

Abstract
Senescent fibroblasts are known to promote tumor growth. However, the exact mechanism remains largely unknown. An important clue comes from recent studies linking autophagy with the onset of senescence. Thus, autophagy and senescence may be part of the same physiological process, known as the autophagy-senescence transition (AST). To test this hypothesis, human fibroblasts immortalized with telomerase (hTERT-BJ1) were stably transfected with autophagy genes (BNIP3, CTSB or ATG16L1). Their overexpression was sufficient to induce a constitutive autophagic phenotype, with features of mitophagy, mitochondrial dysfunction and a shift toward aerobic glycolysis, resulting in L-lactate and ketone body production. Autophagic fibroblasts also showed features of senescence, with increased p21(WAF1/CIP1), a CDK inhibitor, cellular hypertrophy and increased β-galactosidase activity. Thus, we genetically validated the existence of the autophagy-senescence transition. Importantly, autophagic-senescent fibroblasts promoted tumor growth and metastasis, when co-injected with human breast cancer cells, independently of angiogenesis. Autophagic-senescent fibroblasts stimulated mitochondrial metabolism in adjacent cancer cells, when the two cell types were co-cultured, as visualized by MitoTracker staining. In particular, autophagic ATG16L1 fibroblasts, which produced large amounts of ketone bodies (3-hydroxy-butyrate), had the strongest effects and promoted metastasis by up to 11-fold. Conversely, expression of ATG16L1 in epithelial cancer cells inhibited tumor growth, indicating that the effects of autophagy are compartment-specific. Thus, autophagic-senescent fibroblasts metabolically promote tumor growth and metastasis, by paracrine production of high-energy mitochondrial fuels. Our current studies provide genetic support for the importance of “two-compartment tumor metabolism” in driving tumor growth and metastasis via a simple energy transfer mechanism. Finally, β-galactosidase, a known lysosomal enzyme and biomarker of senescence, was localized to the tumor stroma in human breast cancer tissues, providing in vivo support for our hypothesis. Bioinformatic analysis of genome-wide transcriptional profiles from tumor stroma, isolated from human breast cancers, also validated the onset of an autophagy-senescence transition. Taken together, these studies establish a new functional link between host aging, autophagy, the tumor microenvironment and cancer metabolism.
Keywords: ATG16L1, BNIP3, BNIP3L, Beclin1, autophagy, cancer metabolism, cancer-associated fibroblasts, cathepsin B, glycolysis, senescence, tumor stroma
Introduction
We previously proposed a new paradigm to explain the role of autophagy in cancer metabolism.1,2 In this model, tumor cells secrete hydrogen peroxide, which induces oxidative stress in adjacent cancer-associated fibroblasts.3,4 Oxidative stress, in turn, drives the onset of autophagy in cancer-associated fibroblasts, resulting in mitophagy, mitochondrial dysfunction and a shift toward aerobic glycolysis.5,6 Autophagy and mitochondrial dysfunction induces a catabolic state in cancer-associated fibroblasts, resulting in the production of high-energy mitochondrial fuels (such as L-lactate, ketone bodies, glutamine, and free fatty acids).7 These mitochondrial fuels are then transferred to epithelial cancer cells to satisfy their high-energy demands, driving anabolic tumor growth.8 To take advantage of this increased fuel supply, cancer cells amplify their capacity for OXPHOS and oxidative mitochondrial metabolism.9,10 Thus, cancer cells and the tumor stroma are metabolically coupled, resulting in a form of “parasitic” cancer metabolism.7 We have termed this new paradigm (1) the “autophagic tumor stroma model of cancer”11-13 or, more recently, (2) “two-compartment tumor metabolism”14.
This model has important clinical relevance, since protein biomarkers for autophagy and oxidative stress in the tumor stroma effectively predict early tumor recurrence, metastasis, drug-resistance and poor overall survival in human breast cancer patients.15-20 Similar results were also obtained with prostate cancer21 and melanoma patients,22 linking “two-compartment tumor metabolism” with metastasis.
Here, we set out to stringently test the “autophagic tumor stroma model of cancer.” For this purpose, we designed a functional genetic approach to validate the role of stromal autophagy in promoting tumor growth and metastasis. For this purpose, we created a genetically tractable model using hTERT-immortalized human fibroblasts and MDA-MB-231 human breast cancer cells. This compartment-specific approach allowed us to genetically modify the capacity for either stromal fibroblasts or epithelial cancer cells to undergo autophagy and mitophagy. More specifically, we show that genetically engineered autophagic fibroblasts promote tumor growth and metastasis, while constitutive activation of autophagy in cancer cells effectively reduces tumor growth.
Importantly, these constitutively autophagic fibroblasts show a loss of Cav-1 protein expression, consistent with previous studies showing that a loss of stromal Cav-1 is a prognostic biomarker for a “lethal tumor microenvironment”19. Thus, loss of stromal Cav-1 reflects its autophagic digestion by lysosomes, providing a sensitive biomarker for autophagy in the tumor stroma.5,6,19,23
Our current studies also provide new links between autophagy and senescence in cancer pathogenesis. For example, it is now well-accepted that senescent fibroblasts promote tumor growth,24,25 but the mechanism(s) underlying this phenomenon,26 and their physiological relevance, has remained elusive.
Autophagy and senescence may be part of the same biological phenomenon.27,28 In support of this notion, many of the same stimuli that induce autophagy (such as hydrogen peroxide and oxidative stress) also induce senescence,29,30 and myofibroblast differentation.31 This has implications for understanding the metabolic importance of cancer-associated myofibroblasts in tumor growth.
Interestingly, senescent fibroblasts show a progressive increase in lysosomal mass32-34 and a shift toward aerobic glycolysis.35,36 Thus, this senescence phenotype may be secondary to the induction of autophagy and mitophagy, resulting in mitochondrial dysfunction. In further support of this notion, β-galactosidase, an enzyme marker which is currently used as the gold standard for measuring the induction of senescence, is a known lysosomal enzyme.37 Other recent functional studies also directly show that autophagic cells may be more sensitive to the induction of senescence.38-44
We suggest here that this process could be termed the “autophagy-senescence transition (AST),” to better reflect that these two seemingly separate biological phenomena, may be part of a continuum.
In further support of this notion, here we demonstrate that genetically engineered autophagic fibroblasts, with tumor- and metastasis-promoting activity, also show many characteristics of senescence. Specifically, these autophagic-catabolic fibroblasts show increased expression of a CDK inhibitor [namely p21(WAF1/CIP1)], cellular hypertrophy (increased protein mass per cell) and elevated levels of β-galactosidase. Similarly, p21 has been implicated in driving myofibroblast differentiation in cancer-associated fibroblasts45 as well as in the induction of both autophagy and senescence.46
In summary, stromal autophagy and senescence, which reflects catabolism in the tumor microenvironment, may metabolically promote the anabolic growth of cancer cells. Thus, new approaches to target the autophagy-senescence transition could have important implications for preventing tumor growth and metastasis.
Since senescence is thought to reflect biological aging, our studies on autophagy-induced senescence may also explain why cancer incidence dramatically increases with advanced chronological age, by providing a “fertile soil” or metabolically permissive catabolic microenvironment, to support the anabolic growth of “needy” cancer cells.14
Results
Fibroblasts overexpressing BNIP3 show a loss of Cav-1 expression with constitutive activation of the autophagic program
BNIP3 and BNIP3L are two members of the BH3-only subfamily of Bcl-2 family proteins; these proteins form stable homo-dimeric complexes that localize to the outer membrane of the mitochondria after cellular stress. This promotes either apoptosis or the induction of autophagy and/or mitophagy.50
To evaluate the role of BNIP3 and stromal autophagy in tumor development, BNIP3 was stably overexpressed in hTERT fibroblasts. Figure 1A shows that BNIP3 overexpression in fibroblasts is sufficient to cause a marked reduction in caveolin-1 (Cav-1) protein expression. Importantly, loss of stromal Cav-1 expression predicts early tumor recurrence, metastasis, drug-resistance and poor overall clinical outcome in human breast cancer patients.16-20

Figure 1. Fibroblasts overexpressing BNIP3 show a loss of Cav-1 expression, with constitutive activation of the autophagic program. BNIP3 was stably overexpressed in hTERT fibroblasts via transduction with lenti-viral vectors. Lv- represents fibroblasts transduced with the vector alone control, namely Lv-105 (puro). (A) Note that BNIP3 overexpression in fibroblasts is sufficient to cause a dramatic reduction in caveolin-1 (Cav-1) protein expression, seen by immunoblot analysis. (B) Similarly, BNIP3 overexpression drives the upregulation of LC3-I and -II and cathepsin B, under basal cell culture conditions.(C) Overnight starvation drives even stronger expression of BNIP3, as well as Beclin1 and ATG16L1, which are other key markers of autophagy. Thus, BNIP3 overexpression in fibroblasts is indeed sufficient to drive a loss of Cav-1 expression via the activation on an autophagic program. In all three panels, immunoblotting with β-actin is shown as a control for equal protein loading.
Previous studies have shown that Cav-1 loss is mediated via lysosomal degradation, during autophagy.5,6,51 Consistent with this observation, Figures 1B and 1C show that BNIP3 overexpression drives the upregulation of LC3-I and -II and cathepsin B (key markers of autophagy and lysosomes) under basal cell culture conditions. Furthermore, overnight starvation drives even stronger expression of BNIP3 as well as Beclin1 and ATG16L1, which are other key markers of autophagy. Thus, BNIP3 overexpression in fibroblasts is indeed sufficient to drive a loss of Cav-1 expression via the activation on an autophagic program.
