. Finally, there is particular emphasis on the likely pathogenic role of CPPD crystal deposition in degenerative joint disease and the use of targeted anticrystal therapies as potential disease-modifying drugs.
Keywords: pseudogout, CPPD, colchicine, osteoarthritis, inflammasome, crystal, interleukin-1
Introduction
The ‘pseudogout syndrome’ was first described by Kohn and colleagues in 1962 [Kohn et al. 1962] and accurately depicts acute attacks of synovitis induced by calcium pyrophosphate dihydrate (CPPD) crystals, which clinically resemble gouty arthritis due to monosodium urate (MSU) crystal deposition. CPPD deposition may also cause symptoms similar to septic arthritis, polyarticular inflammatory arthritis [often misdiagnosed as rheumatoid arthritis (RA)] or degenerative osteoarthritis (OA) [McCarthy, 2008]. The clinical manifestations, epidemiology, associated metabolic conditions and novel insights into the pathogenesis of CPPD-related joint inflammation are briefly discussed below. Unlike gout, the treatment of CPPD-related arthropathies is often difficult, partly due to patient characteristics, diagnostic uncertainty and the lack of any effective agent to decrease crystal load [Announ and Guerne, 2008]. The purpose of this article is to review current approaches that are used in the treatment of acute pseudogout and the management of chronic CPPD-related arthropathies, with a particular emphasis on the use of available agents to target the ‘inflammasome’ [Martinon et al. 2006], which plays a crucial role in crystal-induced inflammation and the theoretical potential of specific anticrystal therapies.
Clinical manifestations
CPPD crystal deposition disease has the potential to mimic most forms of inflammatory arthritis and a clinical classification of these phenomena has emerged from the studies of McCarty and colleagues [McCarty, 1970, 1976; Rosenthal, 2001]. There are at least six distinct presentations of this disorder, and along with acute pseudogout include asymptomatic or lanthanic chondrocalcinosis, pseudo-OA (with or without acute attacks), pseudo-RA, pseudo-polymyalgia rheumatica (pseudo-PMR) and pseudo-neuropathic arthropathy.
Acute pseudogout
Approximately 25% of patients with CPPD crystal deposition exhibit the classical pseudogout pattern of disease. Compared with true gout, pseudogout attacks may take longer to reach peak intensity and may persist for up to 3 months despite therapy [Masuda and Ishikawa, 1988]. Large joints are more commonly affected and the attacks are characterized by the cardinal signs of inflammation which may involve one or several hot, tender, red and swollen joints [Dieppe et al. 1982]. The knee is the most commonly involved joint, followed by the wrist, ankle, elbow, toe, shoulder and hip. As with gout, these attacks may occur spontaneously or can be provoked by trauma, surgery or severe medical illness [McCarthy, 2008; Rosenthal et al. 2005]. Flares of pseudogout after parathyroidectomy are well described [Bilezikian et al. 1973; Rosenthal et al. 2005] and may reflect abrupt changes in serum calcium levels post surgery. Pamidronate therapy for hypercalcaemia has also been implicated as a trigger for acute attacks of pseudogout [Malnick et al. 1997]. Patients are usually asymptomatic between acute attacks and differentiation from true gout or even a septic joint requires arthrocentesis and synovial fluid (SF) analysis for Gram stain, culture and cytology. Pseudogout is confirmed by the demonstration of CPPD crystals in SF which manifest as rhomboid-shaped rod-like structures that exhibit weakly positive or no birefringence by compensated polarized light microscopy, in contrast to the negatively birefringent needle-shaped MSU crystals found in gout (Figure 1). Gout and pseudogout may also coexist in a single inflammatory effusion, as 20% of patients with CPPD will have hyperuricaemia and a quarter of these will develop gout at some stage [Rosenthal et al. 2005].
Figure 1.

Compensated polarised light microscopy demonstrating coexistent monosodium urate (MSU) and (calcium pyrophosphate dihydrate) CPPD crystals. The MSU crystals (#) are needle shaped and negative birefringent whereas the CPPD crystals (♮) are rhomboid and weakly positively birefringent.
Asymptomatic chondrocalcinosis
Chondrocalcinosis specifically describes radiographic calcification in hyaline and fibrocartilage (see Figure 2). It is commonly present in patients with CPPD deposition disease, but is neither sensitive nor specific for affected patients. While CPPD crystal deposition accounts for approximately 95% of cases of chondrocalcinosis [McCarty et al. 1966], one study reported that a radiographic diagnosis was only made in about 40% of patients who had pathologically proven CPPD crystal deposition from samples obtained at arthroscopy [Fisseler-Eckhoff and Muller, 1992]. Some individuals with radiographic or pathologic evidence of articular chondrocalcinosis have no clinically apparent arthritis and this finding has been referred to as lanthanic CPPD deposition and is of uncertain significance [McCarthy, 2008]. These patients are often of advanced age and have not been rigorously studied to see whether they develop clinically relevant arthritis. However, there are reports that patients with apparently asymptomatic disease, when subjected to closer scrutiny, have a higher frequency of wrist and knee complaints and associated degenerative changes compared with age-matched controls without radiographic chondrocalcinosis [Ellman and Levin, 1975].
Figure 2.
