

Abstract
DNA repair activities at DNA double-strand breaks (DSBs) are under control of regulatory ubiquitylation events governed by the RNF8 and RNF168 ubiquitin-ligases. Defects in this regulatory mechanism, as with mutation of other key DNA damage-response factors, lead to genomic instability and cancer, presumably due to impaired repair of DNA lesions. Recent work revealed that RNF8 and RNF168 also play critical roles at natural chromosome ends, when no longer adequately shielded by telomeres. In contrast to repair of DSBs being needed to maintain genome integrity, repair activities at telomeres create chromosome end-to-end fusions that threaten genome integrity. Upon cell division these telomere fusions give rise to genomic alterations and instability via chromosomal missegregration and initiation of breakage-fusion-bridge cycles. Here, I discuss the role of RNF8 at natural chromosome ends and its (potential) consequences.
Keywords: DNA damage, RNF8, genomic instability, telomeres, ubiquitin
Telomeres to Maintain Genome Integrity
The chance normal cells develop into cancer cells that give rise to life-threatening malignant tumors is greatly increased by genomic instability. While genomic instability can be lethal via loss of essential genes, it also increases the probability that cells accumulate the necessary genetic changes needed for tumor development, such as overexpression of proto-oncogenes or inactivation of tumor suppressor genes.1 To maintain genome integrity, multiple DNA repair mechanisms operate in cells to repair the different kinds of DNA lesions cells acquire from endogenous or exogenous sources. Consequently, people with defects in DNA repair enzymes are predisposed to cancer. In addition to repair activities, proliferation of cells with genetic aberrations is prevented by control mechanisms, including the DNA damage checkpoint that halts cell proliferation until lesions are repaired and the spindle assembly checkpoint that ensures correct separation of sister chromatids.
Experimental animal models as well as studies on human tumors have indicated that a significant degree of genomic instability during tumorigenesis can be attributed to loss of chromosome end protection by telomeres.2,3 Telomeres are nucleoprotein complexes specialized to handle the challenges to genome integrity typically presented by natural chromosome ends.4-6 First there is the risk of loss of genetic information due to the inability of conventional DNA polymerases to replicate the very ends of chromosomes. Second, natural chromosome ends should not be seen and treated as DNA DSBs, as this would lead to loss of proliferation by DNA damage checkpoint activation and inappropriate repair activities that can cause genomic instability (Fig. 1). To deal with these problems chromosome ends are capped by telomeres, consisting of long stretches of TTAGGG-repeats that provide a buffer such that terminal sequence loss will not directly affect juxtaposed genes. Moreover these repeats are essential for binding the telomere-specific protein complex “shelterin,” composed of TRF1, TRF2, RAP1, TIN2, TPP1 and POT1.4-6 Besides consisting of repeat-DNA and a unique set of proteins at high concentrations, telomeres have additional special features that together make them unique chromatin structures. Telomeric chromatin resembles constitutive heterochromatin by containing trimethylated H3K9, trimethylated H4K20 and HP1, all of which affect telomere length.9 Although telomeric nucleosomes consist of canonical core histones and thereby resemble nucleosomes in bulk chromatin, the nucleosomal repeat length appears shorter and telomeric nucleosomes show hypersensitivity to micrococcal nuclease.10-12 Furthermore, in vitro studies indicate that nucleosomes assembled on TTAGGG-repeats show increased mobility.13 In addition, telomeres contain unique structural arrangements in the form of G-quadruplexes and t-loops and are difficult for replication forks to pass through.14

Figure 1. The consequences of loss of chromosome end protection by telomeres. Depicted are the main consequences of loss of TRF2 shelterin activity, namely activation of the ATM-kinase pathway, which leads to p53-dependent senescence or cell death, to degradation of the telomeric single-strand G-overhang and to NHEJ-dependent formation of telomere fusions. Through the generation of unstable dicentric chromosomes these fusions can initiate breakage-fusion-bridge cycles and genomic instability in cells that escape cell cycle arrest or apoptosis. The induction of growth arrest or apoptosis by dysfunctional telomeres is regarded as an important tumor suppressor pathway by restricting the outgrowth of potentially cancerous cells. On the contrary, the repair activities acting at dysfunctional telomeres and consequential genomic instability can facilitate the development of cancer if cells with fused telomeres are allowed to continue through the cell cycle (e.g., due to loss of p53-activity). Not depicted here is activation of the p16Ink4a/Rb pathway, which in human (but not mouse) cells contributes to telomere damage-induced senescence.7,8
In the normal situation of insufficient telomerase, the enzyme capable of adding telomere repeats to counteract terminal sequence loss associated with incomplete end-replication, telomeres in human somatic cells progressively shorten with cell division.15 Eventually telomeres become critically short, which compromises their protective function. Apart from long-term proliferation of cells to provoke telomere deprotection by critical shortening, which is a very asynchronous process, loss of telomere protection can be achieved experimentally by inhibiting shelterin components. This causes well-controlled and relatively synchronous telomere dysfunction that mimics the consequences of critical telomere shortening. Studies relying on interference with shelterin have yielded many valuable insights in the mechanism of telomere protection and the consequences of its loss. These studies revealed that shelterin protects chromosome ends from activating ATM and ATR checkpoint responses, prevents inappropriate repair activities by the homologous recombination (HR) and non-homologous end-joining (NHEJ) repair machineries and controls telomere length and structure.4-6 Different shelterin components appear to play different roles in telomere protection, with for instance TRF2 binding to duplex telomeric DNA mainly repressing ATM and POT1 binding to single-stranded regions of the telomere preventing activation of ATR.
