

Abstract
Previous studies have demonstrated that loss of caveolin-1 (Cav-1) in stromal cells drives the activation of the TGF-β signaling, with increased transcription of TGF-β target genes, such as connective tissue growth factor (CTGF). In addition, loss of stromal Cav-1 results in the metabolic reprogramming of cancer-associated fibroblasts, with the induction of autophagy and glycolysis. However, it remains unknown if activation of the TGF-β / CTGF pathway regulates the metabolism of cancer-associated fibroblasts. Therefore, we investigated whether CTGF modulates metabolism in the tumor microenvironment. For this purpose, CTGF was overexpressed in normal human fibroblasts or MDA-MB-231 breast cancer cells. Overexpression of CTGF induces HIF-1α-dependent metabolic alterations, with the induction of autophagy/mitophagy, senescence, and glycolysis. Here, we show that CTGF exerts compartment-specific effects on tumorigenesis, depending on the cell-type. In a xenograft model, CTGF overexpressing fibroblasts promote the growth of co-injected MDA-MB-231 cells, without any increases in angiogenesis. Conversely, CTGF overexpression in MDA-MB-231 cells dramatically inhibits tumor growth in mice. Intriguingly, increased extracellular matrix deposition was seen in tumors with either fibroblast or MDA-MB-231 overexpression of CTGF. Thus, the effects of CTGF expression on tumor formation are independent of its extracellular matrix function, but rather depend on its ability to activate catabolic metabolism. As such, CTGF-mediated induction of autophagy in fibroblasts supports tumor growth via the generation of recycled nutrients, whereas CTGF-mediated autophagy in breast cancer cells suppresses tumor growth, via tumor cell self-digestion. Our studies shed new light on the compartment-specific role of CTGF in mammary tumorigenesis, and provide novel insights into the mechanism(s) generating a lethal tumor microenvironment in patients lacking stromal Cav-1. As loss of Cav-1 is a stromal marker of poor clinical outcome in women with primary breast cancer, dissecting the downstream signaling effects of Cav-1 are important for understanding disease pathogenesis, and identifying novel therapeutic targets.
Keywords: CTGF, aerobic glycolysis, autophagy, cancer associated fibroblasts, cancer metabolism, caveolin-1, extracellular matrix, senescence, tumor stroma
Introduction
It is now well-established that to fully understand the mechanism(s) driving tumor recurrence, metastasis and clinical outcome in cancer patients, it is necessary to study the role of the tumor microenvironment. In particular, cancer-associated fibroblasts play a crucial role through paracrine interactions with adjacent epithelial cancer cells.1
We and others have recently shown that a loss of caveolin-1 (Cav-1) in stromal cells is a predictor of early tumor recurrence, lymph node metastasis, tamoxifen resistance and poor clinical outcome in human breast cancer patients.2,3 To investigate the downstream effects of a loss of stromal Cav-1, we isolated bone marrow-derived stromal cells from WT and Cav-1(-/-)-null mice and subjected them to metabolomic and proteomic analyses and genome-wide transcriptional profiling. Interestingly, Cav-1(-/-) stromal cells showed significant metabolic alterations, with reprogramming toward glycolysis, induction of autophagy and oxidative stress.4 Indeed, acute knockdown of Cav-1 in fibroblasts induces the expression of pyruvate kinase M2 (PKM2), a glycolytic enzyme sufficient to trigger aerobic glycolysis, and promotes the generation of reactive oxygen species (ROS).5 In addition, we demonstrated that a loss of stromal Cav-1 induces the transcription of ROS-associated genes and of hypoxia-inducible factor 1α (HIF-1α) and NFκB target genes.5 Thus, a loss of Cav-1 in cancer-associated fibroblasts may favor tumor growth via oxidative stress and the stromal activation of HIF-1α and NFκB.6 In a co-culture system of normal fibroblasts and MCF7 breast cancer cells, we demonstrated that MCF7 cells induce ROS production and oxidative stress in adjacent fibroblasts, driving the activation of autophagy/mitophagy and aerobic glycolysis.5,7
The induction of autophagy/mitophagy and glycolysis in stromal cells generates recycled nutrients to feed epithelial cancer cells. Then, increased lactate production derived from glycolysis fuels the mitochondrial metabolism of adjacent cancer cells, leading to high ATP generation in cancer cells and protection against cell death. The induction of the catabolic processes of mitophagy and autophagy in cancer-associated fibroblasts leads to cellular self-digestion, promoting the release of recycled nutrients into the tumor microenvironment, which can be used by adjacent cancer cells as building blocks to support their anabolic growth. In support of this hypothesis, we observed that in a xenograft model, the HIF-1α-dependent activation of autophagy in stromal cells greatly enhanced the tumorigenicity of MDA-MB-231 breast cancer cells. On the contrary, HIF-1α activation in MDA-MB-231 cells suppressed tumor growth.8 As HIF-1α triggers autophagy in both fibroblasts and cancer cells, these data demonstrate that the role of autophagy in driving tumor formation is cell type- and compartment-specific.
Other studies have shown that a loss of Cav-1 in fibroblasts is sufficient to mediate the ligand-independent activation of transforming growth factorβ (TGFβ).1,7 TGFβ is activated during normal wound repair9,10 and in fibrotic skin disorders.11,12 TGFβ determines fibroblast proliferation, increases extracellular matrix deposition and can also induce a reduction of extracellular matrix degradation.13 In a previous study of Cav-1(-/-) stromal cells, we demonstrated the upregulation of 35 transcripts associated with activated TGFβ signaling.14 In particular, one of the most upregulated TGFβ-target genes was connective tissue growth factor (CTGF), with a 2.2-fold induction.4 However, it remains unknown if CTGF plays a critical role in the tumor microenvironment.
CTGF is a TGFβ-target gene and a member of the CCN family of secreted proteins, which consists of four conserved sub-domains: an insulin-like growth factor-binding protein (IGFBP) domain, a von Willebrand factor type C (VWC) domain, a thrombospondin type I (TSP) repeat and a C-terminal cysteine knot (CT) domain.15 CTGF is expressed in several cell types, such as endothelial cells, fibroblasts and leukocytes but especially in osteoblasts and chondrocytes. CTGF is a multi-functional signaling modulator involved in a wide variety of biologic and pathologic processes, including cell proliferation, adhesion, migration and extracellular matrix synthesis. Furthermore, CTGF has been identified as a pro-mitogenic and pro-angiogenic co-factor.9,16-19
While the role of CTGF in tissue fibrosis has been well studied,20 the function of CTGF in cancer pathogenesis remains controversial. Interestingly, CTGF has been identified as an oncogene in a variety of cancer types but is considered a tumor-suppressor gene in other contexts. CTGF overexpression is found in melanoma, sarcoma, chondrosarcoma, acute lymphoblastic leukemia and pancreatic cancer21-25 and is associated with increased invasion, migration, the desmoplastic reaction and with chemo-resistance. However, other studies have shown that CTGF expression inhibits the migration and invasion of ovarian,26 colorectal27 and oral squamous cell28 carcinoma cells.
Thus, the aim of the present study was to evaluate the compartment-specific role of CTGF in breast cancer. In particular, we aimed to explore if CTGF plays a role in the metabolic reprogramming of both cancer cells and their tumor microenvironment. To study the cell type- and compartment-specific effects of CTGF expression, CTGF was overexpressed in fibroblasts as well as in MDA-MB-231 breast cancer cells.
Results
CTGF overexpression in fibroblasts induces an autophagy/mitophagy program.
Loss of stromal Cav-1 drives oxidative stress and the induction of autophagy/mitophagy in the tumor stroma,5,7,29 leading to the generation of recycled nutrients that can be used by adjacent anabolic epithelial cancer cells.5,29 We have previously demonstrated that a loss of stromal Cav-1 induces the ligand-independent activation of the TGFβ pathway7 and that Cav-1(-/-) stromal cells show the upregulation of 35 transcripts associated with activated TGFβ signaling, including the TGFβ target gene CTGF.14
To investigate if CTGF plays a role in breast tumorigenesis, CTGF was stably overexpressed in stromal fibroblasts (Fig. 1A). Empty vector (Lv-105) control fibroblasts were generated in parallel. Then, CTGF overexpressing fibroblasts were analyzed by immunoblot blot analysis with a panel of autophagy/mitophagy markers. Figure 1B shows that CTGF overexpression induces the increased expression of LC3 and Beclin-1 (proteins involved in the formation and maturation of auto-phagosomes), Lamp-1 (a protein associated with auto-lysosomes and lysosomes) and BNIP3 (a mitophagy marker whose expression leads to reductions in mitochondrial mass and respiration).30 Therefore, CTGF expression is sufficient to induce autophagy and mitophagy in fibroblasts, downstream from a loss of stromal Cav-1.