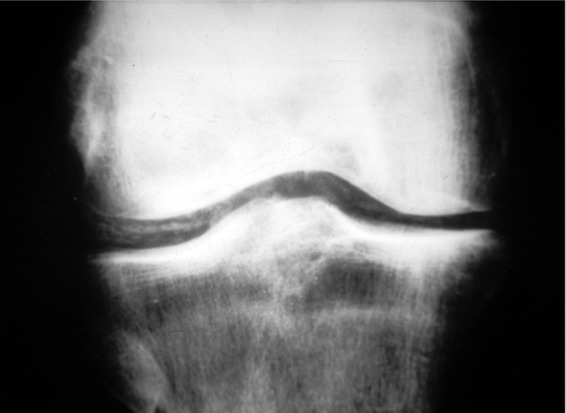
Plain radiograph of right knee demonstrating meniscal chondrocalcinosis due to calcium pyrophosphate dihydrate deposition.
Pseudo-osteoarthritis
A large proportion of patients with clinically apparent CPPD crystal deposition follow a progressive course of articular degeneration in multiple joints in an oddly distributed pattern typically involving the knees, wrists, metacarpophalangeal (MCP) joints, hips, shoulders, spine, elbows and ankles. About half of these patients will experience acute attacks of pseudogout superimposed on their chronic symptoms [McCarthy, 2008], while the remainder will often present with complaints more typical of classical OA. While clinical examination findings are similar to OA, certain features such as the atypical distribution (especially involving the wrists and MCP joints), the presence of contractures and valgus knee deformities should highlight the possibility of CPPD deposition disease. However, when the degenerative process occurs in joints more typical for classical OA, such as the knee and hip, it can be difficult to differentiate from OA and consequently may be significantly under recognized. This is highlighted by the high prevalence of these crystals in samples taken from OA knees at the time of arthroplasty, with one study reporting a third of these specimens displaying definite evidence of CPPD deposition [Derfus et al. 2002]. Furthermore, there is an emerging body of evidence that these and other calcium-containing crystals, such as basic calcium phosphate (BCP), play a pathogenic role in joint degeneration and that calcification of articular cartilage is a feature of OA [Molloy and McCarthy, 2006]. Moreover, there is strong in vitro evidence which demonstrates that such calcium deposition is inhibited by phosphocitrate [Sun et al. 2010], and which is supported by complementary work using an animal model of OA [Cheung et al. 2006].
Pseudo-rheumatoid arthritis
A small proportion (<5%) of patients with CPPD deposition experience a nonerosive, asynchronous, inflammatory arthritis which mimics RA [Rosenthal et al. 2005]. The associated morning stiffness, malaise, synovial thickening, symmetrical involvement, flexion contractures and elevated acute phase inflammatory markers often lead to a misdiagnosis of RA. In addition, 10% of these individuals, who are often of older age, will have low titres of rheumatoid factor thus causing further diagnostic confusion [McCarthy, 2008]. Therefore, the presence of CPPD crystals in synovial fluid and radiographic changes more in keeping with OA favour a diagnosis of pseudo-RA, while elevated titres of anticyclic citrullinated peptide (anti-CCP) antibodies and the presence of bony erosions either on ultrasound or plain film radiographic imaging indicate a diagnosis of classical RA. However, these two conditions may occasionally coexist and the required differences in management approaches can be challenging. A rare subtype of pseudo-RA may also present with prominent systemic features such as fever, leucocytosis, altered mental status and polyarthritis and may mimic sepsis, particularly in older people [Bong and Bennett, 1981].
Pseudo-polymyalgia rheumatica
PMR is classically a disease of older people, presenting with a range of symptoms including fatigue, early morning stiffness and subjective proximal muscle weakness. It is often a considerable challenge for the clinician to distinguish older patients with pure PMR from others presenting with polymyalgia symptoms resembling this condition. Recent work in a large number of patients indicates that CPPD arthropathy should also be included in the wide spectrum of diseases mimicking classical PMR [Pego-Reigosa et al. 2005]. Knee OA, tendinous calcifications and episodic ankle arthritis are proposed as the variables that predict those older patients with PMR features who really have an atypical pattern of CPPD arthropathy [Pego-Reigosa et al. 2005].
Pseudo-neuropathic arthropathy
Some patients with CPPD deposition have a severe destructive monoarthritis that clinically resembles a neuropathic Charcot’s joint [Richards and Hamilton, 1974]. In contrast to the classical description of Charcot’s joints associated with diabetes mellitus, tabes dorsalis, syringomyelia and alcoholic neuropathy, these patients have no underlying neurological disorder yet present with a painful, destructive monoarthritis. The natural history of this condition is not well understood, management options are limited and surgical intervention may often be required.
Epidemiology
CPPD crystal deposition is largely a disease of older people and several radiographic surveys have demonstrated an age-related increase in the prevalence of joint calcification with almost half of all octogenarians displaying some evidence of articular chondrocalcinosis [Ellman and Levin, 1975; Wilkins et al. 1983]. The prevalence of CPPD deposition among younger people is unknown. Although there is no major gender predominance, attacks of acute pseudogout appear more commonly in men, with women more frequently exhibiting the pseudo-OA pattern of the disease [O’Duffy, 1976; Rosenthal et al. 2005].