In support of our current findings, previous studies have demonstrated that ectopic expression of BNIP3 is sufficient to trigger autophagy and mitophagy.52
Fibroblasts overexpressing BNIP3 enhance tumor growth without a measurable increase in angiogenesis
To assess the functional in vivo effects of BNIP3 overexpression in fibroblasts, we next employed a mouse xenograft model. MDA-MB-231 breast cancer cells were co-injected with BNIP3 fibroblasts or vector-alone control fibroblasts into the flanks of athymic nude mice. Figure 2A shows that at 4 weeks post-injection, BNIP3 fibroblasts promoted an ~2.1-fold increase in tumor growth, as compared with vector-alone control fibroblasts. However, Figure 2B shows no differences in tumor neo-vascularization. Therefore, we conclude that autophagic BNIP3 fibroblasts are sufficient to promote tumor growth without increased angiogenesis, further validating a crucial role for autophagic stroma in tumor development.
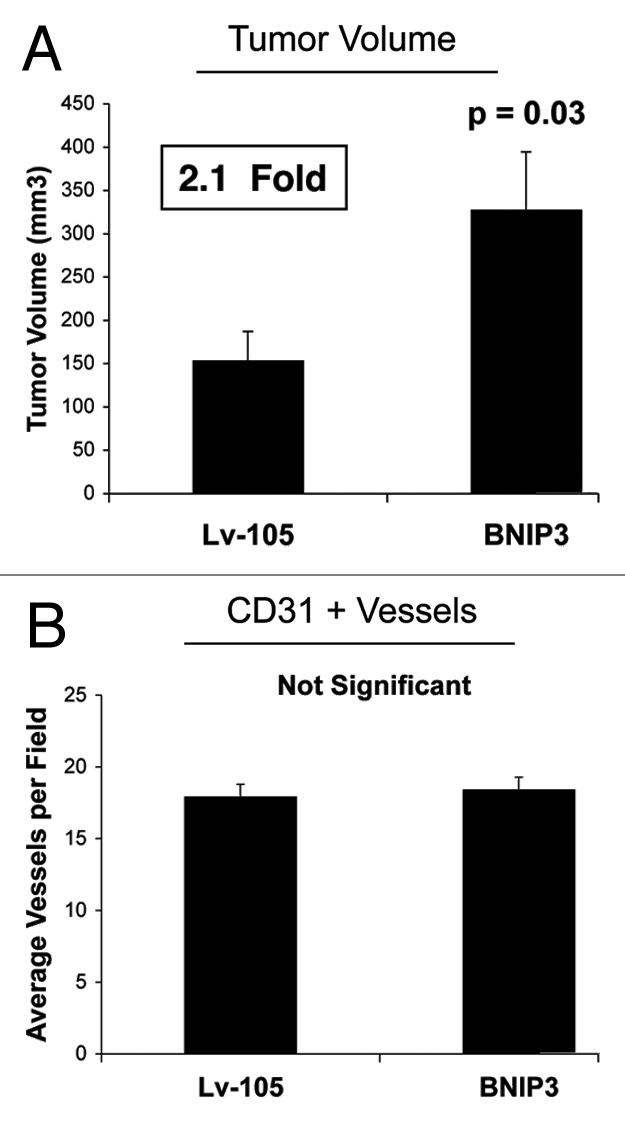
Figure 2. Fibroblasts overexpressing BNIP3 promote tumor growth without a measurable increase in angiogenesis. To assess the functional in vivo effects of BNIP3 overexpression in fibroblasts, we employed a mouse xenograft model. Briefly, MDA-MB-231 breast cancer cells were co-injected with BNIP3 fibroblasts or vector alone control fibroblasts, into the flanks of nude mice. n = 10 per experimental group. Lv-105 represents fibroblasts transduced with the vector alone control, namely Lv-105 (puro). (A) Note that at 4 weeks post-injection, BNIP3 fibroblasts promoted an ~2.1-fold increase in tumor growth. (B) However, no significant differences in tumor neo-vascularization were observed. Thus, autophagic BNIP3 fibroblasts are sufficient to drive tumor growth, without increased angiogenesis.
BNIP3L and Beclin1 overexpression in fibroblasts drives a loss of Cav-1 expression, and promotes tumor growth
As mentioned above, BNIP3L with BNIP3 are members of the Bcl-2 protein family. BNIP3L and BNIP3 are sufficient to initiate the autophagic process, even in the absence of nutrient and oxygen deprivation.53 Like BNIP3, the mechanism proposed for BNIP3L involves the ability of its BH3-domain to displace Beclin1 from the Bcl-2- or Bcl-XL-Beclin1 complexes. Beclin1 is then capable of triggering autophagy, since it is a key regulatory protein in phagophore formation.53
To investigate the involvement of stromal BNIP3L and Beclin1 in tumor formation, we stably overexpressed both genes in hTERT fibroblasts. Figure 3A shows that BNIP3L and Beclin1 overexpression in fibroblasts is sufficient to induce a loss of Cav-1 expression, as predicted.
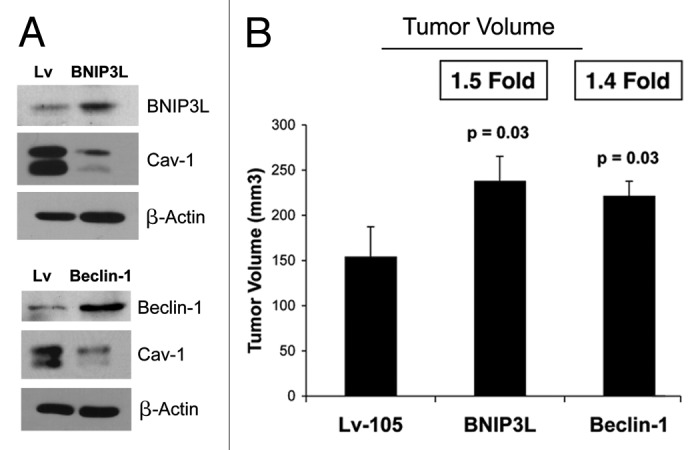
Figure 3. BNIP3L and Beclin1 overexpression in fibroblasts drives a loss of Cav-1 expression, and promotes tumor growth. To investigate the involvement of stromal BNIP3L and Beclin1 in tumor formation, we stably overexpressed both genes in hTERT fibroblasts. Lv- represents fibroblasts transduced with the vector alone control, namely Lv-105 (puro). (A) Note that BNIP3L and Beclin1 overexpression in fibroblasts is sufficient to induce a loss of Cav-1 expression, as observed by immunoblot analysis. Blotting with β-actin is shown as a control for equal protein loading. (B) BNIP3-, Beclin1- or control- fibroblasts were co-injected with MDA-MB-231 epithelial breast cancer cells, into the flanks of nude mice. At 4 weeks post-injection, the mice were sacrificed and the tumors were collected. n = 10 per experimental group. For both BNIP3L- and Beclin1 fibroblasts, a significant increase in tumor growth was observed. Thus, overexpression of both autophagic genes (BNIP3L and Beclin1) is sufficient to drive tumor growth.
In vivo studies were also performed to evaluate if BNIP3L and/or Beclin1 overexpression affects tumor growth. BNIP3, Beclin1 or control fibroblasts were co-injected with MDA-MB-231 epithelial breast cancer cells into the flanks of nude mice. At 4 weeks post-injection, the mice were sacrificed, and the tumors were collected. Figure 3B shows a significant increase in tumor growth of 1.5- and 1.4-fold for BNIP3L and Beclin1, respectively.
Fibroblasts overexpressing Cathepsin B (CTSB) show a loss of Cav-1 expression, with constitutive activation of the autophagic program, and enhanced tumor growth
Cathepsin B is a cysteine protease involved in autophagy that is located primarily within bona fide lysosomes and lysosome-like organelles, where it functions in the degradation and/or processing of lysosomal proteins.54 To investigate if stromal cathepsin B expression plays a significant functional role in breast cancer pathogenesis, we next overexpressed the CTSB gene in fibroblasts, via lenti-viral transduction. Note that both the CTSB-precursor and activated-cleaved form were observed by immunoblot analysis, in stably transfected hTERT fibroblasts (Fig. 4A).

Figure 4. Fibroblasts overexpressing Cathepsin B (CTSB) show a loss of Cav-1 expression, with constitutive activation of the autophagic program. (A) To investigate if stromal cathepsin B expression plays a significant functional role in breast cancer pathogenesis, we overexpressed the CTSB gene in fibroblasts, via lenti-viral transduction. Note that both the CTSB-precursor and activated-cleaved form were observed by immunoblot analysis, in stably transfected hTERT fibroblasts. (B) Cathepsin B expression was also evaluated after 12h and 16h of starvation (in Hepes-buffered HBSS). Note that CTSB continued to accumulate during starvation, consistent with its stabilization within lysosomes, during autophagy. (C) We also evaluated Cav-1 expression after overnight starvation. As expected, we detected a strong reduction in Cav-1 protein levels, validating the connection between autophagy and loss of Cav-1 protein expression. (D) Immunoblot analysis of CTSB fibroblasts demonstrated that cathepsin B overexpression is sufficient to drive the induction of autophagy. Note that cathepsin B overexpression strongly induces several autophagy/mitophagy markers (BNIP3, Lamp1, Beclin1 and ATG16L1). In all panels, β-actin expression was assessed as a control for equal protein loading. Lv- represents fibroblasts transduced with the vector alone control, namely Lv-105 (puro).
We also evaluated CTSB expression after nutrient starvation, a strong natural inducer of autophagy. Figure 4B shows that CTSB continued to accumulate during starvation, consistent with its stabilization within lysosomes during autophagy. Thus, we next evaluated Cav-1 expression after overnight starvation. As expected, we detected a strong reduction in Cav-1 protein levels (Fig. 4C), again validating the connection between autophagy induction and loss of Cav-1 protein expression.