Importantly, loss of telomere protection can have opposite effects on tumorigenesis (Fig. 1). The activation of ATM/ATR-dependent checkpoints by critically short telomeres results in P53/RB-dependent growth arrest or apoptosis and represents an important tumor suppressive mechanism that prevents unlimited outgrowth of incipient cancer cells.2 On the other hand, NHEJ activity at uncapped telomeres results in ligation of chromosome ends, creating dicentric chromosomes. Upon division of cells with ineffective checkpoint responses, these dicentric chromosomes initiate breakage-fusion-bridge cycles, chromosomal missegregation and tetraploidization. Such cells with instable and deviant genomes are at high risk of developing into cancer.2,3
DNA Damage Responses at Dysfunctional Telomeres
Posttranslational protein modifications, including phosphorylation, acetylation, methylation and ubiquitylation, as well as chromatin remodeling, play important roles in DNA damage response activation by DNA DSBs.16,17 Several of the modifications initiated at DSBs are also found at dysfunctional telomeres, such as phosphorylation of ATM and histone variant H2AX.18,19 This prompted the view that dysfunctional telomeres are recognized as if they were DNA DSBs. However, while chromatin decondensation and nucleosome loss have been reported to occur at DSBs16,20-23 it was shown that telomere uncapping is not accompanied by overt chromatin remodeling or nucleosome eviction.24 Recognition of uncapped telomeres by DNA damage response proteins and NHEJ- or HR-mediated repair activities at uncapped telomeres can occur without such chromatin changes, which possibly relates to the observed higher mobility and nuclease sensitivity of telomeric nucleosomes.12,13,24 The unique nature of telomeric chromatin, which impacts on the requirements for DNA damage responses at uncapped telomeres, calls for thorough investigation of how (modification of) telomeric chromatin is involved in controlling recognition and processing of dysfunctional telomeres.