Disease associations
Despite the fact that most cases of CPPD crystal deposition are idiopathic, several metabolic and endocrine disorders, a previous history of joint trauma and the well described hereditary condition of familial chondrocalcinosis are all associated with precocious articular calcification in younger patients [Jones et al. 1992]. Much has been learned about the likely pathogenic processes involved in pathological calcification from studying distinct pedigrees of autosomal dominant familial CPPD deposition and their association with a mutant ANKH gene located on the short arm of chromosome 5 [Pendleton et al. 2002]. This gene encodes a transmembrane transport protein critically involved in generating extracellular inorganic phosphate (PPi) which then acts as a substrate for CPPD crystal formation in the pericellular cartilage matrix [Williams et al. 2002]. This is discussed in more detail below.
Of the other endocrine and metabolic conditions that have been linked with chondrocalcinosis, only hereditary haemochromatosis is associated with the full spectrum of CPPD-related arthropathy [Jones et al. 1992]. Iron accumulation in joint tissues appears to be a key factor, as evidenced by reports of CPPD crystal deposition in patients with secondary iron overload due to transfusion haemosiderosis and haemophilia. Chronic hypomagnesaemia due to Gitelman’s and Bartter’s syndromes has also been associated with both chondrocalcinosis and acute pseudogout, thus forming the rationale for magnesium supplementation to prevent recurrent attacks of pseudogout [Cobeta-Garcia et al. 1998; Punzi et al. 1998].
A clear association with previous joint trauma, particularly involving the knee, lends weight to the likely role of chondrocalcinosis in contributing to the process of joint degeneration [Doherty et al. 1982; Settas et al. 1982]. Hyperparathyroidism, hypophosphatasia, gout and hypocalciuric hypercalcaemia are all associated with acute attacks of pseudogout, while the relationships between acromegaly, Wilson’s disease, diabetes mellitus and CPPD deposition are less clear [Jones et al. 1992].
Pathogenesis
Recent insights into the processes involved in both CPPD deposition and in the mechanisms whereby these crystals can interact with the inflammasome to cause joint inflammation have greatly increased our understanding of the pathogenesis of pseudogout. Several lines of evidence support the hypothesis that elevated levels of PPi in the immediate extracellular environment of chondrocytes provide the substrate for the formation of CPPD crystals, which are then deposited in the cartilaginous matrix [Rachow and Ryan, 1985; Ryan et al. 1984]. The nucleoside triphosphatase pyrophosphohydrolase (NTPPPH) group of ectoenzymes are strongly associated with this process as increased activity of these enzymes catalyses the production of extracellular PPi at the external surface of the chondrocyte cell membrane [Ryan et al. 1984], while deficiency of one of this group [the NTPPPH plasma cell membrane glycoprotein 1 (PC-1) enzyme] leads to excessive mineralization with BCP rather than CPPD crystals in a mouse model of pathological ossification [Okawa et al. 1998]. This is supported by data involving another mouse disorder, murine progressive ankylosis, in which a homozygous nonsense mutation of the aforementioned ANK gene results in reduced extracelluler PPi levels and excessive peripheral and axial skeleton ankylosis with BCP crystal material [Ho et al. 2000]. Thus, one function of extracellular PPi appears to be the inhibition of the growth of BCP crystals while excessive PPi levels, arising from NTPPPH overactivity or gain of function mutations in the ANK transmembrane transporter protein, could provide the substrate for CPPD crystal formation. Matrix vesicles that play a role in normal bone development are also implicated and studies have shown that these vesicles can generate CPPD crystals in vitro [Derfus et al. 1992, 1996]. These matrix vesicles are selectively rich in NTPPPH activity and gene transfer experiments involving the NTPPPH PC-1 enzyme and cultured chondrocytes demonstrate increased chondocyte apoptosis and matrix calcification [Johnson et al. 2001]. A schematic diagram representing some of the aforementioned processes involved in the formation of calcium-containing crystals, mechanisms of inflammation and subsequent joint degeneration is illustrated in Figure 3.
Figure 3.

Schematic diagram of calcium crystal formation [calcium pyrophosphate dihydrate (CPPD) versus basic calcium phosphate (BCP)], with subsequent inflammasome activation via CPPD and synovial inflammation via BCP, and how both can contribute to the vicious cycle of joint degeneration in calcium crystal-related arthropathies. ePPi, extracellular inorganic phosphate; HA, hydroxyapatite; IL-1β, interleukin 1β; MMP, matrix metalloproteinase; MV, matrix vesicle; NALP 3, NACHT, LRR and PYD domains-containing protein 3; NTPPPH, nucleoside triphosphatase pyrophosphohydrolase; PPi, inorganic phosphate.
Acute attacks of pseudogout are believed to represent a dose-related auto-inflammatory response to CPPD crystals shed from cartilaginous tissues into the synovial cavity [McCarthy, 2008]. Recent data describing how CPPD crystals (and indeed MSU crystals) interact with the caspase-1-activating NACHT, LRR and PYD domains-containing protein 3 (NALP-3) inflammasome of the innate immune system provide novel insights into the mechanisms by which these crystals cause episodic bouts of joint inflammation [Martinon et al. 2006]. This results in the production of several proinflammatory cytokines, particularly interleukin (IL)-1β, by macrophages and monocytes, thus initiating an inflammatory cascade with the subsequent influx of neutrophils, fibroblast-like synoviocytes and recruitment of other inflammatory pathways via tumour necrosis factor α (TNFα) [Meng et al. 1997]. Colchicine is used in the treatment of several auto-inflammatory conditions, including gout and pseudogout, and has recently been shown to completely block crystal-induced maturation of IL-1β in vitro, indicating that the drug acts upstream of inflammasome activation [Martinon et al. 2006]. Combining this observation with the known mode of action of colchicine as an inhibitor of microtubule assembly [Goldfinger et al. 1965], it is therefore likely that the drug acts at the level of crystal endocytosis and/or presentation to the inflammasome [Martinon et al. 2006]. Taken together, these data support a pivotal role for the inflammasome in pseudogout and provide a series of novel molecular targets for the treatment and prophylaxis of acute episodes of pseudogout and the other CPPD-related arthropathies.