Immunoblot analysis of CTSB fibroblasts demonstrated that cathepsin B overexpression is sufficient to drive the induction of autophagy. Indeed, Figure 4D shows that cathepsin B overexpression strongly induces several autophagy/mitophagy markers (BNIP3, Lamp1, Beclin1 and ATG16L1).
Finally, the in vivo functional effects of stromal CTSB were evaluated using a xenograft model. Briefly, MDA-MB-231 cells were co-injected with CTSB fibroblasts or vector-alone control fibroblasts into the flanks of nude mice. Figure 5A shows that CTSB fibroblasts promoted a significant ~2.1-fold increase in tumor growth as compared with control fibroblasts. However, quantitation of neo-vascularization, via immunostaining with CD31, did not show any significant increases in tumor angiogenesis (Fig. 5B). Thus, autophagy in tumor stroma can promote tumor growth independently of angiogenesis.
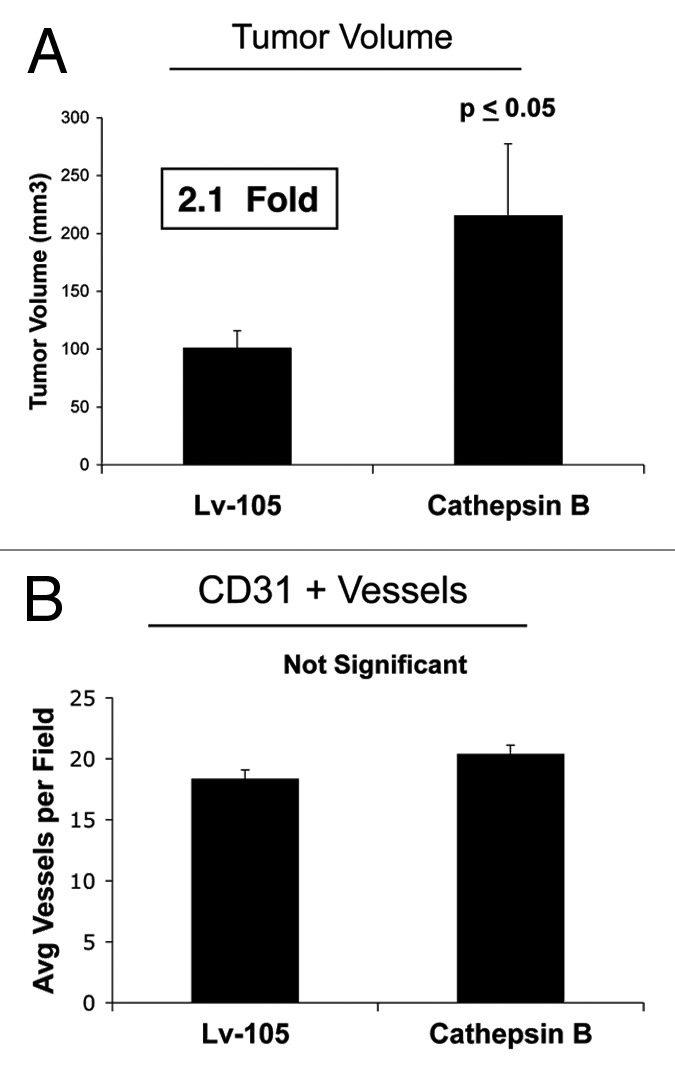
Figure 5. Fibroblasts overexpressing Cathepsin B (CTSB) drive enhanced tumor growth, without a measurable increase in angiogenesis.(A) The in vivo functional effects of stromal CTSB were evaluated using a xenograft model. MDA-MB-231 cells were co-injected with CTSB fibroblasts or vector alone control fibroblasts, into the flanks of nude mice. Note that CTSB fibroblasts promoted a significant ~2.1-fold increase in tumor growth, as compared with control fibroblasts. n = 10 per experimental group. Lv-105 represents fibroblasts transduced with the vector alone control, namely Lv-105 (puro). (B) Quantitation of neo-vascularization, via immunostaining with CD31, did not show any significant increases in tumor angiogenesis. Thus, autophagy in the tumor stroma can promote tumor growth, independently of angiogenesis.
Fibroblasts overexpressing ATG16L1 show constitutive activation of the autophagic program and enhanced tumor growth
ATG16L1 is a key molecular component of a large protein complex essential for autophagy.55 It specifically functions as a clathrin adaptor protein during autophagy, thereby facilitating the recruitment of plasma membrane, driving autophagosome formation via endocytosis.56,57
To assess the functional role of stromal ATG16L1 in breast cancer development, we stably overexpressed ATG16L1 in hTERT fibroblasts (Fig. 6A). We also analyzed the induction of a panel of autophagy and mitophagy markers. Figure 6B and C show that ATG16L1 overexpression induces the upregulation of Beclin1 and Lamp1, under basal cell culture conditions. Similarly, increases in cathepsin B and LC3 were also observed after overnight starvation.
Figure 6.
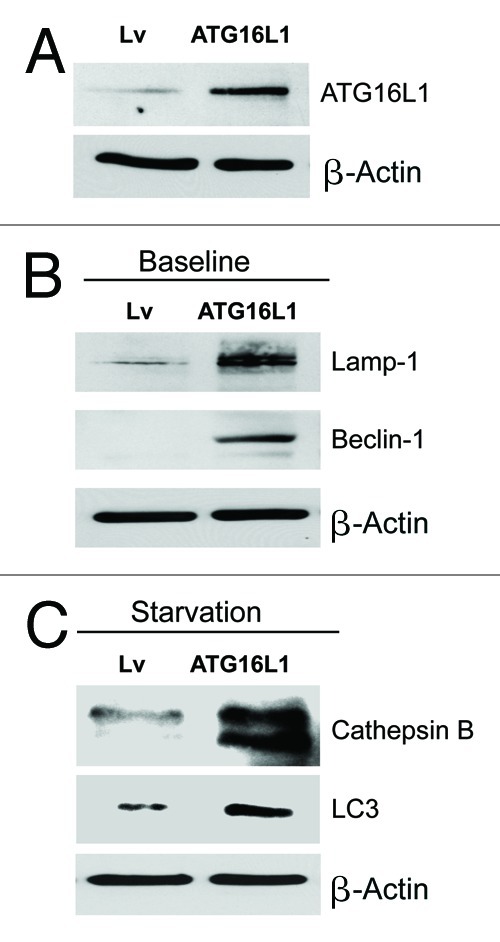
Fibroblasts overexpressing ATG16L1 show constitutive activation of the autophagic program. (A) ATG16L1 functions as a clathrin adaptor protein during autophagy, facilitating the recruitment of plasma membrane, by driving autophagosome formation via endocytosis. To assess the functional role of stromal ATG16L1 in breast cancer development, we stably overexpressed ATG16L1 in hTERT fibroblasts. (B) Note that ATG16L1 overexpression induces the upregulation of Beclin1 and Lamp1, under basal cell culture conditions. (C) Similarly, increases in cathepsin B and LC3 were also observed after overnight starvation (in Hepes-buffered HBSS). In all three panels, β-actin expression was assessed as a control for equal protein loading. Lv- represents fibroblasts transduced with the vector alone control, namely Lv-105 (puro).
In vivo, ATG16L1-fibrolasts also promoted tumor growth, resulting in an ~1.6-fold increase in tumor volume relative to control fibroblasts (Fig. 7A). Again, the increases observed in tumor growth were independent of tumor angiogenesis (Fig. 7B), as we previously observed for BNIP3, and CTSB.
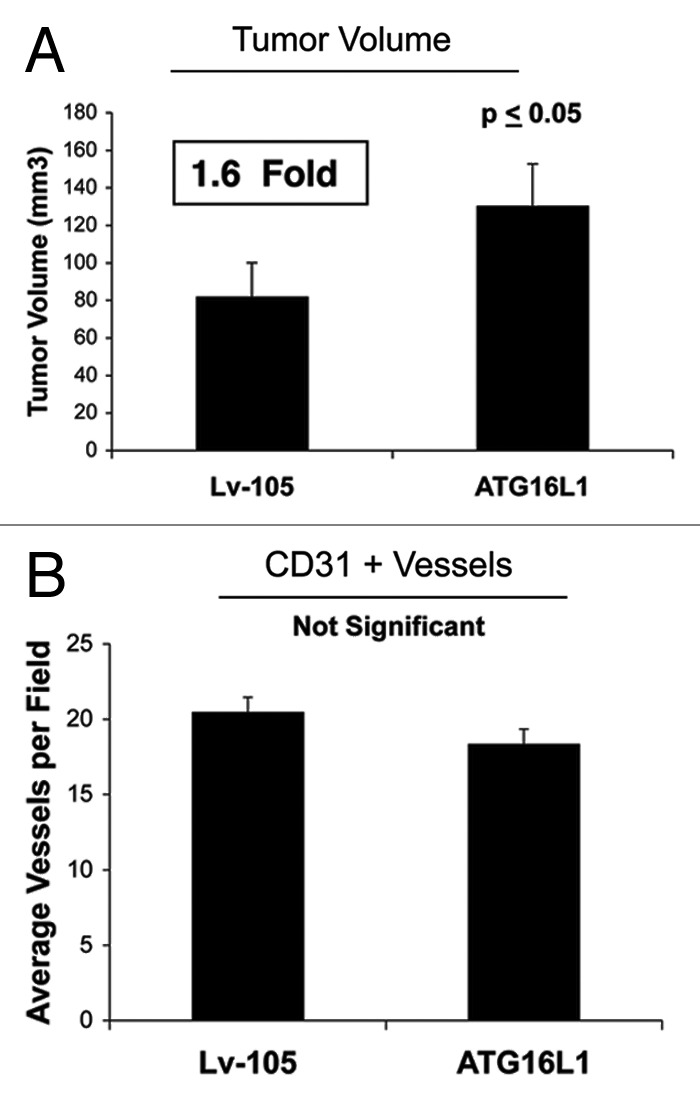
Figure 7. Fibroblasts overexpressing ATG16L1 drive enhanced tumor growth, without a measurable increase in angiogenesis.(A) In vivo, ATG16L1-fibrolasts also promoted tumor growth, resulting in an ~1.6-fold increase in tumor volume, relative to control fibroblasts. n = 10 per experimental group. (B) Increases observed in tumor growth were independent of tumor angiogenesis, as we previously observed for BNIP3, and CTSB. Lv-105 represents fibroblasts transduced with the vector alone control, namely Lv-105 (puro).