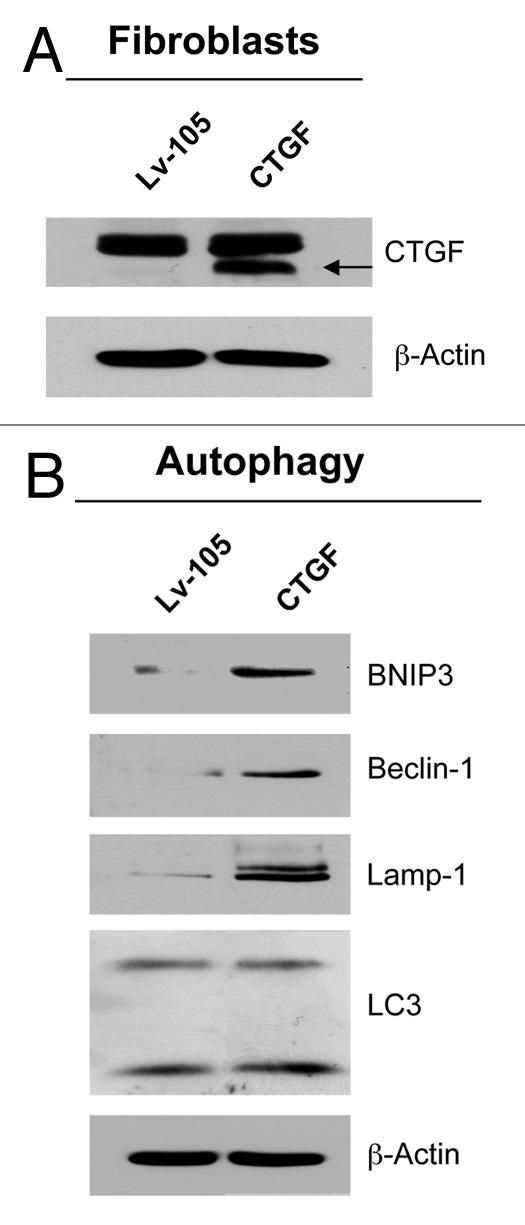
Figure 1. CTGF overexpression induces an autophagy/mitophagy program in fibroblasts. (A) Fibroblasts were stably-transfected with a CTGF or Lv-105 empty vector plasmid, using a lentiviral vector approach. Total proteins were isolated from transfected fibroblasts, and analyzed by immunoblotting to confirm CTGF expression. (B) Immunoblot analysis shows that CTGF overexpression induces the activation of autophagy/mitophagy in fibroblasts, as judged by increased expression levels of BNIP3, Beclin-1, Lamp-1 and LC3, as compared with control cells. The expression of β-actin was assessed for equal protein loading.
CTGF overexpression in fibroblasts induces glycolysis.
Increased BNIP3 expression downregulates mitochondrial mass and respiration by increasing the rate of mitophagy (mitochondrial autophagy), thus compromising ATP production.30 We speculated that CTGF-mediated increases in BNIP3 expression may lead to activation of glycolysis, to compensate for reduced mitochondrial function. Figure 2A shows that CTGF overexpression drives elevated expression of lactate dehydrogenase (LDH) -A, -B and -C, the glycolytic enzymes that convert pyruvate into L-lactate. The induction of aerobic glycolysis was further validated by the increased expression of Enolase 1, another enzyme in the glycolytic pathway.

Figure 2. Fibroblasts overexpressing CTGF show the induction of glycolysis. (A) Immunoblot analysis demonstrates that fibroblasts overexpressing CTGF display the increased expression of several enzymes involved in glycolysis (Enolase-1, LDH-A, LDH-B and LDH-C), relative to empty vector control cells. β-actin was used to assess equal protein loading. (B) An L-lactate assay was performed on cell culture media to confirm the functional role of the increased expression of the glycolytic enzymes. In fibroblasts overexpressing CTGF, the L-lactate production is significantly increased by 20%, as compared with the empty vector control. p < 0.05.
To evaluate the functional role of increased expression of these glycolytic enzymes, we determined the L-lactate content of the fibroblast cell culture media. Figure 2B shows that CTGF fibroblasts display a significant 20% increase in overall L-lactate levels, as compared with control fibroblasts. Increased L-lactate production in stromal cells could stimulate, by a paracrine mechanism, the conversion of lactate into pyruvate in adjacent breast cancer epithelial cells. Then, pyruvate would be used as a substrate for the TCA cycle to promote the oxidative mitochondrial activity of cancer cells.
Consistent with this “paracrine” metabolite hypothesis, we have previouosly shown that treatment with L-lactate is sufficient to induce mitochondrial biogenesis in breast cancer epithelial cells and can functionally enhance their metastastic potential.5,29,31,32
CTGF-induced activation of autophagy/mitophagy and glycolysis is HIF-1α-dependent.
Our previous studies have demonstrated that a loss of stromal Cav-1 causes an accumulation of ROS and the activation of HIF-1α, mimicking a constitutive pseudo-hypoxic state.14 It is well-established that increased ROS levels stabilize HIF-1α expression.33 In order to assess if CTGF overexpression in fibroblasts induces a pseudo-hypoxic condition, we first evaluated the expression of HIF-1α. Figure 3A shows that CTGF-overexpressing fibroblasts display increased levels of HIF-1α compared with control empty vector cells. In addition, a significant increase in ROS production was observed in fibroblasts overexpressing CTGF (Fig. 3B) relative to control cells, indicating that CTGF overexpression in fibroblasts does induce a “pseudo-hypoxic” state.
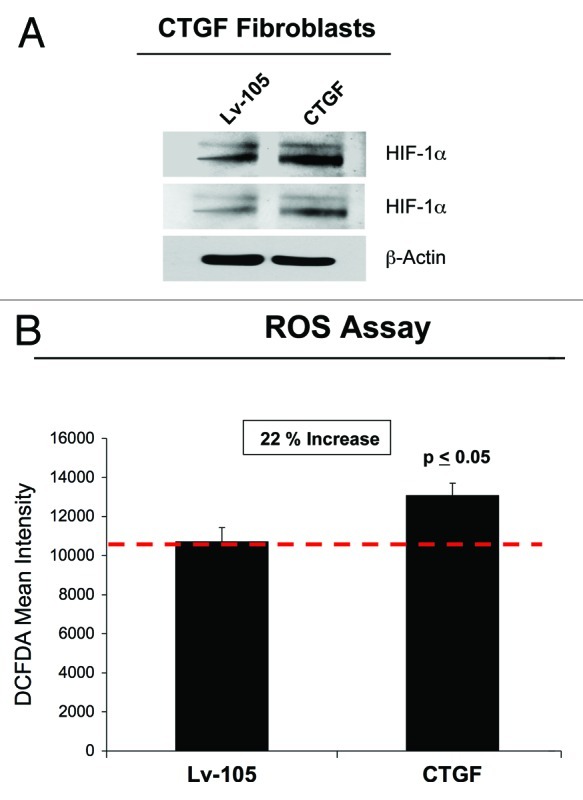
Figure 3. CTGF overexpression in fibroblasts induces a pseudo-hypoxic phenotype. (A) CTGF- and Lv-105 fibroblasts were analyzed by immunoblotting with antibodies against HIF-1α. Note that HIF-1α levels are augmented in CTGF-fibroblasts, suggesting that CTGF induces a pseudo-hypoxic state. Two different exposures are shown. β-actin was used to assess equal protein loading. (B) To evaluate if CTGF expression drives oxidative stress, we performed a ROS assay. CTGF overexpression induces a 20% increase in ROS production, as compared with control cells. p < 0.05.
HIF-1α is a crucial transcription factor for the expression of glycolytic enzymes34 and autophagic proteins.35 To determine if the CTGF-mediated induction of glycolysis and autophagy is HIF-1α-dependent, fibroblasts overexpressing CTGF were treated with the HIF-1α inhibitor echinomycin. Echinomycin blocks the binding of HIF-1α to DNA, thereby inhibiting its transcriptional action. Note that echinomycin treatment decreases HIF-1α expression (Fig. 4A) and greatly reduces the expression levels of autophagy (BNIP3, Lamp-1 and LC3, Fig. 4B) and glycolysis markers (Enolase 1, LDH-A and LDH-C, Fig. 4C). These results clearly indicate that the activation of autophagy, mitophagy and glycolysis in fibroblasts overexpressing CTGF is mediated by HIF-1α stabilization.