Investigations
The definitive diagnosis of pseudogout is most commonly and accurately made by identifying CPPD crystals, by compensated polarized light microscopy, in the SF of affected joints [McCarthy, 2008]. Furthermore, all patients presenting with suspected CPPD deposition can be screened for chondrocalcinosis using certain key radiographs: a nonweight-bearing anteroposterior (AP) view of both knees, an AP view of the pelvis for visualization of the symphysis pubis and hips, and a posteroanterior (PA) view of each hand to include the triangular ligament of the wrists. Changes in the MCP joints, such as squaring of the bone ends, presence of subchondral cysts and hook-like osteophytes, are characteristic features of the arthropathy of haemachromatosis, but are also found in patients with CPPD deposition alone [McCarthy, 2008]. Thus, a probable diagnosis of CPPD arthropathy can be made in patients who present with acute or chronic arthritis of knee, hip, wrist, shoulder or MCP joints in the setting of radiographic features, even without the direct demonstration of CPPD crystals by micropscopy [McCarty, 1994].
Given the aforementioned disease associations, most patients with CPPD deposition, and particularly those who are relatively young, should undergo screening for abnormal serum calcium, phosphate, magnesium, alkaline phosphatase and thyroid-stimulating hormone levels, and any evidence of iron overload by haemetinic analyses (ferritin, iron saturation and transferrin) [Rosenthal et al. 2005]. This is especially true with regard to haemachromatosis in light of the strong association between this condition and all forms of CPPD-related arthropathy, and the deleterious consequences of delayed diagnosis [Jones et al. 1992].
Treatment and management
Available and potential treatment options in the management of pseudogout are summarized in Table 1. Strategies that are currently employed in ameliorating CPPD-related joint disease are broadly limited to the following; those directed against correcting underlying metabolic abnormalities and treating associated conditions, general treatment with nonsteroidal anti-inflammatory drugs (NSAIDs) and/or corticosteroids (either by local intra-articular injection or systemic therapy), and finally, low-dose oral colchicine [Choy, 2005]. Potential future therapies include agents targeted against crystal formation (such as probenecid and phosphocitrate), more potent anti-inflammatory medications such as methotrexate and anticytokine drugs which target the IL-1 pathway [Announ and Guerne, 2008].
Table 1.
Management strategies in the treatment of pseudogout.
| Strategy | Treatment | Comments |
|---|---|---|
| Treating associated metabolic conditions | Decreasing iron overload, treating hypothyroidism | No hard evidence that this improves CPPD-related arthropathy |
| Magnesium supplementation | Only indicated in hypomagnesemic states. Otherwise evidence is largely anecdotal | |
| Conventional anti-inflammatory medications | NSAIDs | Symptomatic relief but may be relatively contraindicated in the older population |
| Corticosteroids | Excellent symptomatic relief with intra-articular injections. Some evidence for short courses of oral/intramuscular steroids for polyarticular flares | |
| Colchicine | Acts upstream of the inflammasome and has an emerging role as a prophylactic agent | |
| Anticrystal therapy | Probenecid | Good theoretical rationale but evidence lacking |
| Phosphocitrate | Strong evidence from animal studies but no safety or efficacy data in humans | |
| Targeting the inflammasome | Methotrexate | Emerging role in the prevention of recurrent attacks |
| Inhibition of IL-1 pathway (anakinra, canakinumab, IL-1 Trap) | May become key agents for refractory polyarticular disease | |
| Anti-TNFα drugs | Poor efficacy in auto-inflammatory conditions and therefore unlikely to be successful in pseudogout |
CPPD, calcium pyrophosphate dehydrate; IL-1, interleukin 1; NSAID, nonsteriodal anti-inflammatory drug; TNFα, tumour necrosis factor α.
Associated conditions
Despite the value of establishing the presence of an associated disorder there is no evidence that targeted treatment of these conditions, such as decreasing iron load with phlebotomy in haemochromatosis, has any effect on CPPD crystal deposition [Hamilton et al. 1981]. However, recent data suggest that reversal of iron overload may at least lead to symptomatic relief in the long term [Harty et al. 2011].
As magnesium acts as a cofactor in the regulation of certain phosphatases, and coupled with the known association between chronic hypomagnesaemic states and chondrocalcinosis [Punzi et al. 1998], magnesium supplementation has often been recommended as a safe prophylactic agent to decrease the frequency of acute attacks of pseudogout. A 10-year follow-up study demonstrated reduced meniscal calcification in patients with familial hypokalaemia/hypomagnesaemia and chondrocalcinosis who were given magnesium supplementation [Smilde et al. 1994]. However, only one small placebo-controlled study in CPPD-related arthropathy, not specifically associated with hypomagnesaemia, using magnesium carbonate supplements showed a tendency to improvement at 6 months, but has never been replicated [Announ and Guerne, 2008].