BNIP3-, CTSB- and ATG16L1-expressing fibroblasts all show mitochondrial dysfunction with increased production of L-lactate or ketone bodies
Autophagy is a process that induces the degradation of several organelles, including mitochondria.58 Therefore, we evaluated the functional consequences of autophagic genes expression on mitochondrial activity in fibroblasts.
First, we determined the levels of mitochondrial enzymes associated with the respiratory chain. Figure 8A shows a strong reduction in key components of complex I, III, and IV for all the genes examined. Furthermore, in CTSB fibroblasts there is a downregulation of the expression of complex II. These data were also corroborated by performing MitoTracker staining, which allows the vital visualization of healthy functional mitochondria. Note that all three types of autophagic fibroblasts (BNIP3, CTSB, and ATG16L1) show dramatic reductions in MitoTracker staining, indicative of a loss of mitochondrial membrane potential (Fig. 8B).
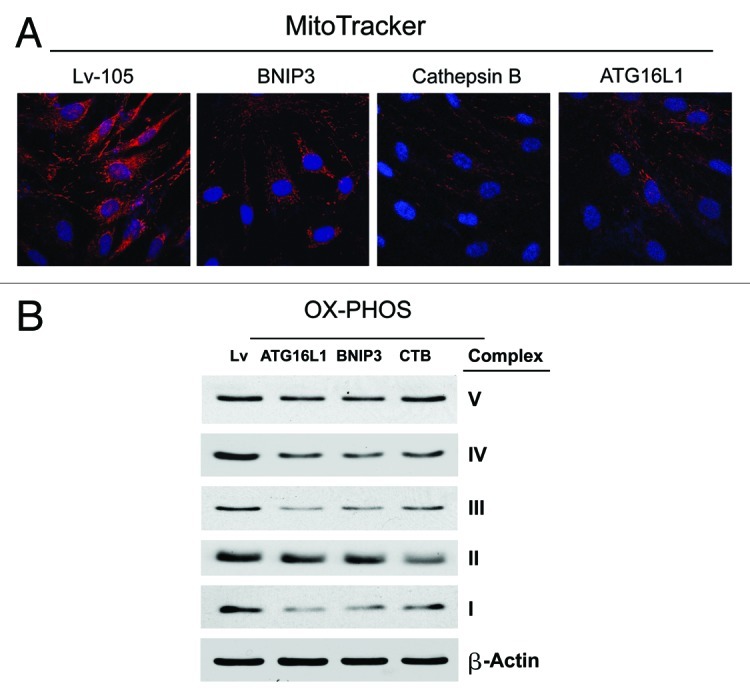
Figure 8. BNIP3-, CTSB- and ATG16L1- fibroblasts all show mitochondrial dysfunction: OXPHOS and MitoTracker. (A) We evaluated the functional consequences of autophagic genes expression on mitochondrial activity in fibroblasts, by determining the levels of mitochondrial enzymes associated with the respiratory chain. Note that a strong reduction in key components of complex I, III, and IV was observed, for all the genes examined. Furthermore, in CTSB fibroblasts there is a downregulation of the expression of complex II. Lv- represents fibroblasts transduced with the vector alone control, namely Lv-105 (puro). (B) Note that all three types of autophagic fibroblasts (BNIP3, CTSB, and ATG16L1) show dramatic reductions in MitoTracker staining, indicative of a loss of mitochondrial membrane potential.
Loss of functional mitochondria is associated with key changes in cell metabolism, leading to the accumulation of L-lactate and/or ketone bodies as end products in the tissue culture media. Figure 9A shows that BNIP3- and CTSB fibroblasts both increase L-lactate production, between 25%-to-37%. However, ATG16L1 fibroblasts did not show any increase in L-lactate accumulation.

Figure 9. BNIP3-, CTSB- and ATG16L1- fibroblasts all show mitochondrial dysfunction, with increased production of L-lactate or ketone bodies. (A) Loss of functional mitochondrial causes changes in cell metabolism, leading to the accumulation of L-lactate. Note that BNIP3- and CTSB fibroblasts both show significant increases in L-lactate production, between 25-to-37%. However, ATG16L1 fibroblasts did not show any increases in L-lactate accumulation. (B, C) Mitochondrial dysfunction can also activate ketone body production, resulting in the accumulation of 3-hydroxy-butyrate. Note that only ATG16L1 fibroblasts showed increases in ketone production, resulting in an up to 2.3-fold accumulation of 3-hydroxy-butyrate. The data were normalized either for cell number or for protein content per well. Thus, BNIP3- and CTSB fibroblasts produce L-lactate, while ATG16L1 fibroblasts produce ketone bodies, as a consequence of autophagy and the resulting mitochondrial dysfunction. (D) Autophagic fibroblasts were co-cultured with MDA-MB-231-GFP cells, and mitochondrial activity was visualized by MitoTracker staining. Note that all three autophagic fibroblast cell lines (BNIP3, CTSB, and ATG16L1) increased the MitoTracker staining (RED; Upper panels) in adjacent MDA-MB-231 cells (GREEN; Lower panels), during co-culture. This is consistent with the notion that autophagic fibroblasts provide mitochondrial fuels, such as L-lactate and ketone bodies, for the anabolic growth of cancer cells.
Mitochondrial dysfunction could activate ketone body production, resulting in the accumulation of 3-hydroxy-butyrate. Interestingly, only ATG16L1 fibroblasts showed increases in ketone production, resulting in an up to 2.3-fold accumulation of 3-hydroxy-butyrate (Fig. 9B and C). Thus, BNIP3 and CTSB fibroblasts produce L-lactate, while ATG16L1 fibroblasts produce ketone bodies, as a consequence of autophagy induction and resulting mitochondrial dysfunction.
Since both L-lactate and 3-hydroxy-butyrate are key substrates for mitochondrial oxidative metabolism, autophagic fibroblasts would be predicted to promote the mitochondrial activity of adjacent cancer cells in a paracrine fashion. To test this hypothesis directly, autophagic fibroblasts were co-cultured with MDA-MB-231-GFP cells, and mitochondrial activity was visualized by MitoTracker staining (Fig. 9D).
As predicted, all three fibroblast cell lines stably expressing autophagy inducers (BNIP3, CTSB, and ATG16L1) increased the MitoTracker staining in adjacent MDA-MB-231 cells during co-culture. This is consistent with the notion that autophagic fibroblasts may provide mitochondrial substrates, such as L-lactate and ketone bodies, for the anabolic growth of cancer cells.
Autophagic fibroblasts promote experimental metastasis and lung colonization
Next, we investigated the ability of autophagic fibroblasts to increase the metastatic potential of epithelial breast cancer cells. For this purpose, we used an experimental metastasis approach, which measures lung colonization as an output. Briefly, MDA-MB-231 cells were co-injected with fibroblasts into the tail vein of nude mice. Importantly, the lung colonization assay showed that all three autophagic fibroblast cell lines (BNIP3, CTSB, and ATG16L1) enhanced the metastatic capacity of MDA-MB-231 cells as compared with vector-alone control fibroblasts (Fig. 10A). For example, BNIP3 fibroblasts and CTSB fibroblasts increased metastasis by ~2.5-fold and ~7-fold, respectively. Interestingly, ATG16L1 fibroblasts showed the largest capacity for increasing metastasis, driving an ~11-fold increase. ATG16L1 fibroblasts also showed increases in myofibroblast markers, such as calponin and vimentin, indicating that autophagy may be also sufficient to promote myofibroblast differentiation (Fig. 10B). Myofibroblasts may play a crucial role in metastasis.59

Figure 10. Autophagic fibroblasts promote experimental metastasis and lung colonization. (A) MDA-MB-231 cells were co-injected with fibroblasts into the tail vein of nude mice. Note that all three autophagic fibroblast cell lines (BNIP3, CTSB, and ATG16L1) enhanced the metastatic capacity of MDA-MB-231 cells, as compared with vector-alone control fibroblasts. BNIP3 fibroblasts and CTSB fibroblasts increased metastasis by ~2.5-fold and ~7-fold, respectively. Interestingly, ATG16L1 fibroblasts showed the largest capacity for increasing metastasis, driving an ~11-fold increase. (B) ATG16L1 fibroblasts also showed increases in myofibroblast markers, such as calponin and vimentin, indicating that autophagy may be also sufficient to promote myofibroblast differentiation. β-actin expression was assessed as a control for equal protein loading.
Consistent with our current findings, a previous study on human oral cancers showed that increased stromal expression of ATG16L1 was associated with both lympho-vascular invasion and lymph-node metastasis.60 However, the authors did not provide any mechanistic insights into the prognostic value of stromal ATG16L1 and its association with metastasis.