Similar to the response to DSBs, the cellular response to uncapped telomeres is believed to start with recognition by the MRE11/RAD50/NBS1 (MRN)-complex and activation of the ATM-kinase, which phosphorylates histone H2AX on its Serine139 residue.5,16,17 Phosphorylated H2AX (γ-H2AX) is recognized and bound by MDC1, which initiates a positive feedback loop by promoting accumulation of MRN and of autophoshorylated ATM on chromatin. This results in extensive spreading of γ-H2AX, visible as subnuclear foci by immunofluorescence microscopy. At DSBs, MDC1 is known to also recruit the ubiquitin-ligase RNF8, which interacts with ATM-phosphorylated motifs in MDC1 via its FHA domain. Ubiquitylation is a post-translational modification process whereby the 76 amino-acid polypeptide ubiquitin is covalently attached to a lysine residue on a target protein by the sequential action of a ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin-ligase (E3). RNF8-ubiquitylation activity is thought to be required for recruitment of the ubiquitin-ligase RNF168. Together RNF8 and RNF168 result in K63-linked poly-ubiquitination of histones H2A and H2AX, which is important for recruitment of downstream factors as 53BP1 and BRCA1 to DSBs. UBC13 has been identified as the critical E2 in this process and its activity is repressed by the deubiquitinating enzyme OTUB1.25
RNF8 Controls Telomere Damage Responses and Telomere Dysfunction-Driven Chromosomal Instability
As it was unknown whether the cellular response to telomere deprotection involves bulky modification of mammalian telomeric histones by ubiquitylation, we recently investigated the potential involvement of the E3-ligase RNF8 in DNA damage response and repair activities at dysfunctional telomeres. Hereto we combined RNA-interference (RNAi)-mediated knockdown of RNF8 with telomere uncapping through rapid temperature-dependent inhibition of TRF2, expression of dominant-negative TRF2 or knockdown of TRF2 or TPP1.26 We found that telomere deprotection results in rapid ubiquitylation of histones H2A and H2AX (< 3 h). H2A ubiquitylation at telomeres, as well as concentrated association of 53BP1 to telomeric chromatin critically depend on the FHA domain-mediated phosphopeptide-binding capacity and the RING E3-ligase domain-mediated ubiquitylation activity of RNF8. Thus, similar as for DSBs, 53BP1 accumulation at uncapped telomeres requires RNF8 ubiquitin-ligase activity and interaction of RNF8 with phosphorylated proteins, in analogy to the DNA damage response, a likely candidate being MDC1. Whereas γ-H2AX focus formation at telomeres appeared not affected by RNF8-knockdown at the time-points examined, RNF8 is involved in ubiquitylation of γ-H2AX in cells undergoing telomere deprotection, consistent with its activity at DSBs.26–28 Moreover, these RNF8 FHA and RING domain-dependent activities at telomeres were found to correlate with the ability of RNF8 to promote telomeric G-overhang degradation and joining of uncapped chromosome ends by NHEJ, which generates dicentric chromosomes and in p53-deficient cells leads to telomere dysfunction-induced genomic instability.26 Too much genomic instability causes crisis, as a result reduced telomere fusion efficiency in RNF8-knockdown cells translates into enhanced survival upon prolonged TRF2 inactivation. Consistent with the detrimental role for RNF8 at uncapped telomeres, RNF8 accumulated at telomeres upon their deprotection. RNF8-mediated control of 53BP1 accumulation and NHEJ at uncapped telomeres were confirmed in a recent publication, which in addition reported on a role for RNF8 in TPP1 stabilization.29
We found that also the ubiquitin-ligase RNF168 contributes to telomere-driven genomic instability. RNF168 depletion by RNAi, like RNF8 or LigaseIV depletion, promoted survival from prolonged TRF2 inactivation.26 As at DSBs, RNF168 might serve to reinforce RNF8-dependent ubiquitylation at uncapped telomeres, but the details of how RNF168 affects telomere-driven genomic instability remain to be addressed, as do the potential involvements of UBC13 and OTUB1.