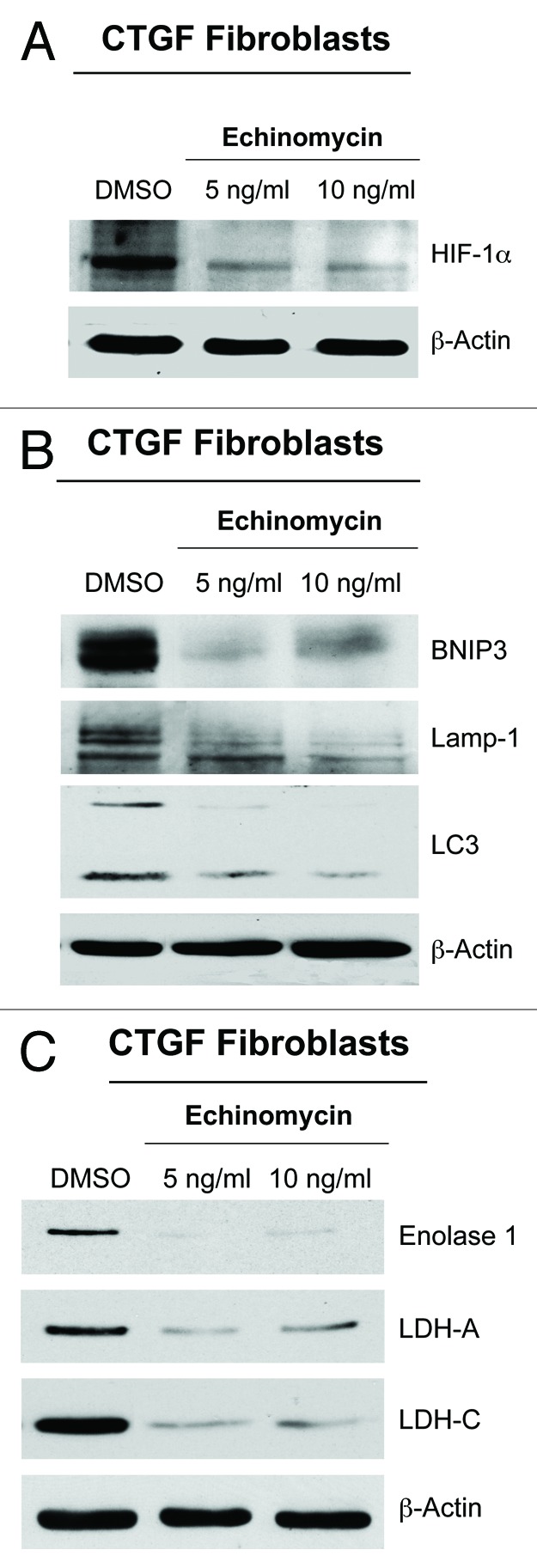
Figure 4. The activation of autophagy and glycolysis in CTGF-fibroblasts is HIF-1α−dependent. To evaluate if the activation of autophagy and glycolysis of CTGF-fibroblasts is HIF-1α dependent, CTGF-fibroblasts were treated with the HIF-1α inhibitor echinomycin (5–10 ng/ml) or vehicle alone (DMSO) for 24 h. Western blot analysis demonstrates that echinomycin treatment reduces the expression of (A) HIF-1α, (B) mitophagy (BNIP3) and autophagy (Lamp-1 and LC3) markers, and (C) glycolytic enzymes (Enolase 1, LDH-A and LDH-C). Equal loading control was assessed using β-actin. These results indicate that CTGF expression enhances autophagy/mitophagy and glycolysis in a HIF-1α-dependent manner.
CTGF overexpression drives cellular senescence in fibroblasts.
Several studies have reported that increased intracellular ROS is involved in induction of senescence. In addition, recent evidence suggests that autophagy may also mediate the acquisition of a senescent phenotype.36,37 To verify if CTGF expression induces a senescent phenotype in fibroblasts, we next analyzed the expression of genes implicated in senescence by immunoblotting. Figure 5A shows that CTGF overexpression drives the upregulation of p21(WAF/CIP1) and p16(INK4A), both inducers of cell cycle arrest. However, no changes were observed in p19(ARF) protein expression. Conversely, CTGF induces an increase of Cyclin D1 expression, likely a compensatory response to senescence. To independently assess if CTGF induces a senescent phenotype, we next performed a β-galactosidase (β-Gal) activity assay by flow cytometry (Fig. 5B) and a β-Gal staining assay (Fig. 5C). Figure 5B shows that CTGF expression increases β-Gal activity, as judged by increased numbers β-Gal-positive cells and increased mean intensity. Similarly, conventional β-Gal staining is augmented in CTGF fibroblasts as compared with control fibroblasts (Fig. 5C), confirming the ability of CTGF to trigger a senescence phenotype.

Figure 5. CTGF overexpression in fibroblasts induces a senescence phenotype. (A) Immunoblot analysis performed on control and CTGF overexpressing fibroblasts demonstrates that CTGF expression induces the upregulation of p21 (CIP1/WAF1) and of p16Ink4A, both inhibitors of cell cycle progression. No changes were detected for p19 expression levels. Conversely, CTGF expression also induces the upregulation of cyclin D1, probably as a compensatory response against senescence. Equal loading was assessed using β-actin. (B) A β-galactosidase Assay was performed by FACS analysis on control and CTGF overexpressing fibroblasts. The number of β-Galactosidase-positive cells (left) and the β-galactosidase intensity mean (right) are both increased in CTGF overexpressing fibroblasts. (C) Conventional β-galactosidase staining was also performed on control and CTGF overexpressing fibroblasts to independently confirm that CTGF overexpression induces senescence. Note that CTGF-fibroblasts have intense blue staining, indicative of increased β-galactosidase activity.
CTGF overexpression in fibroblasts increases breast cancer growth independently of angiogenesis.
To evaluate if CTGF expression in fibroblasts plays a role in breast cancer development, we employed a mouse xenograft model consisting of MDA-MB-231 breast cancer cells co-injected with CTGF or control fibroblasts in the flanks of nude mice. After 4 weeks, mice were sacrificed, and tumor weight and volume were measured. Remarkably, CTGF overexpression in fibroblasts induces an increase of 2-fold in tumor weight and of 2.6-fold in tumor volume, compared with control cells (Fig. 6A). Our previous studies have demonstrated that the autophagic stroma is sufficient to drive tumor development without increased neo-vascularization. Thus, we evaluated tumor vascularity using immuno-staining with antibodies directed against CD31. Figure 6B shows a significant reduction of angiogenesis in CTGF tumors, indicating that CTGF favors breast cancer growth independently of angiogenesis but more likely via the metabolic reprogramming of the tumor stroma.

Figure 6. CTGF overexpression in fibroblasts promotes tumor growth. To evaluate the effects of CTGF on tumor development in vivo, CTGF-fibroblasts or control fibroblasts were co-injected with MDA-MB-231 cells subcutaneously in nude mice. (A) The xenograft model shows that fibroblasts overexpressing CTGF promote tumor growth, as demonstrated by increased tumor weight and tumor volume (respectively 2 and 2.6-fold), compared with Lv-105 control fibroblasts. n = 10 ; p values are as indicated. (B) To investigate if CTGF promotes angiogenesis, tumor xenografts were immuno-stained with CD31 antibodies. Surprisingly, quantification of CD31-positive vessels reveals a significant reduction in angiogenesis in CTFG-tumors, indicating that CTGF stimulates tumor growth independently of angiogenesis. (C) To evaluate the role of extracellular matrix deposition in the CTGF-mediated tumor growth, we performed immuno-histochemistry analysis on tumor xenografts with antibodies against Type I Collagen and Tenascin C. Note that tumors with CTGF overexpressing fibroblasts show higher levels of Type I Collagen and Tenascin C.
It is well known that CTGF stimulates extracellular matrix deposition and activity.38,39 The extracellular matrix activates a variety of signals, which directly influence the growth, migration and differentiation of cells participating in virtually every state of breast cancer pathogenesis. To investigate if the extracellular matrix plays a key role in tumor development in our system, we next evaluated the expression of Type I Collagen and Tenascin C by immunohistochemistry in tumor xenografts. As predicted, the expression levels of both Type I Collagen and Tenascin C were elevated in tumors grown in the presence of CTGF-expressing fibroblasts, as compared with tumors grown with vector-alone control fibroblasts (Fig. 6C). These data suggest that CTGF could also favor tumor growth via the induction of extracellular matrix deposition.
CTGF-activated fibroblasts increase the mitochondrial activity of adjacent MDA-MB-231 cells.
To experimentally evaluate if increased lactate production in fibroblasts stimulates the mitochondrial metabolism of adjacent cancer epithelial cells, we evaluated the expression of ATPase-Inibitor Factor 1 (ATPase-IF1) in a co-culture system of fibroblasts and MDA-MB-231 cells. ATPase-IF1 is an endogenous inhibitor of the mitochondrial ATP synthase, leading to reduced mitochondrial activity. It is known that silencing of ATPase-IF1 activates oxidative phosphorylation. Figure 7 shows that ATPase-IF1 expression is decreased in MDA-MB-231 cells co-cultured with CTGF fibroblasts, as compared with MDA-MB-231 cells grown with control fibroblasts. These results indicate that CTGF expression in fibroblasts stimulates the mitochondrial activity of adjacent cancer cells, in a paracrine way, likely via the generation of high L-lactate levels.

Figure 7. CTGF-fibroblasts greatly decrease the expression of ATPase-IF1 in adjacent MDA-MB-231 cells. To verify if CTGF-fibroblasts exert any paracrine effects on tumor cells, GFP-positive MDA-MB-231 cells were co-cultured with CTGF-fibroblasts or control fibroblasts for 3 d. Cells were then immuno-stained with antibodies against ATPase-IF1 (an endogenous inhibitor of the mitochondrial ATP synthase, RED). Nuclei were stained with DAPI (BLUE). Note that fibroblasts overexpressing CTGF reduce the levels of ATPase-IF in adjacent MDA-MB-231 cells (GFP). The white stars mark the nuclei of MDA-MB-231 cells.These results indicate that CTGF-fibroblasts increase the mitochondrial activity in adjacent breast cancer cells, by reducing ATPase-IF expression.
MDA-MB-231 cells overexpressing CTGF show an increase in autophagy and oxidative stress.