Autophagic fibroblasts show the induction of p21(WAF1/CIP1), a CDK inhibitor, and characteristics of senescence: Cell hypertrophy and β-galactosidase activity
Autophagy has also been linked to the induction of cellular senescence.27,61 In fact, autophagy and senescence may be viewed as a continuum of the same biological process. To begin to assess the induction of a senescence phenotype, we first examined the expression of CDKI’s (cyclin-dependent kinase inhibitors) and cyclin D1 in all three autophagic fibroblast cell lines. BNIP3 fibroblasts showed an induction of p21(WAF1/CIP1) and p16(Ink4a), without any changes in p19(ARF). Conversely, CTSB- and ATG16L1 fibroblasts showed an induction of p21 and p19, with reductions in p16. Thus, the most consistent change observed across all three autophagic fibroblast cell lines was the induction of p21 (Fig. 11A).
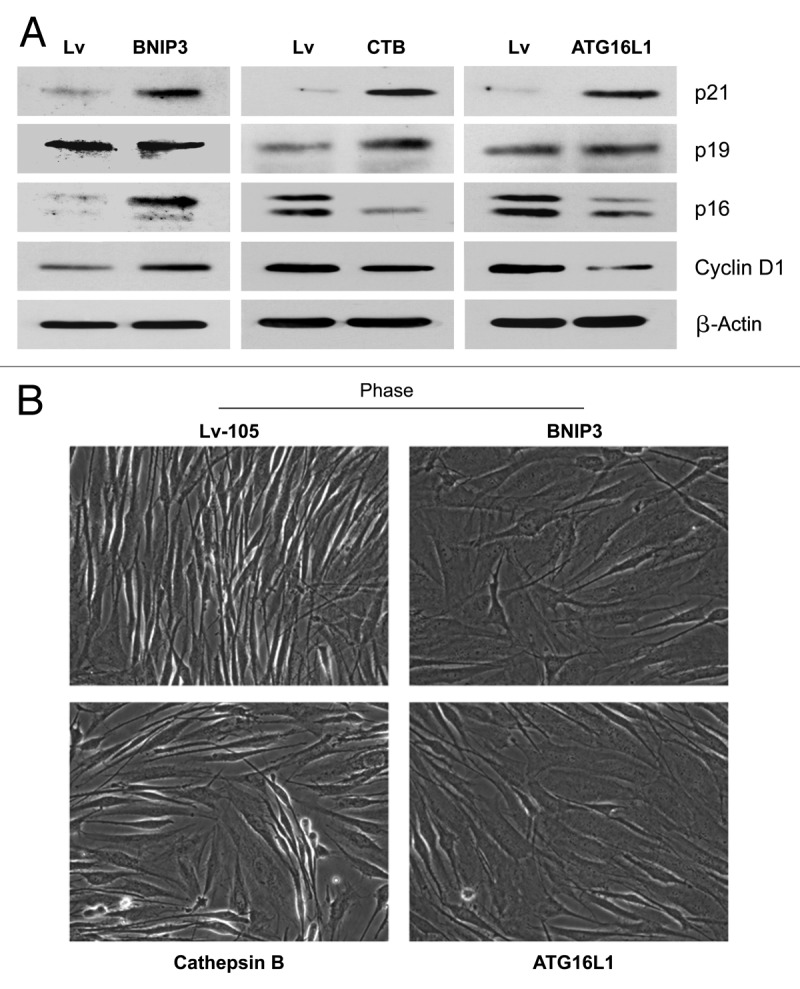
Figure 11. Autophagic fibroblasts show the induction of p21(WAF1/CIP1), a CDK inhibitor, and morphologic cell hypertrophy. (A) To assess the induction of a senescence phenotype, we examined the expression of CDKI’s (cyclin-dependent kinase inhibitors) and cyclin D1, in all three autophagic fibroblast cell lines. BNIP3 fibroblasts showed an induction of p21(WAF/CIP1) and p16(Ink4a), without any changes in p19(ARF). Conversely, CTSB- and ATG16L1 fibroblasts showed an induction of p21 and p19, with reductions in p16. Thus, the most consistent change observed across all three autophagic fibroblast cell lines was the induction of p21. (B) Senescent cells also appear flatter and/or hyper-trophic, as protein synthesis may continue without cell division. Note that BNIP3-, CTSB-, and ATG16L1 fibroblasts all appear more hyper-trophic (“wider”), as seen by light microscopy 72 h after cell plating.
Senescent cells also appear flatter and/or hypertrophic, as protein synthesis may continue without cell division. Indeed, we noted that BNIP3, CTSB and ATG16L1 fibroblasts all appeared more hypertrophic, as seen by light microscopy 72 h after cell plating (Fig. 11B). A more quantitative estimation of cell hypertrophy was achieved by measuring the total amount of protein per cell (Fig. 12A). Previously, studies have demonstrated that an increase in cell protein content is related to the induction of cell cycle arrest and senescence.47 Importantly, BNIP3 and ATG16L1 fibroblasts both showed significant cell hypertrophy (between 30%-to-32%), using this approach, while CTSB fibroblasts also demonstrated a trend toward cell hypertrophy. Increased protein mass per cell (hypertrophy) may simply reflect a compensatory response to constitutive protein degradation (via autophagy),62,63 resulting in increased protein synthesis to avoid cell death.47

Figure 12. Autophagic fibroblasts show increased protein mass per cell, and the induction of β-galactosidase activity. (A) A quantitative view of cell hypertrophy was achieved by measuring the total amount of protein per cell. Previously, it was demonstrated that an increase in cell protein content is related to the induction of senescence. Note that BNIP3- and ATG16L1 fibroblasts both showed significant cell hypertrophy (between 30-to-32%), using this approach, while CTSB fibroblasts also showed a trend toward cell hypertrophy. (B) β-galactosidase activity is the gold standard for measuring the onset of senescence. Thus, we quantitatively measured β-galactosidase activity by FACS analysis. Note that all three autophagic fibroblast cell lines showed an increase in β-galactosidase activity, as reflected by an increase in i) the % of β-Gal-positive cells, and ii) β-Gal-Mean-Intensity. (C) Similar results were obtained with more conventional β-Gal-staining methods, such as in BNIP3 overexpressing fibroblasts.
Finally, the induction of β-galactosidase activity is currently the gold standard for measuring the onset of senescence. Thus, we quantitatively measured the β-galactosidase activity of the autophagic fibroblast cell lines by FACS analysis. Figure 12B directly shows that all three autophagic fibroblast cell lines showed an increase in β-galactosidase activity, as reflected by an increase in (1) the % of β-Gal-positive cells (up to nearly 3-fold) and (2) β-Gal mean intensity (up to ~2.25 fold). Similar results were also obtained with more conventional β-Gal-staining methods, such as in BNIP3 overexpressing fibroblasts (Fig. 12C).
β-galactosidase is predominantly expressed in the tumor stroma of human breast cancer patients
We next investigated the possible compartmentalization of β-galactosidase in a panel of human breast cancer samples, largely derived from patients with a loss of stromal Cav-1 expression. Interestingly, our results directly show that, using specific antibodies directed against β-galactosidase, it is largely confined to the tumor stroma (Fig. 13). Thus, expression of β-galactosidase in vivo may be a stromal phenomenon, reflecting the onset of autophagy and senescence in the tumor stromal compartment. Further studies are warranted to assess the prognostic value of β-galactosidase as a new stromal biomarker for predicting clinical outcome of breast cancer patients.
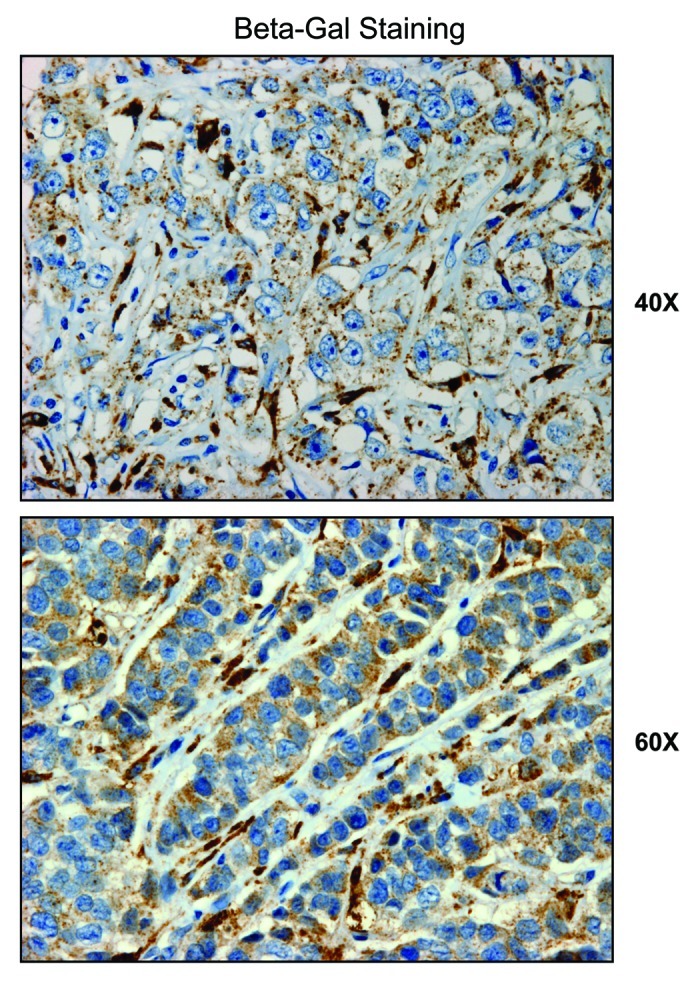
Figure 13. β-galactosidase is predominantly expressed in the tumor stroma of human breast cancer patients. We investigated the compartmentalization of β-galactosidase in a panel of human breast cancer samples, using specific-antibodies directed against β-galactosidase. Note that β-galactosidase is largely confined to the tumor stroma. Thus, expression of β-galactosidase in vivo may be a stromal phenomenon, reflecting the onset of senescence in the tumor microenvironment.