53BP1 was shown to be required for efficient processing and end-joining of uncapped telomeres.30 It appears to facilitate long-range joining of DNA breaks and uncapped telomeres by increasing chromatin mobility.30,31 Thus the defect in NHEJ of uncapped telomeres in RNF8-knockdown cells might be explained by compromised chromatin mobility due to severely diminished association of 53BP1 with uncapped telomeres. 53BP1 localization to uncapped telomeres and its ability to promote NHEJ at telomeres were shown to depend on the H4K20me1/2 binding activity of the 53BP1 Tudor domains as well as on a Tudor domain-independent interaction.30 Our work indicates that efficient recognition of uncapped telomeres by 53BP1 also requires RNF8-mediated protein ubiquitylation and RNF8-FHA domain-dependent interactions. While 53BP1 lacks an obvious ubiquitin-binding domain, RNF8-dependent ubiquitylation might somehow facilitate H4K20me1/2 recognition by 53BP1 and/or recruit another protein with ubiquitin-binding activity that aids 53BP1 recruitment. Although it remains to be seen if a similar mechanism operates at uncapped telomeres, very recent work on the mechanism of 53BP1 recruitment to DSBs suggests this might be mediated via RNF8- and ubiquitin-dependent recruitment of the ATPase VCP, which promotes release of the H4K20me2 binding Polycomb-group protein L3MBTL1, thereby potentially unmasking 53BP1 binding sites.32
Intriguingly, in addition to its well-established importance in controlling 53BP1 accumulation at DNA lesions, and now also dysfunctional telomeres, RNF8 also affects the association of p-ATM to uncapped telomeres.26 This was visible by detection of telomere dysfunction-induced foci (TIFs) using an antibody against ATM phosphorylated on Ser-1987, but importantly, also by differential KCl-extraction of chromatin followed by immunoblotting to address chromatin association of p-ATM. The effect on TIFs detected with a p-ATM antibody was noticeable at all examined time points of telomere deprotection, as early as 30 min after TRF2 inactivation until 24 h (Fig. 2). As was the case for 53BP1, the ability of RNF8 to affect p-ATM accumulation at telomeres was also dependent on the phosphopeptide-binding capacity and ubiquitylation activity of RNF8. Although it is very likely that protein binding via the RNF8 FHA domain is needed for the localization of RNF8, we do not know the target of RNF8-mediated ubiquitylation that contributes to p-ATM foci at uncapped telomeres. Although the pATM effects we observed occur much faster, RNF8 might affect p-ATM accumulation at uncapped telomeres indirectly via its effect on 53BP1, similar to as reported for late-repaired DSBs where 53BP1 concentrates p-ATM via amplification of MRN-complex association.33 In that case, the target(s) of RNF8-mediated ubiquitylation controlling pATM accumulation would be the same as those directing 53BP1 accumulation. Alternatively, RNF8 might affect p-ATM accumulation at uncapped telomeres through a mechanism independent of 53BP1. A possibility, interesting to investigate, is that this involves Lysine-16 acetylation on histone H4, which was recently found to depend on ubiquitylation by RNF8 and its cousin CHFR and suggested to regulate ATM activity at DSBs.34
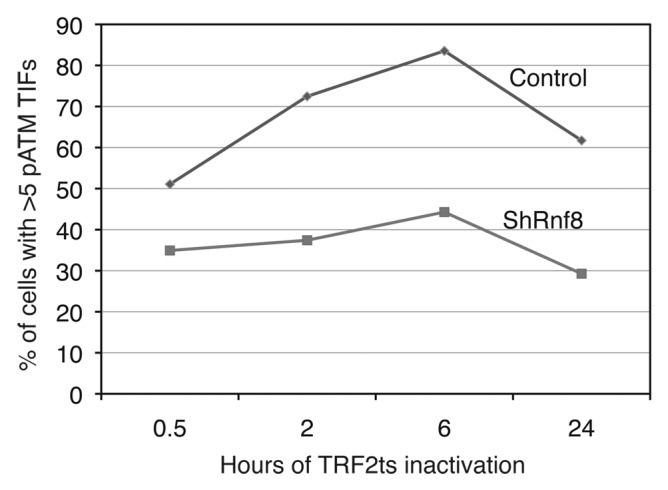
Figure 2. IF-FISH detection of p-ATM focus formation at telomeres (TIF) in control cells vs. cells with RNF8-knockdown at multiple moments after inactivation of temperature-sensitive TRF2 (for details on experimental procedures, see ref. 26). Decreased p-ATM association to chromatin was confirmed by immunoblotting of differentially extracted chromatin at 3 h after TRF2 inactivation.26
ATM was previously demonstrated to be important for efficient processing and end-joining of deprotected telomeres.35 Complete lack of ATM strongly reduced both γ-H2AX and 53BP1 telomeric foci and abolished Chk2 phosphorylation several days after Cre-mediated TRF2 deletion.35 However, at 3 h after TRF2 inactivation under conditions of strongly reduced or mutated RNF8 no significant effect on γ-H2AX foci and only a partial reduction in Chk2 phosphorylation was visible, despite p-ATM accumulation being strongly diminished.26 This difference could be explained by different timing (several days vs. hours) of TIF assessment related to the different methods of TRF2 inactivation, or simply by the fact that upon RNF8-knockdown ATM is not absent, its association with chromatin is only reduced. The remaining ATM-activity could suffice to form γ-H2AX foci at uncapped telomeres. Differences in timing and/or remaining RNF8 or ATM-activity could also explain the recent observation of diminished γ-H2AX foci upon TRF2- or TPP1-knockdown in cells completely lacking RNF8.29 Possibly, RNF8 affects mostly amplification and retention of p-ATM at uncapped telomeres rather than initial activation of p-ATM, which could be sufficient to phosphorylate H2AX and Chk2 at early time points. In addition, the remaining H2AX and Chk2 phosphorylation seen in RNF8-knockdown cells, while telomeric association of p-ATM is strongly reduced, could reflect ATR-kinase activity. ATR is activated by single-stranded (ss) DNA, including ssDNA at telomeres, such as the telomeric G-overhang, and upon telomere deprotection it can contribute to phosphorylation of CHK1, CHK2 and H2AX, and to telomere fusion.35 As also seen before,36 we observed transient phosphorylation of the ATR-target CHK1 after TRF2 inactivation in the temperature-sensitive TRF2 system, both in control and RNF8-knockdown cells, indicating ATR activity. This is explained by partial release of POT1 from telomeres when TRF2 is removed, leading to partial loss of POT1-mediated repression of ATR.36
