

Abstract
Comment on: Colombo E, et al. J Exp Med 2012; 209:521-35.
Keywords: TrkB, astrocyte, multiple sclerosis, neurodegeneration, neurotrophin, nitric oxide
The generation and transmission of the information in the central nervous system (CNS) strongly rely on the structural and functional framework formed by neuronal cells. However, astrocytes, the major glial cell type, may profoundly impact on the performance of the framework, as they give metabolic and trophic support to neurons, and regulate synapse formation and function.1 Under pathological settings astrocytes rapidly respond to injury by promoting the formation of scar tissue and supporting local acute inflammatory reactions–both aspects are essential to confine the lesions, repair the damage and restore tissue homeostasis.1-3 Clearly, dysregulation of astrocyte activities may be dangerous for CNS function.
In multiple sclerosis (MS), an inflammatory neurodegenerative disorder of the CNS, chronic lesions are characterized by permanent demyelination and neurodegeneration. Here, we observed the upregulation of the neurotrophin receptor TrkB on astrocytes (Fig. 1).4
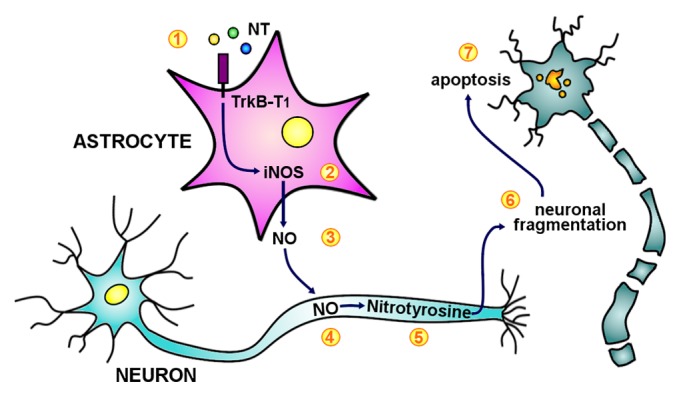
Figure 1. Proposed model for astrocyte TrkB-T1 induced neurodegeneration. (1) Astrocytes upregulate TrkB-T1 in MS and EAE and become responsive to neurotrophins (yellow, blue, green dots for BDNF, NT3, NT4 respectively). TrkB signaling in astrocytes results in enhanced iNOS expression (2) and, consequently, NO synthesis and release (3). Astrocyte NO triggers a secondary NO boost in neurons (4), leading to protein nitrosylation (5), neuronal fragmentation (6) and apoptosis (7).
Neurotrophins constitute a family of growth factors including NGF, BDNF, NT3 and NT4/5. Since they are important for survival, differentiation and function of neurons,5 neurotrophins have been regarded as beneficial for neuroprotection and neuroregeneration.
Similarly to human MS lesions, astrocyte TrkB was strongly upregulated also in experimental autoimmune encephalomyelitis (EAE), the animal model of MS, indicating that neuroinflammation makes astrocytes more sensitive to neurotrophin action. Interestingly, the main TrkB variant detected in astrocytes both in MS and EAE tissues was the truncated TrkB-T1 isoform,4 which results from alternative splicing of the TrkB gene and activates intracellular signaling pathways in glial cells despite lacking the tyrosine kinase domain.6
To address the contribution of astrocyte TrkB to neuroinflammation we analyzed EAE expression in double transgenic mice in which TrkB was excised in astrocytes.7 Surprisingly, transgenic mice showed ameliorated disease, reduced immune cell infiltration and negligible levels of neurodegeneration when compared with control animals, indicating a detrimental role for astrocyte TrkB in neuroinflammation and neurodegeneration.
To investigate the effects of neurotrophins on astrocyte-neuron interaction, we developed an in vitro model in which rat spinal neurons were exposed to conditioned media from human astrocytes. A series of live imaging and immunofluorescence experiments on neuronal cultures showed that low concentrations of the neurotrophin BDNF had no direct effects on neurons, while it activated astrocytes to release factors which induced rapid disruption of the neuronal network and apoptosis. Interestingly, similarly to BDNF, two more TrkB ligands, NT3 and NT4, triggered neurodegeneration when given to astrocytes. This phenomenon depended on astrocyte TrkB, as TrkB-knockout astrocytes were unable to generate media in response to BDNF which induced neuronal fragmentation and death.4
The neurotoxic mediator released by astrocytes was nitric oxide (NO) (Fig. 1). In fact, live imaging experiments on cells loaded with the NO indicator DAF-FM clearly showed TrkB-dependent NO synthesis in BDNF-treated astrocytes.4 Furthermore, neurodegeneration was hampered upon blockade of NO synthases either in astrocytes or in neurons, indicating that the phenomenon is triggered by astrocyte NO and sustained by neuronal NO. NO regulates homeostatic and physiological functions in the CNS, like neuronal survival and differentiation, synaptic activity and neural plasticity. The inducible NO synthase (iNOS), which is physiologically expressed at low levels, is strongly upregulated under inflammatory settings, and excessive levels of NO lead to formation of reactive nitrogen species, tyrosine nitrosylation of intracellular proteins, and cell death.8 As expected, iNOS and nitrosylated proteins were strongly detected in the spinal cord of control EAE mice. In contrast, in vivo depletion of astrocyte TrkB impaired iNOS expression and nitrotyrosine deposition,4 indicating that the TrkB signaling in glial cells is fundamental for the production of NO in injured CNS.
In conclusion, these data suggest that in MS the upregulation of TrkB on astrocytes contributes to neuronal damage via NO production (Fig. 1). The observation that neurotrophins may promote neurodegeneration through the astrocyte raises caution on the potential therapy of CNS disorders with these growth factors. However, it supports the rationale for the development of novel neuroprotective therapies, which target glial cells rather than neurons.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/20798
References
- 1.Sofroniew MV. Trends Neurosci. 2009;32:638–47. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Farina C, et al. Trends Immunol. 2007;28:138–45. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 3.Cordiglieri C, et al. Curr Immunol Rev. 2010;6:150–9. doi: 10.2174/157339510791823655. [DOI] [Google Scholar]
- 4.Colombo E, et al. J Exp Med. 2012;209:521–35. doi: 10.1084/jem.20110698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reichardt LF. Philos Trans R Soc Lond B Biol Sci. 2006;361:1545–64. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fenner BM. Cytokine Growth Factor Rev. 2012;23:15–24. doi: 10.1016/j.cytogfr.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Luikart BW, et al. J Neurosci. 2005;25:3774–86. doi: 10.1523/JNEUROSCI.0041-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calabrese V, et al. Antioxid Redox Signal. 2009;11:2717–39. doi: 10.1089/ars.2009.2721. [DOI] [PubMed] [Google Scholar]