To evaluate if the role of CTGF in tumorigenesis is compartment-specific, we overexpressed CTGF in MDA-MB-231 cells (Fig. 8A). We next investigated whether CTGF also induces autophagy/mitophagy in epithelial cancer cells. Immunoblot analysis demonstrated that MDA-MB-231 cells overexpressing CTGF show the upregulation of several autophagy/mitophagy markers under basal condition (BNIP3, Lamp-1 and cathepsin B) (Fig. 8B) or upon nutrient-starvation (Beclin-1 and LC3) (Fig. 8C), indicating that CTGF can activate autophagy also in breast cancer epithelial cells. However, no changes in L-lactate production were observed in MDA-MB-231 cells overexpressing CTGF (data not shown).

Figure 8. MDA-MB-231 cells overexpressing CTGF display the induction of autophagy/mitophagy, with HIF-1α activation. To study if CTGF expression has any compartment-specific effects, CTGF was overexpressed in MDA-MB-231 cells. (A) Immunoblot analysis was performed on total protein lysates to confirm CTGF overexpression. (B, C) MDA-MB-231 cells overexpressing CTGF show activation of an autophagy/mitophagy program. Immunoblot analysis on CTGF- and control-MDA-MB-231 cells reveals that CTGF significantly increases the expression levels of several proteins involved in mitophagy (BNIP3) and autophagy (Lamp-1, Beclin-1, cathepsin B and LC3) under (B) basal condition or (C) after 2h of nutrient starvation. (D) Immunblot analysis of total protein lysates from control and CTGF-MDA-MB-231 cells shows that CTGF expression induces the activation of HIF-1α in MDA-MB-231 cells. Two exposures are shown. For all, β-actin was used as control for equal protein loading. (E) ROS assay performed on control- and CTGF-MDA-MB-231 cells reveals that CTGF expression induces a 20% increase in ROS production.
Since we demonstrated that CTGF induces autophagy in fibroblasts via HIF-1α (Fig. 3), we evaluated if the same mechanism operates in MDA-MB-231 cells. Although HIF-1α expression is only slightly increased in MDA-MB-231 cells overexpressing CTGF (Fig. 8D), a significant 20% increase in ROS production in MDA-MB-231 cells overexpressing CTGF was observed as compared with control cells (Fig. 8E). Therefore, we believe that the CTGF induction of autophagy also depends on increased oxidative stress in breast cancer cells. Thus, we conclude that CTGF induces an autophagic program in both cell types, fibroblasts and MDA-MB-231 cells, by inducing oxidative stress, HIF-1α activation and a “pseudo-hypoxic” phenotype.
CTGF overexpression does not modulate the expression of senescence markers in breast cancer cells.
In order to understand if CTGF activate the same processes activated in fibroblasts in epithelial breast cancer cells, we evaluated the expression of proteins involved in cell cycle regulation. Figure 9 demonstrates that CTGF overexpression in MDA-MB-231 cells does not induce markers of senescence. Indeed, no significant changes were detected in p16(INK4A), p19(ARF) and Cyclin D1 expression levels, whereas p21(WAF1/CIP1) was undetectable.
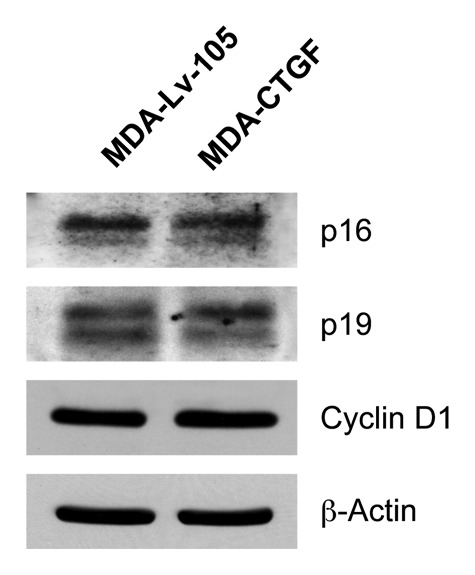
Figure 9. Overexpression of CTGF in MDA-MB-231 does not induce senescence. Protein lysates from Lv-105 and CTGF-MDA-MB-231 cells were analyzed by immunoblotting with antibodies against cell cycle proteins. No differences in p16, p19 and cyclin D1 expression levels were detected, while p21 was undetectable. β-actin was used as control for equal loading.
CTGF overexpression in breast cancer cells inhibits tumor growth.
To investigate the effects of CTGF overexpression in breast cancer cells in vivo, CTGF-MDA-MB-231 and control MDA-MB-231 cells were injected into the flanks of athymic nude mice. Surprisingly, Figure 10A shows that after 3 weeks, CTGF overexpression caused a nearly 3-fold reduction in tumor growth. Also in this context, we did not detect any differences in tumor neo-vascularization, as assessed by quantification of CD31-positive vessels (Fig. 10B). These results clearly indicate that CTGF plays a compartment- and cell type-specific role during tumor formation and exerts opposing effects depending on which cell type expresses it.
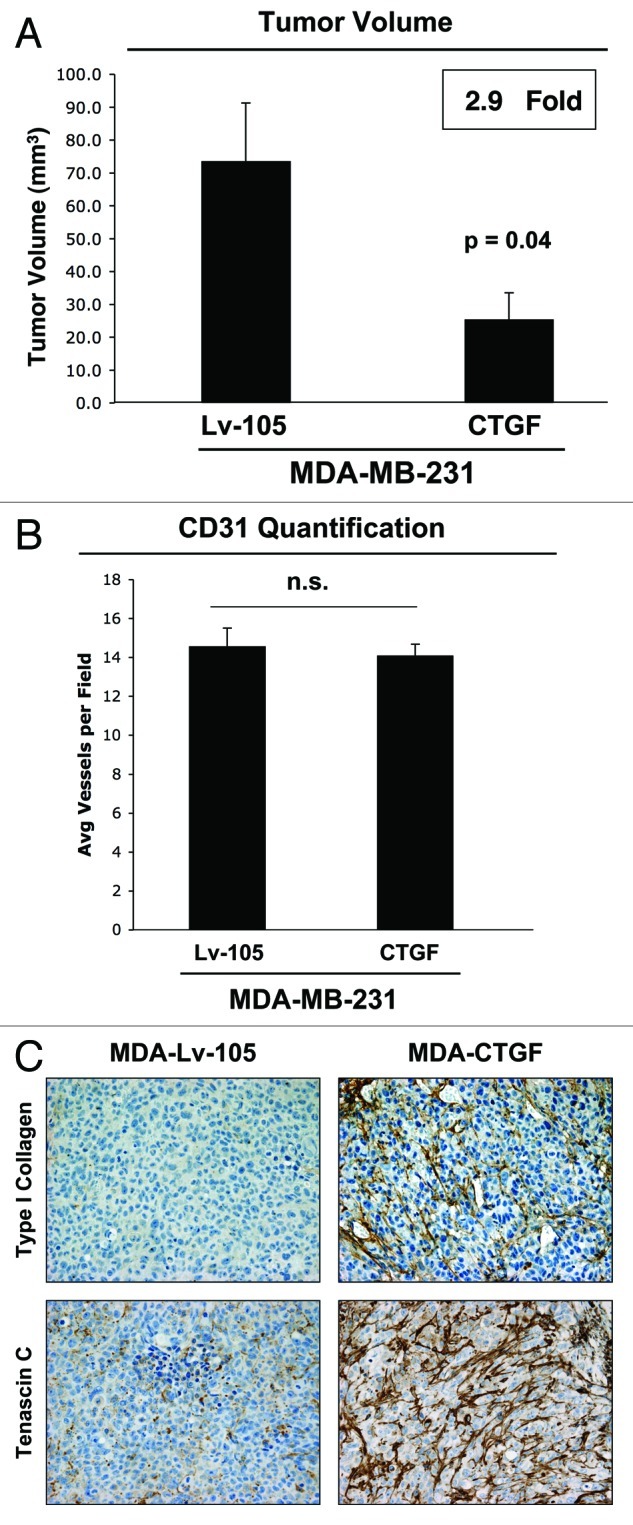
Figure 10. CTGF overexpression in breast cancer cells inhibits tumor growth. (A) Lv-105- and CTGF-MDA-MB-231 cells were injected subcutaneously in nude mice. Tumors were then harvested after 3 weeks. Surprisingly, CTGF overexpression in MDA-MB-231 cells induces a nearly 3-fold reduction in tumor growth, as compared with empty vector controls. n = 10, p values are as indicated. (B) CD31 staining and quantification on tumor xenografts shows no differences in tumor neo-vascularization. (C) Tumor xenografts were immunostained with antibodies against Type I Collagen and Tenascin C. Interestingly, the expression of Type I Collagen and Tenascin C are greatly increased in MDA-MB-231 tumors with CTGF overexpression, although tumor size is decreased.
Finally, to evaluate the role of extracellular matrix deposition in CTGF-mediated tumorigenesis, we evaluated the expression of Type I Collagen and Tenascin C in MDA-MB-231 tumor xenografts. Interestingly, the expression levels of Type I Collagen and Tenascin C are increased in tumors formed by MDA-MB-231 cells overexpressing CTGF, relative to control tumors (Fig. 10C). These results indicate that in our model, the role of CTGF in tumorigenesis is independent on its role in extracellular matrix remodeling. In fact, we observed increased extracellular matrix deposition in tumors both when CTGF was expressed by cancer cells and by fibroblasts, despite the fact that the CTGF effects on tumor growth are just the opposite.