ATM has been demonstrated to specifically facilitate repair of DSBs in heterochromatin by providing sufficient elasticity of chromatin by phosphorylating KAP1 and diminishing its interactions with chromatin.37 In addition, ATM-kinase activity has recently been shown to contribute to repair of DSBs via a pathway independent of H2AX, by directing mono-ubiquitylation of histone H2B via phosphorylation of RNF20-RNF40 heterodimers.38,39 H2B ubiquitylation is thought to promote repair by inducing chromatin decondensation.40 Also RNF8 has been shown to play a role in H2B ubiquitylation.41 To what extent KAP1- or H2B-Ub-dependent mechanisms operate at uncapped telomeres remains to be investigated. Nevertheless, our results suggest that RNF8 affects the processing and end-joining of deprotected telomeres in at least two ways (Fig. 3). RNF8 facilitates DNA repair at telomeres by promoting the accumulation of DNA repair proteins like 53BP1, through ubiquitylation of histone H2A, H2AX and possibly other substrates, thereby promoting long range end-joining by increasing chromatin mobility. In addition, RNF8 facilitates DNA repair by promoting p-ATM association with deprotected telomeres. P-ATM at telomeres might subsequently, independently of H2AX phosphorylation, enhance processing and repair at uncapped telomeres via increasing chromatin accessibility to DNA repair proteins.

Figure 3. Representation of how the ubiquitin E3-ligase RNF8 facilitates chromosome-end-to-end fusion via ubiquitylation of telomeric histones and possibly other substrates, and control of both 53BP1 and p-ATM accumulation. These activities of RNF8 depend on its RING domain, responsible for its ubiquitin-ligase activity, and on its FHA domain, which at DNA DSBs is known to mediate interaction with phosphorylated MDC1. On cell division telomere fusions can lead to genomic instability, which if too severe, compromises viability, but at sub-lethal levels increases cancer risk in cells that remain viable.
Future Perspectives
The identification of RNF8 as a critical component of the telomere damage response provides new insights into the consequences of telomere uncapping and how loss of telomere protection leads to genome instability. Furthermore, it unveils an additional level of complexity as to what consequences altered levels of RNF8 might have for tumorigenesis. As observed for several other proteins involved in DNA repair, mice deficient in RNF8 show that RNF8 has tumor suppressor activity, expected to relate to its role in mediating repair of DSBs and thereby preventing genomic instability.42,43 However, our recent work suggests that RNF8 might also affect cancer development via its crucial role in facilitating the generation of telomere fusions, thereby promoting genome instability in DNA damage checkpoint-deficient cells that fail to arrest upon telomere uncapping (Figs. 1, 3). Compromised RNF8-activity, while increasing cancer risk due to impaired DSB repair, might simultaneously exert a tumor suppressive effect by preventing telomere-driven genomic instability resulting from fusion between uncapped telomeres. On the other hand, it is interesting to speculate on what the consequences could be of RNF8 overexpression. In this regard it is interesting to note that besides frequent cases of LOH, copy number analyses conducted within the context of the Cancer Genome Project by the Sanger Centre revealed high level amplifications of RNF8 in lung cancer and lymphoma cell lines. In addition, according to the Oncomine database, elevated RNF8 copy numbers and/or mRNA are found in multiple cancers, especially in breast carcinoma and prostate cancer. These data suggest that here RNF8 might act as an oncogene rather than as a tumor suppressor gene. With the current knowledge it is difficult to foresee to what extent overexpression of RNF8 might affect normal DNA repair functions and in this way affect tumor development. However, an intriguing possibility is that RNF8 overexpression enhances cancer development by promoting telomere fusion and thereby telomere-driven genomic instability. For one, further insights in whether RNF8 might affect tumorigenesis via control of telomere-induced genomic instability could come from studying the impact of RNF8 on cancer development in mouse models for telomere uncapping.