ATG16L1 expression in human breast cancer cells drives autophagy and reduces tumor growth
To understand the compartment-specific role of autophagy in tumor growth, we also stably overexpressed ATG16L1 in human breast cancer cells. ATG16L1 overexpression in MDA-MB 231 cells by itself is indeed sufficient to drive autophagy and mitophagy under baseline cell culture conditions, as evidenced by the loss of Cav-1 expression and the upregulation of BNIP3 (Fig. 14A and B). Similarly, Figure 14A and B show that MDA-MB 231 cells harboring ATG16L1 display the upregulation of certain autophagy markers (Beclin1 and CTSB) after overnight starvation.
Figure 14.

ATG16L1 expression in human breast cancer cells drives autophagy and reduces tumor growth. (A, B) To understand the compartment-specific role of autophagy in tumor growth, we also stably overexpressed ATG16L1 in human breast cancer cells. ATG16L1 overexpression in MDA-MB 231 cells, by itself, is indeed sufficient to drive autophagy and mitophagy under baseline cell culture conditions, as evidenced by the loss of Cav-1 expression and the upregulation of BNIP3 (B). Similarly, MDA-MB 231 cells harboring ATG16L1 display the upregulation of certain autophagy markers (Beclin1 and CTSB), after overnight starvation (A). (C) Note that ATG16L1 overexpression in MDA-MB-213 cells induces p21 expression, but no significant changes were detected in p19 and/or Cyclin D1 protein levels. (D) We also assessed the capacity of autophagic MDA-MD-231 cells to undergo tumor growth in vivo, after implantation in the flanks of nude mice. Note that ATG16L1 overexpression caused a near 2-fold reduction in tumor volume. (E) However, we did not observe any differences in tumor neo-vascularization, as assessed by quantitation of CD31-positive vessels.
Next, we evaluated the expression of key cell cycle regulators: CDK inhibitors and cyclin D1. Figure 14C shows that ATG16L1 overexpression in MDA-MB-213 cells induces p21 expression but no significant changes were detected in p19 and/or Cyclin D1 protein levels.
Finally, we assessed the capacity of autophagic MDA-MD-231 cells to undergo tumor growth in vivo after implantation in the flanks of nude mice. Figure 14D shows that ATG16L1 overexpression caused a near 2-fold reduction in tumor volume. These “tumor suppressor” effects were independent of angiogenesis, since we did not observe any differences in tumor neo-vascularization, as assessed by quantitation of CD31-positive vessels (Fig. 14E).
Thus, the effects of autophagy on tumor cell growth are clearly cell type- and compartment-specific and may exert opposing effects depending in which cell type the autophagic program is activated.
Validation of the autophagy-senescence transition (AST) by analysis of existing transcriptional profiles of laser-captured tumor stroma isolated from human breast cancer patient materials
Previously, Morag Park and colleagues48 laser-captured the normal stroma from human breast tissue (n = 38 patients) and the tumor stroma from human breast cancers (n = 53 patients). Then, they subjected these patient materials to genome-wide transcriptional profiling. Of the breast cancer patients analyzed (n = 53), a significant number of the patients analyzed underwent tumor recurrence (n = 11) or metastasis (n = 25).
We retrieved these stromal transcriptional profiles from existing public databases49 and interrogated them for evidence of autophagy and/or senescence in the tumor stroma. These results are summarized briefly in Tables 1 and 2. P-values for these associations are as shown. Table 1 includes a list of autophagy-associated genes that are selectively upregulated in the tumor stroma (relative to normal stroma). Note that many of these gene transcripts are also specifically upregulated in the tumor stroma of patients that subsequently underwent tumor recurrence and/or metastatsis (relative to breast cancer patients that did not undergo recurrence or metastasis).
Table 1. Increased expression of autophagy markers and telomerase in the tumor stroma of human breast cancers.
| Gene stroma | Description | Tumor stroma | Recurrence stroma | Metastasis stroma |
|---|---|---|---|---|
|
Atg16l1 |
autophagy-related 16-like 1 (yeast) |
3.75E-15 |
1.64E-04 |
|
| Atg9b |
ATG9 autophagy related 9 homolog B (S. cerevisiae) |
1.97E-20 |
1.58E-03 |
|
| Atg7 |
autophagy-related 7 (yeast) |
7.64E-14 |
|
|
| Atg4b |
autophagy-related 4B (yeast) |
3.49E-05 |
2.75E-02 |
|
| Atg4a |
autophagy-related 4A (yeast) |
6.21E-03 |
|
|
| Atg3 |
autophagy-related 3 (yeast) |
1.01E-02 |
3.08E-03 |
|
| |
|
|
|
|
|
Bnip3 |
BCL2/adenovirus E1B interacting protein 3 |
|
8.08E-03 |
|
|
Bnip3l |
BCL2/adenovirus E1B interacting protein 3-like |
2.85E-08 |
|
|
|
Becn1 |
beclin 1, autophagy related |
7.50E-05 |
|
|
|
Ctsb |
cathepsin B |
4.11E-36 |
3.27E-02 |
1.39E-02 |
| Ctsz |
cathepsin Z |
5.62E-24 |
|
|
| Ctsk |
cathepsin K |
3.28E-20 |
|
|
| Ctso |
cathepsin O |
1.49E-19 |
|
|
| Ctse |
cathepsin E |
1.66E-19 |
3.78E-03 |
|
| Ctss |
cathepsin S |
3.70E-18 |
|
|
| Ctsw |
cathepsin W |
1.94E-15 |
|
|
| Ctsf |
cathepsin F |
7.03E-14 |
|
|
| Ctsh |
cathepsin H |
7.21E-12 |
|
|
| Ctsg |
cathepsin G |
1.32E-11 |
4.03E-04 |
|
| |
|
|
|
|
|
Tert |
telomerase reverse transcriptase |
1.34E-11 |
1.23E-02 |
2.17E-02 |
| Tep1 |
telomerase associated protein 1 |
2.21E-07 |
5.05E-05 |
|
| Rtel1 |
regulator of telomere elongation helicase 1 |
6.03E-06 |
|
|
| Terf1 |
telomeric repeat binding factor 1 |
|
|
4.49E-02 |
| Terf2 | telomeric repeat binding factor 2 | 3.56E-08 | 1.63E-02 |
Data Mining from the Morag Park Data Set: Finak et al. Nature Medicine 2008; 14:518–27. Certain key molecules are highlighted in BOLD, which we recombinantly overexpressed in human fibroblasts. p-values are as shown.
Table 2. Increased expression of CDK inhibitors and beta-gal (senescence markers) in the tumor stroma of human breast cancers.
| Gene stroma | Description | Tumor stroma | Recurrence stroma | Metastasis stroma |
|---|---|---|---|---|
| |
|
|
|
|
|
Cdkn1a |
cyclin-dependent kinase inhibitor 1A (p21, WAF/CIP1) |
3.73E-19 |
|
|
| Ciz1 |
CDKN1A interacting zinc finger protein 1 |
2.13E-02 |
4.59E-03 |
|
| Cdkn1b |
cyclin-dependent kinase inhibitor 1B (p27, Kip1) |
|
|
4.82E-02 |
| |
|
|
|
|
|
Cdkn2a |
cyclin-dependent kinase inhibitor 2A (p16, INK4A) |
5.71E-29 |
|
|
| Cdkn2b |
cyclin-dependent kinase inhibitor 2B (p15, inhibits CDK4) |
4.47E-07 |
|
9.38E-03 |
| Cdkn2c |
cyclin-dependent kinase inhibitor 2C (p18, inhibits CDK4) |
1.04E-05 |
|
|
| Cdkn3 |
cyclin-dependent kinase inhibitor 3 |
|
|
4.12E-02 |
| Ccnb1 |
cyclin B1 |
1.53E-04 |
8.59E-03 |
|
|
Ccnd1 |
cyclin D1 |
1.42E-14 |
3.67E-02 |
4.21E-02 |
| |
|
|
|
|
| Glb1 | galactosidase, beta1 | 1.91E-23 |
Data Mining from the Morag Park Data Set: Finak et al. Nature Medicine 2008; 14:518–27. Certain key molecules are highlighted in BOLD, which we monitored in autophagic fibroblasts overexpressing either BNIP3, CTSB or ATG16L1. p-values are as shown.
This genes list also includes many of the same genes that we functionally analyzed by recombinant expression in fibroblasts, such as ATG16L1, BNIP3, BNIP3L, Beclin1 and CTSB as well as hTERT. Importantly, this supporting bioinformatics data provides direct evidence for the physiological relevance of the new genetically tractable models that we have created to study two-compartment tumor metabolism.
Similarly, Table 2 shows a list of senescence-associated genes that are specifically upregulated in the tumor stroma of primary human breast cancers, including p21(WAF1/CIP1), p16 (INK4A), cyclin D1/B1 and β-galactosidase. Upregulation of cyclin D1 and cyclin B1 may represent compensatory responses to cell cycle arrest. A more complete list of senescence-associated genes that are upregulated in the tumor stroma is included as Table S1.
Thus, this data mining exercise provides additional in vivo evidence for the existence of the autophagy-senescence transition (AST) in the tumor microenvironment of human breast cancers.