Thus, we speculate that in our experimental system, CTGF’s effects are likely due to its intracellular action and not to its extracellular action. The cells overexpressing CTGF undergo metabolic reprogramming and become more autophagic. The induction of CTGF-mediated autophagy in epithelial tumor cells will cause self-digestion and inhibition of tumor growth. Conversely, the induction of CTGF-mediated autophagy in tumor fibroblasts will generate building blocks for the anabolic growth of cancer cells.
Discussion
Previous studies have demonstrated that a loss of Cav-1 in stromal cells induces the ligand-independent activation of the TGFβ pathway,7,40-42 with the increased transcription of the TGFβ target gene CTGF.1,4,14,43 It is now well known that CTGF induces tissue fibrosis, and that alterations in the extracellular matrix influence tumor growth and clinical outcome.21,44 It has also been demonstrated that a loss of stromal Cav-1 induces the metabolic reprogramming of cancer-associated fibroblasts with the induction of glycolysis and autophagy. However, it remains unknown if the activation of the TGFβ/CTGF pathway plays a role in the metabolic reprogramming of stromal cells induced by a loss of stromal Cav-1.
Therefore, the purpose of this study was to investigate if the TGFβ-target gene CTGF plays a role in the metabolic remodeling of the tumor microenvironment. In particular, we aimed to study if the cell type-specific expression of CTGF differentially affects tumor development.
The role of CTGF in breast cancer remains controversial. Elevated CTGF mRNA levels were found in human invasive ductal carcinomas and mouse mammary tumors and were confined to the fibrous tumor stroma.44 In another breast cancer study, overexpression of CTGF was positively associated with age, tumor size, stage and lymph node metastasis.45 Mechanistically, the tumor-promoting role of CTGF is supported by data showing that CTGF enhances tumor cell migration and angiogenesis46 and confers drug resistance.47,48 Conversely, other studies have indicated that CTGF may act as a tumor suppressor and have reported that low levels of CTGF are associated with increased metastasis and poor prognosis in breast cancer patients.49 For example, Hishikawa et al. demonstrated that forced overexpression of CTGF in MCF7 cells induces apoptosis.50
In our current studies, we propose a novel viewpoint to explain the controversial role of CTGF in breast cancer. Our data clearly indicate that CTGF exerts compartment-specific actions, and that its effects on tumor growth are opposite depending on the cell type producing CTGF. In fact, surprisingly, overexpression of CTGF in breast cancer epithelial cells inhibits tumor growth, but the opposite, tumor-promoting effect was observed when CTGF is overexpressed in the tumor fibroblast compartment.
We show for the first time that the overexpression of CTGF drives the induction of autophagy in both cell types, fibroblasts and breast cancer cells. Thus, CTGF-induced autophagy in fibroblasts can drive stromal cell digestion, leading to the release of chemical building blocks into the tumor microenvironment. These nutrients could be used as fuel for the anabolic growth of breast cancer cells, driving increased tumor mass independently of angiogenesis. Furthermore, we show that CTGF overexpression in stromal cells triggers the induction of glycolysis. The final product of glycolysis, L-lactate, could act in a paracrine way on breast cancer cells. Increased L-lactate uptake by breast cancer cells could activate LDH in cancer cells. At high lactate concentrations, LDH converts L-lactate into pyruvate, which is a substrate of the Krebs cycle, driving an increase in mitochondrial metabolic activity. Consistent with this hypothesis, we detected reductions in ATPase-IF1 expression in MDA-MB-231 cells co-cultured with CTGF fibroblasts compared with the control fibroblasts. Mechanistically, we show that the CTGF-mediated induction of autophagy occurs via increased oxidative stress and HIF-1α stabilization. Our results are consistent with previous studies showing that CTGF induces HIF-1α upregulation.51 However, the mechanism(s) by which CTGF induces HIF-1α activation is currently unknown.
Conversely, we show that forced CTGF overexpression in breast cancer cells inhibits tumor growth. We demonstrate that CTGF overexpression in epithelial breast cancer cells induces autophagy. Activation of autophagy in cancer cells increases tumor cell self-digestion, with a consequent decrease in tumor mass. Mechanistically, we propose that CTGF overexpression leads to increased oxidative stress, which, in turn, stabilizes HIF-1α. In fact, we have previously demonstrated that HIF-1α activation in breast cancer cells drives the induction of autophagy and inhibits tumor growth.8
Several studies have reported that increased intracellular ROS is involved in the induction of senescence. Two mechanisms have been proposed to explain ROS action on senescence. The first possibility is that ROS can lead to random damage to cellular components, thus acting as a non-specific senescence mediator. For example, an increase in ROS levels causes DNA damage, leading to activation of p53, which, in turn, drives cell cycle arrest via induction of p21. The second explanation is that ROS can function as messenger molecules that activate specific redox-dependent targets, and those could induce senescence.52
Recent evidence also links autophagy to cellular senescence. In particular, it has been demonstrated that ULK-3, the human homolog of the yeast ATG1 is essential for the initial building of the autophagosome, is highly expressed in senescent cells, and that ULK-3 overexpression induces autophagy and senescence. Furthermore, the knockdown of ATG5 or ATG7 reduces β-galactosidase activity, the most widely used marker of senescence.37 Inhibition of autophagy delays the senescence phenotype. Thus, the induction of autophagy in fibroblasts promotes the acquisition of the senescent phenotype.37 Recently, a new mechanism by which autophagy can lead to pre-mature senescence, has been proposed. Goligorsky et al. have demonstrated that stress-induced lysosomal membrane permeabilization drives the release of cathepsin B in the cytosol. Cathepsin B is a lysosomal cysteine protease, which induces SIRT1 depletion leading to autophagy-induced premature senescence.36 Thus, autophagy and senescence may be part of the same physiological process, known as the autophagy-senescence transition (AST) (Fig. 11).

Figure 11. CTGF drives the autophagy-senescence transition (ast) in cancer associated fibroblasts. Here, we show that activation of TGF-β signaling, via CTGF expression in normal fibroblasts, is sufficient to confer the cancer associated fibroblast phenotype. Briefly, CTGF expression in stromal fibroblasts leads to ROS production, resulting in the stabilization of the HIF1-transcription factor. HIF1-activation then promotes the induction of autophagy, mitophagy, and glycolysis. Autophagy drives the onset of senescence, via the autophagy-senescence transition (AST), resulting in the upregulation of CDK-inhibitors [p21(WAF1/CIP1) and p16(INK4A)], as well as β-galactosidase (a lysosomal enzyme and marker of senescence). Oxidative stress, autophagy, and senescence may also contribute to CTGF-induced fibrosis and extracellular matrix (ECM) remodeling.
Cellular senescence is a reversible process that limits proliferation of cells at risk for neoplastic transformation and contributes to aging.53-56 On the other hand, although the mechanisms have not been fully elucidated yet but are likely to contrast aging, the induction of senescence leads to the secretion of several mitogenic substances, including growth factors, cytokines and extracellular matrix components,53,57 that alter the tumor microenvironment and favor tumor growth.58
In our study, we show for the first time that CTGF has the ability to induce senescence. Interestingly, CTGF-mediated induction of senescence is cell-type specific, as it occurs only in fibroblasts but not in breast cancer cells. It might be that the induction of senescence in fibroblasts could constitute an additional mechanism through which CTGF overexpression in stromal cells drives tumor growth. Although the molecular mechanism(s) that link autophagy with senescence are still unclear, we propose that systemic induction of autophagy and increased protein turnover could lead stromal cells to establish a senescent-like phenotype to protect them from further self-digestion.
Our results indicate that the tumor-promoting effects of CTGF may be independent of its well-known role in extracellular matrix remodeling. We unexpectedly observed that CTGF has opposite effects when it is produced by stromal cells or by breast cancer cells. This suggests that the CTGF effects are not due to its extracellular secretion; otherwise, we should observe the same results, independently of the cell type producing CTGF. Thus, our data clearly indicate that CTGF acts via an intracellular mechanism, likely through the metabolic reprogramming of the CTGF-producing cells. In support of this notion, we observed increased extracellular matrix deposition in tumor xenografts generated by CTGF-MDA-MB-231 cells and by CTGF-fibroblasts. Indeed, we observed increased extracellular matrix, which is usually considered a marker of tumor aggressiveness, in the CTGF-MDA-MB-231 xenogafts as well when the tumor mass was reduced. These data demonstrate that CTGF can still be secreted, but that the main CTGF tumor-promoting effects are due to its ability to drive metabolic reprogramming within cells. This is the first time that CTGF has been shown to modulate the metabolic status of stromal cells within the tumor microenvironment.
In conclusion, we propose a new compartment-specific role for CTGF in tumor formation, which is mediated via intracellular metabolic rearrangements. The overexpression of CTGF in breast cancer epithelial cells leads to autophagy activation, tumor cell digestion and inhibition of tumor growth. On the other hand, overexpression of CTGF in fibroblasts similarly drives the induction of autophagy, but in this case, enhances the release of recycled chemical building blocks into the tumor microenvironment, which can be used as “fuel” by anabolic tumor cells. Finally, the overexpression of CTGF drives a senescence phenotype in fibroblasts, which may further promote tumor growth.