While ubiquitylation of telomeric chromatin by RNF8 and potentially additional E3-ligases, including RNF168, clearly control the cellular responses to telomere uncapping upon loss of shelterin function, shelterin subunits themselves are also subject to ubiquitylation. Changes in ubiquitin-mediated control of shelterin stability lead to aberrantly high or low shelterin protein levels that affect telomere maintenance. For instance, the stability of TRF1, TRF2 and TIN2 have been shown to be controlled by respectively the SCF(FBX4) ubiquitin-ligase and USP22 ubiquitin-specific protease, and the SIAH1 and SIAH2 ubiquitin-ligases.44-48 Interestingly, most recently RNF8 was also implicated in control of shelterin stability. RNF8 was found to interact with TPP1 and increase its stability via UBC13-dependent K63-linked polyubiquitylation of TPP1.29 The absence of this mechanism in Rnf8-knockout mouse cells causes telomere shortening and telomere fusion.
Apart from direct effects on telomere maintenance via ubiquitylation of shelterin proteins, an intriguing possibility worth exploring is whether control of DNA repair activities by RNF8-mediated ubiquitylation not only affects genomic instability upon telomere uncapping, but also affects telomere maintenance. DNA damage responses and repair activities are not only important in the event of telomere dysfunction but they for instance also play a role in the late S/G2 phase of the cell cycle when they assist telomeres in completing replication and acquiring a structure essential for telomere function.49 RNF8 could play a role in regulating these activities important for telomere maintenance and function. Along similar lines of thought, RNF8 could be a potential candidate for controlling the activity of repair factors involved in telomere recombination during telomere maintenance via ALT. ALT refers to the telomerase-independent mechanism of telomere maintenance that relies on the activity of proteins involved in HR, but is poorly understood.50 Approximately 10–15% of human tumors, including some with particularly poor outcome employ the ALT mechanism to maintain telomeres, making ALT an important target for cancer therapy. Interestingly, ALT cells commonly show short telomeres that elicit a DNA damage response but somehow repress telomere fusion.50 It will be interesting to address if RNF8 affects this, given its role in facilitating telomere fusion. Together, these possibilities and the recently discovered roles of RNF8 in telomere dysfunction-driven genomic instability and shelterin stability make the potential contribution of RNF8 to cancer via its activities at telomeres highly interesting to address (and challenging).
Acknowledgments
I thank Marieke Peuscher for contributing the time-course experiment on p-ATM accumulation at telomeres in the presence or absence of RNF8 presented in Figure 2. Work in my group is supported by project grants from the Dutch Cancer Society and a personal VIDI grant from the Innovational Research Incentives Scheme of the Dutch Organization for Scientific Research.
Footnotes
Previously published online: www.landesbioscience.com/journals/nucleus/article/19322
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Artandi SE, DePinho RA. Telomeres and telomerase in cancer. Carcinogenesis. 2010;31:9–18. doi: 10.1093/carcin/bgp268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davoli T, de Lange T. The causes and consequences of polyploidy in normal development and cancer. Annu Rev Cell Dev Biol. 2011;27:585–610. doi: 10.1146/annurev-cellbio-092910-154234. [DOI] [PubMed] [Google Scholar]
- 4.de Lange T. How telomeres solve the end-protection problem. Science. 2009;326:948–52. doi: 10.1126/science.1170633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Lange T. How shelterin solves the telomere end-protection problem. Cold Spring Harb Symp Quant Biol. 2010;75:167–77. doi: 10.1101/sqb.2010.75.017. [DOI] [PubMed] [Google Scholar]
- 6.O’Sullivan RJ, Karlseder J. Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol. 2010;11:171–81. doi: 10.1038/nrm2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacobs JJ, de Lange T. Significant role for p16INK4a in p53-independent telomere-directed senescence. Curr Biol. 2004;14:2302–8. doi: 10.1016/j.cub.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 8.Jacobs JJ, de Lange T. p16INK4a as a second effector of the telomere damage pathway. Cell Cycle. 2005;4:1364–8. doi: 10.4161/cc.4.10.2104. [DOI] [PubMed] [Google Scholar]