Discussion
The role of autophagy in tumorigenesis is complex; its action may be compartment- and cell type-specific.23 Autophagy was originally linked to reduced tumor growth when Beclin1, a crucial autophagy regulator, was first reported to be deleted in many epithelial cancers, indicating that autophagy can function as a tumor suppressor.64 In dramatic support of this simple concept, recombinant expression of Beclin1 in epithelial cancer cells drives autophagy and effectively suppresses tumorigenesis.65
Similarly, deficiencies in other autophagy regulators have been linked to a pro-tumorigenic phenotype.66 More specifically, autophagy-deficient cancer cells show increased DNA damage and genomic instability as well as increased ROS levels and oxidative stress.67 Several oncogenes have been shown to repress autophagy in cancer cells, supporting the overall idea that catabolic processes might be suppressed in tumor cells in order to favor the accumulation of cell biomass.68
However, just the opposite effects may be occurring in the tumor stromal microenvironment (Fig. 15). Thus, a catabolic tumor stroma, driven by autophagy in cancer-associated fibroblasts, could “energize” the anabolic growth of cancer cells by providing essential nutrients and mitochondrial fuels (L-lactate, ketones, glutamine and free fatty acids) in a paracrine fashion.2,69 We have previously termed this hypothesis the “autophagic tumor stroma model of cancer metabolism”2,7.
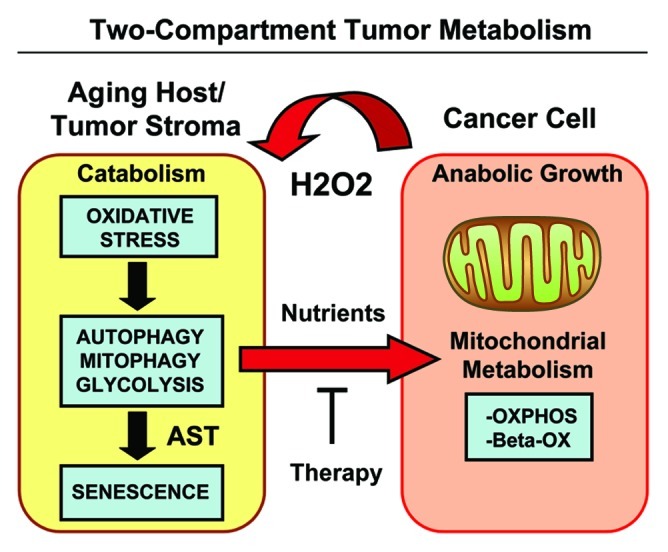
Figure 15. Two-compartment tumor metabolism is fueled by the autophagy-senescence transition (AST). In this model, cancer cells secrete hydrogen peroxide (H2O2), which induces oxidative stress in neighboring normal fibroblasts. Oxidative stress in stromal fibroblasts is then sufficient to confer the cancer-associated fibroblast phenotype, resulting in autophagy, mitophagy, and a shift toward aerobic glycolysis. Autophagy also drives the onset of senescence, via the autophagy-senescence transition. Autophagic-senescent fibroblasts then produce high-energy nutrients (L-lactate, ketone bodies, glutamine, and free fatty acids), which “fuel” mitochondrial metabolism (OXPHOS and β-OX; oxidative phosphorylation and β-oxidation) in adjacent cancer cells, resulting in the onset of anabolic tumor growth. This simple model could explain why chronological aging is one of the most significant risk factors for the development of cancer. AST, autophagy-senescence transition.
More specifically, we have demonstrated that oncogene-induced activation of H2O2 production in cancer cells drives oxidative stress in cancer-associated fibroblasts (CAFs), which then undergo autophagy and mitophagy.3,4,70 Loss of mitochondrial function increases aerobic glycolysis, driving the production and release of micro-nutrients, such as L-lactate and ketone bodies into the tumor microenvironment.11,49,71-73
Ketones and L-lactate, in turn, can act by a paracrine mechanism on cancer cells, driving mitochondrial biogenesis and OXPHOS (oxidative phosphorylation) in tumor cells, generating a positive feedforward loop to support anabolic tumor growth.9,10,74-76
Here, we developed a genetically tractable model system to directly study the compartment-specific role of autophagy in tumor growth and metastasis. First, we expressed autophagy-associated genes (such as BNIP3, CTSB, and ATG16L1) in hTERT-immortalized fibroblasts to genetically generate constitutively autophagic fibroblasts. Then, we validated that these genetically modified fibroblasts undergo autophagy, mitophagy, develop significant mitochondrial dysfunction and metabolically shift toward glycolysis, with increased L-lactate and ketone body production. Importantly, these autophagic fibroblasts effectively promoted tumor growth and metastasis, mimicking the behavior of cancer-associated fibroblasts.
Interestingly, we demonstrated that overexpression of a single autophagy gene is sufficient to drive autophagy induction and promote tumor growth. Our study establishes that autophagy, by itself, is sufficient to enhance tumor growth, independently of neo-vascularization. Autophagy action is clearly compartment-specific, since we also show that autophagy induction in human breast cancer cells greatly diminishes tumor growth. Thus, the induction of autophagy in stroma cells releases nutrients into the tumor microenvironment, which may support cancer cell growth. On the other hand, activation of autophagy in cancer cells drives the consumption of cellular components and effectively reduces tumor growth in MDA-MB-231 cells harboring ATG16L1.
A loss of stromal Cav-1 expression, specifically in cancer-associated fibroblasts, is a new important prognostic marker that predicts early tumor recurrence, lymph node metastasis and tamoxifen resistance, as well as poor clinical outcome in breast cancer patients.15-20 The proposed mechanism underlying a loss of stromal Cav-1 in cancer-associated fibroblasts is that Cav-1 undergoes lysosomal degradation, due to the induction of autophagy, secondary to oxidative stress.5,6,23,51
Our current data mechanistically validated this hypothesis, as genetic induction of autophagy/mitophagy with numerous autophagy-associated genes was indeed sufficient to drive a loss of Cav-1 protein expression in fibroblasts. Thus, the autophagic destruction of Cav-1 is biomarker for poor prognosis, as the autophagic tumor stroma provides recycled nutrients to fuel anabolic tumor growth.
We also show that autophagy-induced mitochondrial dysfunction drives a shift toward glycolysis in stromal fibroblasts, with associated increases L-lactate or ketone body production. Remarkably, autophagic fibroblasts also promoted tumor cell metastasis, likely via the production of high-energy mitochondrial fuels, such as L-lactate and ketone bodies, which could be “burned” by OXPHOS in adjacent cancer cells. In this regard, ketogenic ATG16L1 fibroblasts showed the highest efficiency in promoting distant metastasis, resulting in an ~11-fold increase in lung colonization. Consistent with these findings, ketones are a more powerful mitochondrial fuel as compared with L-lactate. Ketones can produce more energy than L-lactate, as they require substantially less oxygen consumption and can be “burned” by mitochondria under ischemic and/or hypoxic conditions.77,78 Thus, there may be a direct connection between ketone body production/utilization and cancer cell metastasis.
Ketone bodies are a group of organic molecules are produced both in the cytosol and in mitochondria, which are always present at a low level in healthy individuals. However, dietary manipulations and certain pathological conditions can increase the levels of these compounds in vivo. Pathologies leading to abnormal glucose metabolism, such as poorly controlled diabetes mellitus, can drive increased ketone body production.79
Epidemiologic studies suggest that people suffering from diabetes are at a higher risk of many forms of cancer (including breast, colon, pancreas and etc.) and diabetes is also associated with reduced survival after cancer.80,81 Breast cancer mortality rates remain high, primarily due to the metastasis of primary tumors to distant organs, such as the lungs.82 In diabetic patients, it is possible that breast cancer metastasis may be augmented by metabolic dysfunction, such as a tendency toward ketone body production. Although these data need to be confirmed with additional studies, they suggest that ketone bodies could play a crucial role in breast cancer metastasis.
Recently, several studies have linked autophagy with senescence. For example, ULK-3, a homolog of ATG1, induces autophagy in fibroblasts, conferring the acquisition of a senescence phenotype.27,61 Another protein proposed as a link between autophagy and senescence is cathepsin B. More specifically, CTSB is able to proteolytically cleave SIRT1, allowing for SIRT1 inactivation and driving the establishment of a senescence phenotype.83 Thus, autophagy and senescence may be viewed as a continuum or two related phases of the same overall biological process.
Our current results also directly support a molecular link between autophagy and senescence in the tumor stroma. For example, all the autophagic fibroblast cell lines we examined (overexpressing either BNIP3, CTSB, or ATG16L1) showed key features of a senescence phenotype, such as (1) induction of p21(WAF1/CIP1), a well-characterized CDK inhibitor, (2) a change in cell shape, consistent with cellular hypertrophy and (3) the upregulation of β-galactosidase activity. To confirm the clinical relevance of our findings, we also examined the distribution of β-galactosidase in human breast cancers by immunohistochemistry, using specifc antibody probes. Interestingly, the distribution of β-galactosidase (and thus the senescence phenotype) was largely confined to the tumor stroma and absent from epithelial cancer cells. Thus, senescence may be primarily confined to the tumor stromal compartment. Interestingly, β-galactosidase is also a known lysosomal enzyme and senescent cells show an increase in lysosomal mass,32,37 also supporting the general idea that senescence and autophagy are two sides of the same coin. Senescent cells also show a shift toward aerobic glycolysis,35,36 probably secondary to the onset of mitophagy.