Materials and Methods
Materials.
Antibodies were as follow: CTGF (6B13) (Santa Cruz, sc-101586); β-Actin, (Sigma-Aldrich, A5441); Beclin-1 (Novus Biologicals, NBP1-00085); BNIP3 (Abcam, ab10433); Cathepsin B (FL-339) (Santa Cruz, sc-13985); Lamp-1 (E-5) (Santa Cruz, sc-17768); LC3 (Abcam, ab48395); Enolase 1 (Cell Signaling, 3810); LDH-A (Cell Signaling, 2012); LDH-B (Sigma-Aldrich, AV48210); LDH-C (Sigma-Aldrich, AV53602); HIF-1α, (BD Transduction Laboratories, 610959); p21 (H-164) (Santa Cruz, sc-756); p19 (DCS-240) (Santa Cruz, sc-53639); p16 (H-156) (Santa Cruz, sc-759); cyclin D1 (Thermo Scientific, MS-210); ATPase-IF1 (Abcam, ab811061); Tenascin C (Leica Microsystems, NCL-TENAS-C); and Type-I-Collagen (Cell Sciences, PS065).
Cell culture.
GFP-positive MDA-MB-231 breast cancer cells (gift of Dr. Fatatis, Drexel University) and human foreskin immortalized fibroblast (hTERT-BJ1, Clontech) were grown in DMEM supplemented with 10% fetal bovine serum (FBS) and PS (100 units/mL penicillin, 100 µg/mL streptomycin). Cells were maintained at 37°C, in a 5% CO2 incubator. Starvation was performed using Hank’s Balanced salt solution (HBSS) supplemented with 40 mM Hepes and 1% PS. For the echinomycin-related experiments, cells were treated with 5 or 10 ng/ml echinomycin (Enzo life Sciences) dissolved in DMSO for 24 h. For co-culture experiments, fibroblasts and MDA-MB-231 cells were plated at a ratio 5:1 on glass coverslips in 12-well plates in complete media. After 24 h, the media was changed to DMEM with 10% NuSerum (BD Biosciences). Cells were maintained in co-culture for 3 d.
Lenti-viruses.
Lentiviral plasmids, packaging cells and reagents were from Genecopoeia. Lenti-viruses were prepared by transfection of 293Ta packaging cells. For this purpose, 1.7 million 293Ta cells were seeded in 10-cm plates. After 48 h, 293Ta cells were transfected with the lentiviral plasmids EX-NEG-Lv105 (Empty Vector) and EX-A0312-Lv105 (encoding CTGF) using EndoFectin. After 48 h, the viruses were collected, centrifuged and filtered. Target cells (fibroblasts or MDA-MB-231 cells) were infected with the viruses and then selected with puromycin (1.5 µg/ml for fibroblasts and 2 µg/ml for MDA-MB-231 cells).
Immunoblot analysis.
Cells were scraped into lysis buffer containing 10 mM TRIS-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100 and 60 mM N-octyl-glucoside supplemented with protease (Roche Diagnostics) and phosphatase (Sigma) inhibitor cocktails. After rotating for 40 min, samples were centrifuged 10 min at 13,000x g at 4°C, and the supernatants were collected. For nuclear proteins, RIPA buffer (50 mM TRIS-HCl pH 7.5, 150mM NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS plus protease and phosphatase inhibitors) was used. After lysis, cells were sonicated and centrifuged at 12,000x g for 10 min at 4°C to pellet insoluble debris. Protein concentrations were evaluated with BCA kit (Pierce). To detect HIF-1α, cells were lysed in Urea Buffer (6.7 M urea, 10% glycerol, 1% SDS, 10 mM TRIS-HCl pH 6.6, 1% Triton X-100 containing protease and phosphatase inhibitors). Cells were homogenized, incubated on ice for 10 min and centrifuged at 12,000x g for 10 min at 4°C. Protein content was determined by Bradford assay (BioRad). Proteins were separated by SDS-PAGE (10 to 15% acrylamide) and transferred to a nitrocellulose membrane. After blocking in 5% milk, membranes were incubated with primary antibodies, followed by incubation with peroxidase-coupled secondary antibodies. Bound antibodies were detected using enhanced chemiluminescence substrate (Thermo Scientific).
Lactate assay.
105 cells were plated into 12-well plates in complete media. After 24 h, the media was changed to DMEM containing 2% FBS. After 48 h, the media was collected, and the lactate concentration was measured using the EnzyChromTM L-Lactate Assay Kit (cat #ECLC-100, BioAssay Systems) according to the manufacturer’s instructions. The L-lactate concentration was normalized to the cellular protein content per well.
ROS assay.
Cells were seeded in 12-well plates in complete media. The next day, the media was changed to DMEM containing 10% NuSerum and 1% PS. ROS assay was performed after 48 h. Fibroblasts were incubated with 10 µM CM-H2DCFDA (Invitrogen, C6827) for 15 min at 37°C. Then, cells were washed with PBS and incubated in complete media for 15 min at 37°C. GFP-positive MDA-MB-231 cells were incubated with CellROX Deep Red Reagent (Invitrogen, C10422) at a final concentration of 5 µM in complete media for 30 min at 37°C. To evaluate ROS content, cells were washed, trypsined, resuspended in HBSS and analyzed by flow-cytometry.
Senescence-associated β-galactosidase staining.
To detect β-galactosidase, the senescence β-Galactosidase Staining Kit (Cell Signaling 9860) was used. Cells were plated into 6-well plates in complete media; after 24 h, the media was changed to DMEM 10% NuSerum. After 48 h, cells were washed with PBS and fixed for 15 min at room temperature with fixative solution (2% formaldehyde, 0.2% glutaraldehyde in PBS). Afterwards, cells were washed two times with PBS and incubated over night at 37°C in a dry incubator without CO2 with the β-galactosidase staining solution. Then, cells were observed under a microscope.
Senescence-associated β-galactosidase activity by flow cytometry.
The senescence β-Galactosidase Activity Kit (Invitrogen, F1930) was used according to the manufacturer’s instructions. Briefly, cells were seeded in 6-well plates in DMEM supplemented with 10% FBS and 1% PS. After 24 h, the media was changed to DMEM with 10% NuSerum. After 48 h, cells were trypsinezed, centrifuged and counted. Then, cells were resuspended with staining media to obtain 107 cells/mL, and 100 µl of samples were transferred to flow cytometer tubes and placed on ice. 100 µl of pre-warmed FDG solution (2 mM Fluorescein β-D-galactopyranoside) was added to the pre-warmed cells. Tubes were placed in the 37°C water bath for exactly 1 min. The reaction was stopped by adding ice-cold staining media. Cells were analyzed by flow cytometry.
Animal studies.
All in vivo studies were performed under National Institutes of Health (NIH) guidelines and were approved by the Institutional Animal Care and Use Committee (IACUC) of Thomas Jefferson University. Briefly, CTGF fibroblasts or control fibroblasts (300,000 cells) were co-injected with MDA-MB-231 (1 million cells) in the flanks of athymic NCr nude mice (NCRNU; Taconic Farms; 6–8 weeks of age). In other experiments, CTGF-MDA-MB-231 or control MDA-MB-231 (1 million cells) were injected in the flank of nude mice. Tumor size and weight was recorded over a 2–4 week period. Tumors were dissected out to determine weight and size using calipers. Tumor volume was calculated using the formula (x2y)/2, where x and y are the short and long tumor dimensions, respectively. Tumors were either fixed with 10% formalin or flash-frozen in liquid nitrogen-cooled isopentane.
Quantification of tumor angiogenesis.
Six µm tumor frozen sections were fixed with 4% paraformaldehyde in PBS for 10 min at 4°C and washed three times with PBS. A three-step biotin-streptavidin-horseradish peroxidase method was used for antibody detection. After fixation, the sections were blocked with 10% rabbit serum and incubated overnight at 4°C with rat monoclonal CD31 antibody (550274, BD Biosciences). The sections were then incubated with biotinylated rabbit anti-rat IgG (Vector Labs) and streptavidin-HRP (Dako). Immunoreactivity was revealed with 3,3'-diaminobenzidine. For quantitation of vessels, CD31-positive vessels were counted in 8–10 fields within the central area of each tumor using a 20x objective lens and an ocular grid (0.25 mm2 per field). The total numbers of vessel per unit area was calculated, and the data was represented graphically.
Immunohistochemistry.
Formalin-fixed paraffin-embedded tumor sections were de-paraffinized, rehydrated and washed in PBS. Antigen retrieval was performed in 10 mM sodium citrate, pH 6.0 for 10 min using a pressure cooker. After blocking with 3% hydrogen peroxide for 10 min, sections were incubated with 10% goat serum for 1 h. Then, sections were incubated with primary antibodies overnight at 4°C. Antibody binding was detected using a biotinylated secondary (Vector Labs) followed by strepavidin-HRP (Dako). Immunoreactivity was revealed using 3,3'-diaminobenzidine. Finally, samples were counter-stained with hematoxylin, dehydrated, mounted and observed under the microscope.