- 9.Blasco MA. The epigenetic regulation of mammalian telomeres. Nat Rev Genet. 2007;8:299–309. doi: 10.1038/nrg2047. [DOI] [PubMed] [Google Scholar]
- 10.Lejnine S, Makarov VL, Langmore JP. Conserved nucleoprotein structure at the ends of vertebrate and invertebrate chromosomes. Proc Natl Acad Sci U S A. 1995;92:2393–7. doi: 10.1073/pnas.92.6.2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Makarov VL, Lejnine S, Bedoyan J, Langmore JP. Nucleosomal organization of telomere-specific chromatin in rat. Cell. 1993;73:775–87. doi: 10.1016/0092-8674(93)90256-P. [DOI] [PubMed] [Google Scholar]
- 12.Tommerup H, Dousmanis A, de Lange T. Unusual chromatin in human telomeres. Mol Cell Biol. 1994;14:5777–85. doi: 10.1128/mcb.14.9.5777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pisano S, Marchioni E, Galati A, Mechelli R, Savino M, Cacchione S. Telomeric nucleosomes are intrinsically mobile. J Mol Biol. 2007;369:1153–62. doi: 10.1016/j.jmb.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 14.Giraud-Panis MJ, Pisano S, Poulet A, Le Du MH, Gilson E. Structural identity of telomeric complexes. FEBS Lett. 2010;584:3785–99. doi: 10.1016/j.febslet.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 15.Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–60. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 16.Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25:409–33. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lukas J, Lukas C, Bartek J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat Cell Biol. 2011;13:1161–9. doi: 10.1038/ncb2344. [DOI] [PubMed] [Google Scholar]
- 18.Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13:1549–56. doi: 10.1016/S0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- 19.d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–8. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 20.van Attikum H, Fritsch O, Hohn B, Gasser SM. Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell. 2004;119:777–88. doi: 10.1016/j.cell.2004.11.033. [DOI] [PubMed] [Google Scholar]
- 21.Tsukuda T, Fleming AB, Nickoloff JA, Osley MA. Chromatin remodelling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature. 2005;438:379–83. doi: 10.1038/nature04148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kruhlak MJ, Celeste A, Dellaire G, Fernandez-Capetillo O, Müller WG, McNally JG, et al. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J Cell Biol. 2006;172:823–34. doi: 10.1083/jcb.200510015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berkovich E, Monnat RJ, Jr., Kastan MB. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat Cell Biol. 2007;9:683–90. doi: 10.1038/ncb1599. [DOI] [PubMed] [Google Scholar]
- 24.Wu P, de Lange T. No overt nucleosome eviction at deprotected telomeres. Mol Cell Biol. 2008;28:5724–35. doi: 10.1128/MCB.01764-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakada S, Tai I, Panier S, Al-Hakim A, Iemura S, Juang YC, et al. Non-canonical inhibition of DNA damage-dependent ubiquitination by OTUB1. Nature. 2010;466:941–6. doi: 10.1038/nature09297. [DOI] [PubMed] [Google Scholar]
- 26.Peuscher MH, Jacobs JJ. DNA-damage response and repair activities at uncapped telomeres depend on RNF8. Nat Cell Biol. 2011;13:1139–45. doi: 10.1038/ncb2326. [DOI] [PubMed] [Google Scholar]
- 27.Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, et al. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007;131:901–14. doi: 10.1016/j.cell.2007.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, et al. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007;131:887–900. doi: 10.1016/j.cell.2007.09.040. [DOI] [PubMed] [Google Scholar]
- 29.Rai R, Li JM, Zheng H, Lok GT, Deng Y, Huen MS, et al. The E3 ubiquitin ligase Rnf8 stabilizes Tpp1 to promote telomere end protection. Nat Struct Mol Biol. 2011;18:1400–7. doi: 10.1038/nsmb.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dimitrova N, Chen YC, Spector DL, de Lange T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature. 2008;456:524–8. doi: 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Difilippantonio S, Gapud E, Wong N, Huang CY, Mahowald G, Chen HT, et al. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature. 2008;456:529–33. doi: 10.1038/nature07476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Acs K, Luijsterburg MS, Ackermann L, Salomons FA, Hoppe T, Dantuma NP. The AAA-ATPase VCP/p97 promotes 53BP1 recruitment by removing L3MBTL1 from DNA double-strand breaks. Nat Struct Mol Biol. 2011;18:1345–50. doi: 10.1038/nsmb.2188. [DOI] [PubMed] [Google Scholar]