Anti-inflammatory medications
NSAIDs, corticosteroids and colchicine have all been used clinically for managing acute pseudogout and the episodic inflammatory arthritis that is regularly seen in pseudo-OA [Choy, 2005]. NSAIDs still represent the mainstay of treatment, but are often of insufficient efficacy and also relatively contraindicated in a predominately older population due to the increased risk of gastrointestinal haemorrhage and associated renal impairment [Announ and Guerne, 2008]. Alternatively, and particularly when only one joint such as the knee is involved, aspiration and intra-articular injection of corticosteroids can be extremely effective [Rosenthal et al. 2005]. Short courses of oral steroids (0.5–1 mg/kg) with a rapid taper or even intramuscular injections are also useful for recurrent flares of polyarticular CPPD-related arthritis [Roane et al. 1997].
There is some evidence that hydroxychloroquine inhibits matrix metalloproteinase activity in an animal model of CPPD arthritis, and while it does not have a role in the management of acute pseudogout, current European League Against Rheumatism guidelines recommend its use as an adjunctive agent in patients with chronic CPPD arthropathy [Zhang et al. 2011].
In addition to the aforementioned inhibitory effects of colchicine on crystal-induced IL-1β production by the inflammasome, its disruption of normal cytoskeletal function leads to the impairment of a variety of cellular functions, including the secretion of other chemokines and mediators, and reduced cell migration and division [Nuki, 2008]. These effects, when coupled with its plasma half-life of 20–30 h, large volume of distribution, partial renal excretion (20%), and preferential accumulation in red blood cells and neutrophils result in a relatively narrow therapeutic index and significant potential for toxicity, especially regarding bone marrow suppression [Nuki, 2008]. Therefore, colchicine is nowadays only used in low-dose preparations (0.5 mg twice daily) for both gout and pseudogout. While there have been no major trials of colchicine in the treatment of acute pseudogout, it is commonly used clinically for this purpose [Choy, 2005]. However, there is some evidence for its role in the prevention of both acute attacks of pseudogout and recurrent episodes of the other CPPD-related arthritides [Alvarellos and Spilberg, 1986], and the recent mechanistic insights into its effects on crystal uptake into cells and inhibition of their subsequent interaction with the inflammasome have generated renewed interest in this medication as a potential anchor drug in the prophylaxis of pseudogout.
Anticrystal agents
Unlike gout, there are no available agents which directly target crystal load in CPPD deposition disease. Probenecid is a transmembrane-transport inhibitor which reduces the production of extracellular PPi in vitro [Rosenthal and Ryan, 1994], but to date has never been demonstrated to decrease CPPD crystal deposition in humans. The clear contribution of CPPD crystals to the acceleration of joint degeneration in the various CPPD-related arthropathies and the possible pathological role of these crystals in OA [Macmullan and McCarthy, 2010] has resulted in significant interest in the potential of phosphocitrate to directly target crystal deposition in both of these diseases [Sun et al. 2010]. Calcification of articular cartilage is now well recognized as an indissociable feature of OA [Molloy and McCarthy, 2006]. Furthermore, there is strong in vitro evidence that such calcium deposition is inhibited by phosphocitrate [Sun et al. 2010]. Taken together with previous work demonstrating that CPPD crystals accelerate joint degeneration in a rabbit model of OA [Fam et al. 1995], this suggests that agents which directly target articular calcification may not only have the potential to treat CPPD-related arthropathy but also OA itself.
Targeting the inflammasome
Alongside the recent advances in the understanding of the mechanisms involved in crystal-induced joint inflammation, specific attention has been focused on the ability of methotrexate to decrease the frequency and intensity of recurrent attacks of pseudogout, particularly in relation to polyarticular disease [Chollet-Janin et al. 2007]. Despite the fact that this agent is relatively contraindicated in patients with haemochromatosis-associated arthropathy (because of concomitant liver disease and the potential for increased toxicity), due to its broad-spectrum ability to inhibit most cytokine pathways involved in joint inflammation, this generally well tolerated molecule represents an attractive alternative in the treatment and prophylaxis of the entire spectrum of ‘pseudogout syndrome’ [Announ and Guerne, 2008]. The anti-inflammatory effect of methotrexate is mediated chiefly by an increase in extracellular adenosine levels [Cronstein et al. 1993], which activates membrane receptors on macrophages and other inflammatory cells, thereby inhibiting the release of several cytokines, most notably IL-1β [Morabito et al. 1998]. Thus, by acting in this proximal manner, methotrexate has a potent indirect effect on the ability of the inflammasome to generate IL-1β. Multicentre clinical trials are currently underway to further assess the potential impact of this widely available and affordable medication in this area.
Specific insights into the role of IL-1β in a variety of auto-inflammatory diseases have resulted in a number of case reports documenting the efficacy of anakinra, an IL-1 receptor antagonist, in difficult-to-treat polyarticular pseudogout [McGonagle et al. 2008]. Furthermore, a newly developed monoclonal antibody to IL-1β, canakinumab, has proven to be very effective in treating refractory cases of gouty arthritis [So et al. 2010], and a novel soluble fusion protein, IL-1 Trap, has recently been licensed for the treatment of several auto-inflammatory conditions [Ratner, 2008]. Taken together with the recent data on the role of the inflammasome in generating IL-1β in response to crystal endocytosis, these drugs may find a new place as disease-modifying agents in the treatment of CPPD-crystal-related arthropathy. However, their immunosuppressive effects in a largely older patient population and relative high cost ensure that, at this point, their use should be reserved for particularly difficult cases.