Immunofluorescence.
Cells were fixed with 2% PFA for 30 min, rinsed and permeabilized with 0.1% Triton X-100, 0.2% BSA in PBS for 10 min. To quench free aldehyde groups, cells were incubated with 25 mmol/L NH4Cl in PBS for 10 min. Then, cells were rinsed with PBS, incubated with ATPase-IF1 antibodies, followed by fluorescently labeled secondary antibodies. Samples were incubated with DAPI, mounted and analyzed using a confocal microscope.
Statistical analysis.
Data were analyzed with the Student t-test. p-values lower than 0.05 were considered statistically significant.
Acknowledgments
F.S. and her laboratory were supported by grants from the Breast Cancer Alliance (BCA) and the American Cancer Society (ACS). U.E.M. was supported by a Young Investigator Award from the Margaret Q. Landenberger Research Foundation. M.P.L. was supported by grants from the NIH/NCI (R01-CA-080250; R01-CA-098779; R01-CA-120876; R01-AR-055660), and the Susan G. Komen Breast Cancer Foundation. R.G.P. was supported by grants from the NIH/NCI (R01-CA-70896, R01-CA-75503, R01-CA-86072, and R01-CA-107382) and the Dr. Ralph and Marian C. Falk Medical Research Trust. The Kimmel Cancer Center was supported by the NIH/NCI Cancer Center Core grant P30-CA-56036 (to R.G.P.). Funds were also contributed by the Margaret Q. Landenberger Research Foundation (to M.P.L.). This project is funded, in part, under a grant with the Pennsylvania Department of Health (to M.P.L. and F.S.). The Department specifically disclaims responsibility for any analyses, interpretations or conclusions. This work was also supported, in part, by a Centre grant in Manchester from Breakthrough Breast Cancer in the UK (to A.H.) and an Advanced ERC Grant from the European Research Council.
Glossary
Abbreviations:
- Cav-1
caveolin-1
- CTGF
connective tissue growth factor
- HIF1α
hypoxia-inducible factor 1-alpha
- LDH
lactate dehydrogenase
- ROS
reactive oxygen species
- TGF-β
transforming growth factor beta
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/20717
References
- 1.Sotgia F, Del Galdo F, Casimiro MC, Bonuccelli G, Mercier I, Whitaker-Menezes D, et al. Caveolin-1-/- null mammary stromal fibroblasts share characteristics with human breast cancer-associated fibroblasts. Am J Pathol. 2009;174:746–61. doi: 10.2353/ajpath.2009.080658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Witkiewicz AK, Dasgupta A, Sotgia F, Mercier I, Pestell RG, Sabel M, et al. An absence of stromal caveolin-1 expression predicts early tumor recurrence and poor clinical outcome in human breast cancers. Am J Pathol. 2009;174:2023–34. doi: 10.2353/ajpath.2009.080873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sloan EK, Ciocca DR, Pouliot N, Natoli A, Restall C, Henderson MA, et al. Stromal cell expression of caveolin-1 predicts outcome in breast cancer. Am J Pathol. 2009;174:2035–43. doi: 10.2353/ajpath.2009.080924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8:3984–4001. doi: 10.4161/cc.8.23.10238. [DOI] [PubMed] [Google Scholar]
- 5.Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle. 2010;9:3256–76. doi: 10.4161/cc.9.16.12553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Loss of stromal caveolin-1 leads to oxidative stress, mimics hypoxia and drives inflammation in the tumor microenvironment, conferring the “reverse Warburg effect”: a transcriptional informatics analysis with validation. Cell Cycle. 2010;9:2201–19. doi: 10.4161/cc.9.11.11848. [DOI] [PubMed] [Google Scholar]
- 7.Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Daumer KM, Milliman JN, Chiavarina B, et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle. 2010;9:2423–33. doi: 10.4161/cc.9.12.12048. [DOI] [PubMed] [Google Scholar]
- 8.Chiavarina B, Whitaker-Menezes D, Migneco G, Martinez-Outschoorn UE, Pavlides S, Howell A, et al. HIF1-alpha functions as a tumor promoter in cancer associated fibroblasts, and as a tumor suppressor in breast cancer cells: Autophagy drives compartment-specific oncogenesis. Cell Cycle. 2010;9:3534–51. doi: 10.4161/cc.9.17.12908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Surmann-Schmitt C, Sasaki T, Hattori T, Eitzinger N, Schett G, von der Mark K, et al. The Wnt antagonist Wif-1 interacts with CTGF and inhibits CTGF activity. J Cell Physiol. 2012;227:2207–16. doi: 10.1002/jcp.22957. [DOI] [PubMed] [Google Scholar]
- 10.Levine JH, Moses HL, Gold LI, Nanney LB. Spatial and temporal patterns of immunoreactive transforming growth factor beta 1, beta 2, and beta 3 during excisional wound repair. Am J Pathol. 1993;143:368–80. [PMC free article] [PubMed] [Google Scholar]
- 11.Peltonen J, Kähäri L, Jaakkola S, Kähäri VM, Varga J, Uitto J, et al. Evaluation of transforming growth factor beta and type I procollagen gene expression in fibrotic skin diseases by in situ hybridization. J Invest Dermatol. 1990;94:365–71. doi: 10.1111/1523-1747.ep12874491. [DOI] [PubMed] [Google Scholar]
- 12.Smith EA, LeRoy EC. A possible role for transforming growth factor-beta in systemic sclerosis. J Invest Dermatol. 1990;95(Suppl):125S–7S. doi: 10.1111/1523-1747.ep12874998. [DOI] [PubMed] [Google Scholar]
- 13.Frazier K, Williams S, Kothapalli D, Klapper H, Grotendorst GR. Stimulation of fibroblast cell growth, matrix production, and granulation tissue formation by connective tissue growth factor. J Invest Dermatol. 1996;107:404–11. doi: 10.1111/1523-1747.ep12363389. [DOI] [PubMed] [Google Scholar]
- 14.Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Transcriptional evidence for the “Reverse Warburg Effect” in human breast cancer tumor stroma and metastasis: similarities with oxidative stress, inflammation, Alzheimer’s disease, and “Neuron-Glia Metabolic Coupling”. Aging (Albany NY) 2010;2:185–99. doi: 10.18632/aging.100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bork P. The modular architecture of a new family of growth regulators related to connective tissue growth factor. FEBS Lett. 1993;327:125–30. doi: 10.1016/0014-5793(93)80155-N. [DOI] [PubMed] [Google Scholar]
- 16.Friedrichsen S, Heuer H, Christ S, Winckler M, Brauer D, Bauer K, et al. CTGF expression during mouse embryonic development. Cell Tissue Res. 2003;312:175–88. doi: 10.1007/s00441-003-0712-6. [DOI] [PubMed] [Google Scholar]
- 17.Igarashi A, Okochi H, Bradham DM, Grotendorst GR. Regulation of connective tissue growth factor gene expression in human skin fibroblasts and during wound repair. Mol Biol Cell. 1993;4:637–45. doi: 10.1091/mbc.4.6.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Babic AM, Chen CC, Lau LF. Fisp12/mouse connective tissue growth factor mediates endothelial cell adhesion and migration through integrin alphavbeta3, promotes endothelial cell survival, and induces angiogenesis in vivo. Mol Cell Biol. 1999;19:2958–66. doi: 10.1128/mcb.19.4.2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Safadi FF, Xu J, Smock SL, Kanaan RA, Selim AH, Odgren PR, et al. Expression of connective tissue growth factor in bone: its role in osteoblast proliferation and differentiation in vitro and bone formation in vivo. J Cell Physiol. 2003;196:51–62. doi: 10.1002/jcp.10319. [DOI] [PubMed] [Google Scholar]
- 20.Leask A, Denton CP, Abraham DJ. Insights into the molecular mechanism of chronic fibrosis: the role of connective tissue growth factor in scleroderma. J Invest Dermatol. 2004;122:1–6. doi: 10.1046/j.0022-202X.2003.22133.x. [DOI] [PubMed] [Google Scholar]
- 21.Wenger C, Ellenrieder V, Alber B, Lacher U, Menke A, Hameister H, et al. Expression and differential regulation of connective tissue growth factor in pancreatic cancer cells. Oncogene. 1999;18:1073–80. doi: 10.1038/sj.onc.1202395. [DOI] [PubMed] [Google Scholar]
- 22.Kubo M, Kikuchi K, Nashiro K, Kakinuma T, Hayashi N, Nanko H, et al. Expression of fibrogenic cytokines in desmoplastic malignant melanoma. Br J Dermatol. 1998;139:192–7. doi: 10.1046/j.1365-2133.1998.02354.x. [DOI] [PubMed] [Google Scholar]
- 23.Steffen CL, Ball-Mirth DK, Harding PA, Bhattacharyya N, Pillai S, Brigstock DR. Characterization of cell-associated and soluble forms of connective tissue growth factor (CTGF) produced by fibroblast cells in vitro. Growth Factors. 1998;15:199–213. doi: 10.3109/08977199809002117. [DOI] [PubMed] [Google Scholar]