- 33.Noon AT, Shibata A, Rief N, Löbrich M, Stewart GS, Jeggo PA, et al. 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nat Cell Biol. 2010;12:177–84. doi: 10.1038/ncb2017. [DOI] [PubMed] [Google Scholar]
- 34.Wu J, Chen Y, Lu LY, Wu Y, Paulsen MT, Ljungman M, et al. Chfr and RNF8 synergistically regulate ATM activation. Nat Struct Mol Biol. 2011;18:761–8. doi: 10.1038/nsmb.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Denchi EL, de Lange T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature. 2007;448:1068–71. doi: 10.1038/nature06065. [DOI] [PubMed] [Google Scholar]
- 36.Konishi A, de Lange T. Cell cycle control of telomere protection and NHEJ revealed by a ts mutation in the DNA-binding domain of TRF2. Genes Dev. 2008;22:1221–30. doi: 10.1101/gad.1634008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goodarzi AA, Noon AT, Deckbar D, Ziv Y, Shiloh Y, Löbrich M, et al. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol Cell. 2008;31:167–77. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 38.Moyal L, Lerenthal Y, Gana-Weisz M, Mass G, So S, Wang SY, et al. Requirement of ATM-dependent monoubiquitylation of histone H2B for timely repair of DNA double-strand breaks. Mol Cell. 2011;41:529–42. doi: 10.1016/j.molcel.2011.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakamura K, Kato A, Kobayashi J, Yanagihara H, Sakamoto S, Oliveira DV, et al. Regulation of homologous recombination by RNF20-dependent H2B ubiquitination. Mol Cell. 2011;41:515–28. doi: 10.1016/j.molcel.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 40.Fierz B, Chatterjee C, McGinty RK, Bar-Dagan M, Raleigh DP, Muir TW. Histone H2B ubiquitylation disrupts local and higher-order chromatin compaction. Nat Chem Biol. 2011;7:113–9. doi: 10.1038/nchembio.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu J, Huen MS, Lu LY, Ye L, Dou Y, Ljungman M, et al. Histone ubiquitination associates with BRCA1-dependent DNA damage response. Mol Cell Biol. 2009;29:849–60. doi: 10.1128/MCB.01302-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Santos MA, Huen MS, Jankovic M, Chen HT, López-Contreras AJ, Klein IA, et al. Class switching and meiotic defects in mice lacking the E3 ubiquitin ligase RNF8. J Exp Med. 2010;207:973–81. doi: 10.1084/jem.20092308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li L, Halaby MJ, Hakem A, Cardoso R, El Ghamrasni S, Harding S, et al. Rnf8 deficiency impairs class switch recombination, spermatogenesis, and genomic integrity and predisposes for cancer. J Exp Med. 2010;207:983–97. doi: 10.1084/jem.20092437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chang W, Dynek JN, Smith S. TRF1 is degraded by ubiquitin-mediated proteolysis after release from telomeres. Genes Dev. 2003;17:1328–33. doi: 10.1101/gad.1077103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee TH, Perrem K, Harper JW, Lu KP, Zhou XZ. The F-box protein FBX4 targets PIN2/TRF1 for ubiquitin-mediated degradation and regulates telomere maintenance. J Biol Chem. 2006;281:759–68. doi: 10.1074/jbc.M509855200. [DOI] [PubMed] [Google Scholar]
- 46.Atanassov BS, Evrard YA, Multani AS, Zhang Z, Tora L, Devys D, et al. Gcn5 and SAGA regulate shelterin protein turnover and telomere maintenance. Mol Cell. 2009;35:352–64. doi: 10.1016/j.molcel.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fujita K, Horikawa I, Mondal AM, Jenkins LM, Appella E, Vojtesek B, et al. Positive feedback between p53 and TRF2 during telomere-damage signalling and cellular senescence. Nat Cell Biol. 2010;12:1205–12. doi: 10.1038/ncb2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bhanot M, Smith S. TIN2 stability is regulated by the E3 ligase Siah2. Mol Cell Biol. 2012;32:376–84. doi: 10.1128/MCB.06227-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Verdun RE, Karlseder J. The DNA damage machinery and homologous recombination pathway act consecutively to protect human telomeres. Cell. 2006;127:709–20. doi: 10.1016/j.cell.2006.09.034. [DOI] [PubMed] [Google Scholar]
- 50.Cesare AJ, Reddel RR. Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet. 2010;11:319–30. doi: 10.1038/nrg2763. [DOI] [PubMed] [Google Scholar]