Although several reports exist of gout being successfully treated with anti-TNFα drugs, published cases of pseudogout successfully treated with these agents are lacking [Announ and Guerne, 2008]. As with many auto-inflammatory conditions, it seems that CPPD-related arthritis responds poorly to this class of medication. To date, the experimental data indicating that TNFα, unlike in RA, appears to exert its influence downstream of IL-1β in the acute inflammatory cascade associated with pseudogout would appear to be borne out clinically.
Conclusion
Pseudogout and the associated CPPD-related arthropathies are likely to increase in prevalence due to the demographic drift towards an aging population. Thus, the management of these conditions will continue to present particular challenges in the future. The use of regular colchicine as a prophylactic agent against acute attacks of pseudogout will probably become more widespread. Similarly, drugs targeting the IL-1 pathway offer considerable potential in severe refractory disease. However, in contrast to MSU crystals in gout, there is no practical way to remove CPPD crystals from the joint. Therefore, current management strategies remain symptomatic but not disease modifying. Further research into ways of reducing the CPPD crystal burden is very necessary.
Footnotes
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
The authors declare no conflicts of interest in preparing this article.
References
- Alvarellos A., Spilberg I. (1986) Colchicine prophylaxis in pseudogout. J Rheumatol 13: 804–805 [PubMed] [Google Scholar]
- Announ N., Guerne P.A. (2008) Treating difficult crystal pyrophosphate dihydrate deposition disease. Curr Rheumatol Rep 10: 228–234 [DOI] [PubMed] [Google Scholar]
- Bilezikian J.P., Connor T.B., Aptekar R., Freijanes J., Aurbach G.D., Pachas W.N., et al. (1973) Pseudogout after parathyroidectomy. Lancet 1: 445–446 [DOI] [PubMed] [Google Scholar]
- Bong D., Bennett R. (1981) Pseudogout mimicking systemic disease. JAMA 246: 1438–1440 [PubMed] [Google Scholar]
- Cheung H.S., Sallis J.D., Demadis K.D., Wierzbicki A. (2006) Phosphocitrate blocks calcification-induced articular joint degeneration in a guinea pig model. Arthritis Rheum 54: 2452–2461 [DOI] [PubMed] [Google Scholar]
- Chollet-Janin A., Finckh A., Dudler J., Guerne P.A. (2007) Methotrexate as an alternative therapy for chronic calcium pyrophosphate deposition disease: an exploratory analysis. Arthritis Rheum 56: 688–692 [DOI] [PubMed] [Google Scholar]
- Choy G. (2005) An update on the treatment options for gout and calcium pyrophosphate deposition. Expert Opin Pharmacother 6: 2443–2453 [DOI] [PubMed] [Google Scholar]
- Cobeta-Garcia J.C., Gascon A., Iglesias E., Estopinan V. (1998) Chondrocalcinosis and Gitelman’s syndrome. A new association? Ann Rheum Dis 57: 748–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronstein B.N., Naime D., Ostad E. (1993) The antiinflammatory mechanism of methotrexate. Increased adenosine release at inflamed sites diminishes leukocyte accumulation in an in vivo model of inflammation. J Clin Invest 92: 2675–2682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derfus B.A., Kurian J.B., Butler J.J., Daft L.J., Carrera G.F., Ryan L.M., et al. (2002) The high prevalence of pathologic calcium crystals in pre-operative knees. J Rheumatol 29: 570–574 [PubMed] [Google Scholar]
- Derfus B.A., Kurtin S.M., Camacho N.P., Kurup I., Ryan L.M. (1996) Comparison of matrix vesicles derived from normal and osteoarthritic human articular cartilage. Connect Tissue Res 35: 337–342 [DOI] [PubMed] [Google Scholar]
- Derfus B.A., Rachow J.W., Mandel N.S., Boskey A.L., Buday M., Kushnaryov V.M., et al. (1992) Articular cartilage vesicles generate calcium pyrophosphate dihydrate-like crystals in vitro. Arthritis Rheum 35: 231–240 [DOI] [PubMed] [Google Scholar]
- Dieppe P.A., Alexander G.J., Jones H.E., Doherty M., Scott D.G., Manhire A., et al. (1982) Pyrophosphate arthropathy: a clinical and radiological study of 105 cases. Ann Rheum Dis 41: 371–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty M., Watt I., Dieppe P.A. (1982) Localised chondrocalcinosis in post-meniscectomy knees. Lancet 1: 1207–1210 [DOI] [PubMed] [Google Scholar]
- Ellman M.H., Levin B. (1975) Chondrocalcinosis in elderly persons. Arthritis Rheum 18: 43–47 [DOI] [PubMed] [Google Scholar]
- Fam A.G., Morava-Protzner I., Purcell C., Young B.D., Bunting P.S., Lewis A.J. (1995) Acceleration of experimental lapine osteoarthritis by calcium pyrophosphate microcrystalline synovitis. Arthritis Rheum 38: 201–210 [DOI] [PubMed] [Google Scholar]