- 24.Yang DH, Kim HS, Wilson EM, Rosenfeld RG, Oh Y. Identification of glycosylated 38-kDa connective tissue growth factor (IGFBP-related protein 2) and proteolytic fragments in human biological fluids, and up-regulation of IGFBP-rP2 expression by TGF-beta in Hs578T human breast cancer cells. J Clin Endocrinol Metab. 1998;83:2593–6. doi: 10.1210/jc.83.7.2593. [DOI] [PubMed] [Google Scholar]
- 25.Vorwerk P, Wex H, Hohmann B, Oh Y, Rosenfeld RG, Mittler U. CTGF (IGFBP-rP2) is specifically expressed in malignant lymphoblasts of patients with acute lymphoblastic leukaemia (ALL) Br J Cancer. 2000;83:756–60. doi: 10.1054/bjoc.2000.1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barbolina MV, Adley BP, Kelly DL, Shepard J, Fought AJ, Scholtens D, et al. Downregulation of connective tissue growth factor by three-dimensional matrix enhances ovarian carcinoma cell invasion. Int J Cancer. 2009;125:816–25. doi: 10.1002/ijc.24347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin BR, Chang CC, Chen RJ, Jeng YM, Liang JT, Lee PH, et al. Connective tissue growth factor acts as a therapeutic agent and predictor for peritoneal carcinomatosis of colorectal cancer. Clin Cancer Res. 2011;17:3077–88. doi: 10.1158/1078-0432.CCR-09-3256. [DOI] [PubMed] [Google Scholar]
- 28.Yang MH, Lin BR, Chang CH, Chen ST, Lin SK, Kuo MY, et al. Connective tissue growth factor modulates oral squamous cell carcinoma invasion by activating a miR-504/FOXP1 signalling. Oncogene. 2012;31:2401–11. doi: 10.1038/onc.2011.423. [DOI] [PubMed] [Google Scholar]
- 29.Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFκB activation in the tumor stromal microenvironment. Cell Cycle. 2010;9:3515–33. doi: 10.4161/cc.9.17.12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle. 2010;9:3506–14. doi: 10.4161/cc.9.17.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinez-Outschoorn UE, Prisco M, Ertel A, Tsirigos A, Lin Z, Pavlides S, et al. Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer: achieving personalized medicine via Metabolo-Genomics. Cell Cycle. 2011;10:1271–86. doi: 10.4161/cc.10.8.15330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000;275:25130–8. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 34.Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, et al. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem. 1996;271:32529–37. doi: 10.1074/jbc.271.51.32529. [DOI] [PubMed] [Google Scholar]
- 35.Semenza GL. Hypoxia-inducible factor 1: regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim Biophys Acta. 2011;1813:1263–8. doi: 10.1016/j.bbamcr.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen J, Xavier S, Moskowitz-Kassai E, Chen R, Lu CY, Sanduski K, et al. Cathepsin cleavage of sirtuin 1 in endothelial progenitor cells mediates stress-induced premature senescence. Am J Pathol. 2012;180:973–83. doi: 10.1016/j.ajpath.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Phanish MK, Winn SK, Dockrell ME. Connective tissue growth factor-(CTGF, CCN2)–a marker, mediator and therapeutic target for renal fibrosis. Nephron, Exp Nephrol. 2010;114:e83–92. doi: 10.1159/000262316. [DOI] [PubMed] [Google Scholar]
- 39.Brigstock DR. The connective tissue growth factor/cysteine-rich 61/nephroblastoma overexpressed (CCN) family. Endocr Rev. 1999;20:189–206. doi: 10.1210/er.20.2.189. [DOI] [PubMed] [Google Scholar]
- 40.Razani B, Zhang XL, Bitzer M, von Gersdorff G, Böttinger EP, Lisanti MP. Caveolin-1 regulates transforming growth factor (TGF)-beta/SMAD signaling through an interaction with the TGF-beta type I receptor. J Biol Chem. 2001;276:6727–38. doi: 10.1074/jbc.M008340200. [DOI] [PubMed] [Google Scholar]
- 41.Del Galdo F, Lisanti MP, Jimenez SA. Caveolin-1, transforming growth factor-beta receptor internalization, and the pathogenesis of systemic sclerosis. Curr Opin Rheumatol. 2008;20:713–9. doi: 10.1097/BOR.0b013e3283103d27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Del Galdo F, Sotgia F, de Almeida CJ, Jasmin JF, Musick M, Lisanti MP, et al. Decreased expression of caveolin 1 in patients with systemic sclerosis: crucial role in the pathogenesis of tissue fibrosis. Arthritis Rheum. 2008;58:2854–65. doi: 10.1002/art.23791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pavlides S, Vera I, Gandara R, Sneddon S, Pestell RG, Mercier I, et al. Warburg Meets Autophagy: Cancer-Associated Fibroblasts Accelerate Tumor Growth and Metastasis via Oxidative Stress, Mitophagy, and Aerobic Glycolysis. Antioxid Redox Signal. 2012;16:1264–84. doi: 10.1089/ars.2011.4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frazier KS, Grotendorst GR. Expression of connective tissue growth factor mRNA in the fibrous stroma of mammary tumors. Int J Biochem Cell Biol. 1997;29:153–61. doi: 10.1016/S1357-2725(96)00127-6. [DOI] [PubMed] [Google Scholar]
- 45.Xie D, Nakachi K, Wang H, Elashoff R, Koeffler HP. Elevated levels of connective tissue growth factor, WISP-1, and CYR61 in primary breast cancers associated with more advanced features. Cancer Res. 2001;61:8917–23. [PubMed] [Google Scholar]
- 46.Chien W, O’Kelly J, Lu D, Leiter A, Sohn J, Yin D, et al. Expression of connective tissue growth factor (CTGF/CCN2) in breast cancer cells is associated with increased migration and angiogenesis. Int J Oncol. 2011;38:1741–7. doi: 10.3892/ijo.2011.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lai D, Ho KC, Hao Y, Yang X. Taxol resistance in breast cancer cells is mediated by the hippo pathway component TAZ and its downstream transcriptional targets Cyr61 and CTGF. Cancer Res. 2011;71:2728–38. doi: 10.1158/0008-5472.CAN-10-2711. [DOI] [PubMed] [Google Scholar]
- 48.Wang MY, Chen PS, Prakash E, Hsu HC, Huang HY, Lin MT, et al. Connective tissue growth factor confers drug resistance in breast cancer through concomitant up-regulation of Bcl-xL and cIAP1. Cancer Res. 2009;69:3482–91. doi: 10.1158/0008-5472.CAN-08-2524. [DOI] [PubMed] [Google Scholar]
- 49.Jiang WG, Watkins G, Fodstad O, Douglas-Jones A, Mokbel K, Mansel RE. Differential expression of the CCN family members Cyr61, CTGF and Nov in human breast cancer. Endocr Relat Cancer. 2004;11:781–91. doi: 10.1677/erc.1.00825. [DOI] [PubMed] [Google Scholar]
- 50.Hishikawa K, Oemar BS, Tanner FC, Nakaki T, Lüscher TF, Fujii T. Connective tissue growth factor induces apoptosis in human breast cancer cell line MCF-7. J Biol Chem. 1999;274:37461–6. doi: 10.1074/jbc.274.52.37461. [DOI] [PubMed] [Google Scholar]
- 51.Nishida T, Kondo S, Maeda A, Kubota S, Lyons KM, Takigawa M. CCN family 2/connective tissue growth factor (CCN2/CTGF) regulates the expression of Vegf through Hif-1alpha expression in a chondrocytic cell line, HCS-2/8, under hypoxic condition. Bone. 2009;44:24–31. doi: 10.1016/j.bone.2008.08.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu T, Finkel T. Free radicals and senescence. Exp Cell Res. 2008;314:1918–22. doi: 10.1016/j.yexcr.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Campisi J. Cancer, aging and cellular senescence. In Vivo. 2000;14:183–8. [PubMed] [Google Scholar]
- 54.Campisi J. Aging and cancer: the double-edged sword of replicative senescence. J Am Geriatr Soc. 1997;45:482–8. doi: 10.1111/j.1532-5415.1997.tb05175.x. [DOI] [PubMed] [Google Scholar]
- 55.Smith JR, Pereira-Smith OM. Replicative senescence: implications for in vivo aging and tumor suppression. Science. 1996;273:63–7. doi: 10.1126/science.273.5271.63. [DOI] [PubMed] [Google Scholar]
- 56.Campisi J. Replicative senescence: an old lives’ tale? Cell. 1996;84:497–500. doi: 10.1016/S0092-8674(00)81023-5. [DOI] [PubMed] [Google Scholar]
- 57.Rinehart CA, Torti VR. Aging and cancer: the role of stromal interactions with epithelial cells. Mol Carcinog. 1997;18:187–92. doi: 10.1002/(SICI)1098-2744(199704)18:4<187::AID-MC1>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 58.Martinez-Outschoorn UE, Whitaker-Menezes D, Lin Z, Flomenberg N, Howell A, Pestell RG, et al. Cytokine production and inflammation drive autophagy in the tumor microenvironment: role of stromal caveolin-1 as a key regulator. Cell Cycle. 2011;10:1784–93. doi: 10.4161/cc.10.11.15674. [DOI] [PMC free article] [PubMed] [Google Scholar]