- Fisseler-Eckhoff A., Muller K.M. (1992) Arthroscopy and chondrocalcinosis. Arthroscopy 8: 98–104 [DOI] [PubMed] [Google Scholar]
- Goldfinger S.E., Howell R.R., Seegmiller J.E. (1965) Suppression of metabolic accompaniments of phagocytosis by colchicine. Arthritis Rheum 8: 1112–1122 [DOI] [PubMed] [Google Scholar]
- Hamilton E.B., Bomford A.B., Laws J.W., Williams R. (1981) The natural history of arthritis in idiopathic haemochromatosis: progression of the clinical and radiological features over ten years. Q J Med 50: 321–329 [PubMed] [Google Scholar]
- Harty L., Lai D., Connor S., Dunne A., Ali M., Ryan J., et al. (2011) Prevalence and progress of joint symptoms in hereditary hemochromatosis and symptomatic response to venesection. Clin Rheumatol 17: 220–222 [DOI] [PubMed] [Google Scholar]
- Ho A.M., Johnson M.D., Kingsley D.M. (2000) Role of the mouse ank gene in control of tissue calcification and arthritis. Science 289: 265–270 [DOI] [PubMed] [Google Scholar]
- Johnson K., Pritzker K., Goding J., Terkeltaub R. (2001) The nucleoside triphosphate pyrophosphohydrolase isozyme PC-1 directly promotes cartilage calcification through chondrocyte apoptosis and increased calcium precipitation by mineralizing vesicles. J Rheumatol 28: 2681–2691 [PubMed] [Google Scholar]
- Jones A.C., Chuck A.J., Arie E.A., Green D.J., Doherty M. (1992) Diseases associated with calcium pyrophosphate deposition disease. Semin Arthritis Rheum 22: 188–202 [DOI] [PubMed] [Google Scholar]
- Kohn N.N., Hughes R.E., McCarty D.J., Jr, Faires J.S. (1962) The significance of calcium phosphate crystals in the synovial fluid of arthritic patients: the ‘pseudogout syndrome’. II. Identification of crystals. Ann Intern Med 56: 738–745 [DOI] [PubMed] [Google Scholar]
- Macmullan P.A., McCarthy G.M. (2010) The meniscus, calcification and osteoarthritis: a pathologic team. Arthritis Res Ther 12: 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malnick S.D., Ariel-Ronen S., Evron E., Sthoeger Z.M. (1997) Acute pseudogout as a complication of pamidronate. Ann Pharmacother 31: 499–500 [DOI] [PubMed] [Google Scholar]
- Martinon F., Petrilli V., Mayor A., Tardivel A., Tschopp J. (2006) Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440: 237–241 [DOI] [PubMed] [Google Scholar]
- Masuda I., Ishikawa K. (1988) Clinical features of pseudogout attack. A survey of 50 cases. Clin Orthop Relat Res (229): 173–181 [PubMed] [Google Scholar]
- McCarthy G. (2008) Calcium phosphate dihydrate, hydroxyapatite, and miscellaneous crystals. In: Klippel J.H., Sone J.H., Crofford L.J., White P.H. (eds), Primer on the Rheumatic Diseases. 13th ed. Berlin: Springer, pp; 263–270 [Google Scholar]
- McCarty D.J. (1970) Crystal-induced inflammation of the joints. Annu Rev Med 21: 357–366 [DOI] [PubMed] [Google Scholar]
- McCarty D.J. (1976) Calcium pyrophosphate dihydrate crystal deposition disease – 1975. Arthritis Rheum 19(Suppl. 3): 275–285 [DOI] [PubMed] [Google Scholar]
- McCarty D.J. (1994) Crystals and arthritis. Dis Mon 40: 255–299 [DOI] [PubMed] [Google Scholar]
- McCarty D.J., Jr, Hogan J.M., Gatter R.A., Grossman M. (1966) Studies on pathological calcifications in human cartilage. I. Prevalence and types of crystal deposits in the menisci of two hundred fifteen cadavera. J Bone Joint Surg Am 48: 309–325 [PubMed] [Google Scholar]
- McGonagle D., Tan A.L., Madden J., Emery P., McDermott M.F. (2008) Successful treatment of resistant pseudogout with anakinra. Arthritis Rheum 58: 631–633 [DOI] [PubMed] [Google Scholar]
- Meng Z.H., Hudson A.P., Schumacher H.R., Jr, Baker J.F., Baker D.G. (1997) Monosodium urate, hydroxyapatite, and calcium pyrophosphate crystals induce tumor necrosis factor-alpha expression in a mononuclear cell line. J Rheumatol 24: 2385–2388 [PubMed] [Google Scholar]
- Molloy E.S., McCarthy G.M. (2006) Calcium crystal deposition diseases: update on pathogenesis and manifestations. Rheum Dis Clin North Am 32: 383–400, vii [DOI] [PubMed] [Google Scholar]
- Morabito L., Montesinos M.C., Schreibman D.M., Balter L., Thompson L.F., Resta R., et al. (1998) Methotrexate and sulfasalazine promote adenosine release by a mechanism that requires ecto-5’-nucleotidase-mediated conversion of adenine nucleotides. J Clin Invest 101: 295–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuki G. (2008) Colchicine: its mechanism of action and efficacy in crystal-induced inflammation. Curr Rheumatol Rep 10: 218–227 [DOI] [PubMed] [Google Scholar]
- O’Duffy J.D. (1976) Clinical studies of acute pseudogout attacks: comments on prevalence, predispositions, and treatment. Arthritis Rheum 19(Suppl. 3): 349–352 [DOI] [PubMed] [Google Scholar]
- Okawa A., Nakamura I., Goto S., Moriya H., Nakamura Y., Ikegawa S. (1998) Mutation in Npps in a mouse model of ossification of the posterior longitudinal ligament of the spine. Nat Genet 19: 271–273 [DOI] [PubMed] [Google Scholar]
- Pego-Reigosa J.M., Rodriguez-Rodriguez M., Hurtado-Hernandez Z., Gromaz-Martin J., Taboas-Rodriguez D., Millan-Cachinero C., et al. (2005) Calcium pyrophosphate deposition disease mimicking polymyalgia rheumatica: a prospective followup study of predictive factors for this condition in patients presenting with polymyalgia symptoms. Arthritis Rheum 53: 931–938 [DOI] [PubMed] [Google Scholar]
- Pendleton A., Johnson M.D., Hughes A., Gurley K.A., Ho A.M., Doherty M., et al. (2002) Mutations in ANKH cause chondrocalcinosis. Am J Hum Genet 71: 933–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punzi L., Calo L., Schiavon F., Pianon M., Rosada M., Todesco S. (1998) Chondrocalcinosis is a feature of Gitelman’s variant of Bartter’s syndrome. A new look at the hypomagnesemia associated with calcium pyrophosphate dihydrate crystal deposition disease. Rev Rhum Engl Ed 65: 571–574 [PubMed] [Google Scholar]
- Rachow J.W., Ryan L.M. (1985) Adenosine triphosphate pyrophosphohydrolase and neutral inorganic pyrophosphatase in pathologic joint fluids. Elevated pyrophosphohydrolase in calcium pyrophosphate dihydrate crystal deposition disease. Arthritis Rheum 28: 1283–1288 [DOI] [PubMed] [Google Scholar]
- Ratner M. (2008) IL-1 trap go-ahead. Nat Biotechnol 26: 485. [DOI] [PubMed] [Google Scholar]
- Richards A.J., Hamilton E.B. (1974) Destructive arthropathy in chondrocalcinosis articularis. Ann Rheum Dis 33: 196–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roane D.W., Harris M.D., Carpenter M.T., Finger D.R., Jarek M.J., Alloway J.A., et al. (1997) Prospective use of intramuscular triamcinolone acetonide in pseudogout. J Rheumatol 24: 1168–1170 [PubMed] [Google Scholar]
- Rosenthal A.K. (2001) Pathogenesis of calcium pyrophosphate crystal deposition disease. Curr Rheumatol Rep 3: 17–23 [DOI] [PubMed] [Google Scholar]
- Rosenthal A.K., Ryan L.M. (1994) Probenecid inhibits transforming growth factor-beta 1 induced pyrophosphate elaboration by chondrocytes. J Rheumatol 21: 896–900 [PubMed] [Google Scholar]
- Rosenthal A., Ryan L.M., McCarty D.J. (2005) Calcium pyrophosphate deposition disease, pseudogout, and articular chondrocalcinosis. In: Koopman W.J., Moreland L.W. (eds), Arthritis and Allied Conditions. 15th ed. Philadelphia: Lippincott Wiliams & Wilkins, p. 273 [Google Scholar]
- Ryan L.M., Wortmann R.L., Karas B., McCarty D.J., Jr (1984) Cartilage nucleoside triphosphate (NTP) pyrophosphohydrolase. I. Identification as an ecto-enzyme. Arthritis Rheum 27: 404–409 [DOI] [PubMed] [Google Scholar]
- Settas L., Doherty M., Dieppe P. (1982) Localised chondrocalcinosis in unstable joints. Br Med J (Clin Res Ed) 285: 175–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smilde T.J., Haverman J.F., Schipper P., Hermus A.R., van Liebergen F.J., Jansen J.L., et al. (1994) Familial hypokalemia/hypomagnesemia and chondrocalcinosis. J Rheumatol 21: 1515–1519 [PubMed] [Google Scholar]
- So A., De Meulemeester M., Pikhlak A., Yucel A.E., Richard D., Murphy V., et al. (2010) Canakinumab for the treatment of acute flares in difficult-to-treat gouty arthritis: results of a multicenter, phase II, dose-ranging study. Arthritis Rheum 62: 3064–3076 [DOI] [PubMed] [Google Scholar]
- Sun Y., Mauerhan D.R., Honeycutt P.R., Kneisl J.S., Norton H.J., Zinchenko N., et al. (2010) Calcium deposition in osteoarthritic meniscus and meniscal cell culture. Arthritis Res Ther 12: R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins E., Dieppe P., Maddison P., Evison G. (1983) Osteoarthritis and articular chondrocalcinosis in the elderly. Ann Rheum Dis 42: 280–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams C.J., Zhang Y., Timms A., Bonavita G., Caeiro F., Broxholme J., et al. (2002) Autosomal dominant familial calcium pyrophosphate dihydrate deposition disease is caused by mutation in the transmembrane protein ANKH. Am J Hum Genet 71: 985–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Doherty M., Pascual E., Barskova V., Guerne P.A., Jansen T.L., et al. (2011) EULAR recommendations for calcium pyrophosphate deposition. Part II: management. Ann Rheum Dis 70: 571–575 [DOI] [PubMed] [Google Scholar]