Importantly, Judy Campisi’s group has previously shown that senescent fibroblasts promote tumor growth.24-26,84 Her laboratory attributed the growth-promoting activity of senescent fibroblasts to the senescence-associated secretory phenotype (SASP), which results in the secretion of specific growth factors and cytokines, as well as extracellular matrix remodeling.24,25 However, she estimated that the SASP accounts for only ~50% of the tumor-promoting activity of senescent fibroblasts.24,25 The other 50% still remains unaccounted for.24,25 Importantly, many of the same stimuli that she used to generate senescent fibroblasts, such as acute exposure to hydrogen peroxide and oxidative stress, are now known to be inducers of autophagy, and are also inducers of myofibroblast differentiation.29-31 Thus, we propose here that the other 50% of the tumor-promoting activity of senescent fibroblasts may actually reflect their autophagic/catabolic phenotype, mitochondrial dysfunction and a shift toward aerobic glycolysis, which results in production of mitochondrial fuels (such as L-lactate, ketones, glutamine and free-fatty acids). These mitochondrial fuels can then “feed” the TCA cycle and OXPHOS in adjacent cancer cells, allowing for the anabolic growth of tumor cells. In accordance with our new “metabolic” hypothesis, certain sub-types of senescent fibroblasts promote tumor growth but do not show the senescence-associated secretory phenotype (SASP).26
Thus, inhibition of autophagy and senescence in cancer-associated fibroblasts is now a new attractive metabolic therapeutic target for “cutting off the fuel supply” to rapidly proliferating cancer cells that are energetically dependent on the tumor stroma.
Materials and Methods
Materials
Commercially available antibodies used were as follows: β-actin (Sigma-Aldrich, #A5441); Cav-1 (BD Transduction Laboratories, #610406); Beclin1 (Novus Biologicals, #NBP1–00085); BNIP3 (Abcam, #ab10433); Cathepsin B (FL-339) (Santa Cruz, #sc-13985); Lamp-1 (E-5) (Santa Cruz, #sc-17768); LC3 (Abcam, ab48395); ATG16L1 (Abgent, #AP1817a); p21 (H-164) (Santa Cruz #, sc-756); p19 (DCS-240) (Santa Cruz, #sc-53639); p16 (H-156) (Santa Cruz, #sc-759); cyclin D1 (Thermo Scientific, #MS-210); OXPHOS (MitoSciences, #MS601); Calponin 1/2/3 (FL-297) (Santa Cruz, #sc-28545); Vimentin (BD Pharmigen, #550513). Lentiviral vectors containing full-length cDNAs encoding various autophagy genes (BNIP3, BNIP3L, Beclin1, CTSB, and ATG16L1) were all obtained commercially from GeneCopoeia (Rockville, MD).
Cell culture
Immortalized human fibroblasts (hTERT-BJ1 cells) and human breast cancer cells (MDA-MB-231-GFP), were cultured in DMEM implemented with 10% fetal bovine serum (FBS) and penicillin (100 units/mL) / streptomycin (100 μg/mL) (P/S). Hank’s Balanced salt solution (HBSS) supplemented with 40 mM Hepes and 1% P/S was used to initiate cell starvation. For co-culture studies, hTERT fibroblasts and MDA-MB-231-GFP cells were plated onto glass coverslips in 12-well plates in complete media at a cellular ratio of 5:1. The next day, the media was switched to DMEM supplemented with 10% NuSerum. Cells were maintained in co-culture for 3 d.
Lenti-virus production and cell line derivation
For lenti-virus production, 293Ta cells were transfected with different plasmids, in particular, EX-neg-Lv105 (the empty vector control), EX-T1853-Lv105 (encoding BNIP3), EX-U0307-Lv105 (encoding BNIP3L), EX-M0768-Lv105 (encoding Beclin1), EX-Z7498-Lv105 (encoding Beclin1), EX-5654-Lv105 (encoding Cathepsin B), and EX-H2109-Lv105 (encoding ATG16L1). All plasmids also contained a puromycin resistance marker. The resulting lenti-viruses were then used to infect hTERT fibroblasts or MDA-MB-231-GFP cells to obtain stable cell lines overexpressing the target gene of interest.
Immunoblot analysis
Immunoblotting was performed on samples obtained through OG buffer extraction. For this purpose, cells were scraped in OG lyses buffer (10 mM Tris, pH 7.5, 150 mM NaCl, 1% Triton X-100 and 60 mM n-octyl-glucoside) supplemented with a protease inhibitor cocktail (Roche Diagnostics) and a phosphatase inhibitor cocktail (Sigma). The samples were rotated for 40 min, then centrifuged for 10 min at 13,000 x g at 4°C. Finally, supernatants were collected. To determine the expression of cell cycle-related proteins, the cells were scraped into RIPA buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS, plus protease inhibitor and phosphatase inhibitor cocktails) and sonicated for 20 sec, then the samples were centrifuge 13,000 x g for 10 min at 4°C, and the supernatants were collected. The protein concentrations were determined using the BCA protein assay kit (Thermo scientific, #23225). Protein lysates were then separated by SDS-PAGE (using a 10-to-15% acrylamide gel) and transferred to nitrocellulose membranes. To detect expression of the protein of interest, specific primary antibodies and peroxidase-conjugated secondary antibodies were used. Bound antibodies were revealed using enhanced chemiluminescence (ECL) substrates (Thermo Scientific).
L-lactate assays
L-lactate levels were assessed according to the manufacturer’s instructions, using the EnzyChromTM L-Lactate Assay Kit (cat #ECLC-100, BioAssay Systems). For this purpose, cells were seeded in 12-well plates in complete media. The next day, the media was switched to DMEM containing 2% FBS. After 48 h, the media was collected and the concentration of L-lactate was measured. Results were normalized for total cell number.
Mitochondrial vital staining
Cells were seeded onto coverslips in 12-well plates in DMEM containing 10% FBS. After 24 h, the media was change to DMEM supplemented with 10% Nu-serum and 1% P/S. After 48 h, the mitochondria were labeled by incubating cells with a pre-warmed (37°C) staining solution containing the MitoTracker Red probe (25 nM for 12 min at 37°C). Then, the cells were washed with PBS, fixed in 2% PFA and observed under a fluorescence microscope.
Ketone Body Production.
Cells were seeded in 12-well plates in DMEM supplemented with 10% FBS. The next day, the media was switched to DMEM without red-phenol, containing 2% FBS. After 48 h, the media was collected, and the keto-acid concentration was measured according the manufacturer’s instructions using the β-Hydroxy-butyrate (β-HB) Assay Kit (Biovision, #K632). Results were normalized for either total cell number or total cellular protein per well, depending on the experiment.
β-galactosidase flow-cytometry assay
Approximately 400,000 cells were seeded per well in 6-well plates in DMEM with 10% FBS and 1% P/S. The next day, the media was changed to DMEM with 10% Nu-serum. Cells were then incubated for 48 h at 37°C with 5% CO2, under normal conditions. Then, the cells were trypsinized, centrifuged and counted to obtain 106 cells. Afterwards, cells were treated according the manufacturer’s instructions, using the FluoReporter lacZ Flow Cytometry Kit (Molecular probes, #F-1930). Assay results were evaluated by flow-cytometry analysis (FACS).
β-galactosidase staining assay
β-galactosidase activity was also detected by using a Senescence β-Galactosidase Staining Kit (Cell Signaling, #9860). For this purpose, cells were seeded into 6-well plates in complete media. After 24 h, the media was changed to DMEM supplemented with 10% Nu-serum. After 48 h, the cells were fixed and incubated overnight at 37°C in a dry incubator without CO2, with the β-galactosidase staining solution. Afterwards, cells were observed under the microscope.
Cellular hypertrophy assay
Approximately 100,000 cells were seeded per well in 12-well plates in DMEM containing 10% FBS. The next day, the media was switched to DMEM, supplemented with 10% Nu-serum. After 24 h, cells were counted and protein lysates were prepared with OG buffer. Protein quantification was performed using the BCA protein assay kit (Thermo scientific, #23225). The values were obtained were expressed as a ratio between the total amount of protein per well and the total number of cells per well (protein/cell number). An increased protein/cell number ratio would be indicative of cellular hypertrophy.47
Animal studies
To evaluate the in vivo tumor promoting effects of autophagy-related genes, flank injections were performed on athymic NCr nude mice (NCRNU; Taconic Farms; at 6–8 weeks of age). Experiments were performed according to the National Institutes of Health (NIH) guidelines, with the consensus of the Institutional Animal Care and Use Committee (IACUC) of Thomas Jefferson University. MDA-MB-231-GFP cells (alone or plus hTERT fibroblasts) were injected into the flanks of nude mice. More specifically, MDA-MB-231-GFP cancer cells (1 million cells) plus hTERT-BJ1 fibroblasts (300,000 cells) were re-suspended in 100 μl of sterile PBS, just prior to co-injection into the flank. After 4 weeks post-injection, the tumors were collected, and tumor growth was quantitatively measured.
Experimental metastasis assay
To evaluate the functional effects of autophagic fibroblasts on distant metastasis, a lung colonization assay was performed. Briefly, MDA-MB-231-GFP breast cancer cells were co-injected with fibroblasts in the tail vein of athymic NCr nude mice (NCRNU; Taconic Farms; at 6–8 weeks of age). More specifically, MDA-MB-231-GFP cancer cells (1 million cells) plus hTERT-BJ1 fibroblasts (300,000 cells) were re-suspended in 100 μl of sterile PBS, just prior to co-injection into the tail vein. After 7 weeks post-injection, the lungs were insuflated with 15% India ink dye, washed in water and bleached in Fekete’s solution (70% ethanol, 3.7% para-formaldehyde, 0.75 M glacial acetic acid). A low-power stereo-microscope was then used to determine the number of negatively stained metastatic colonies per lung lobe.
Quantitation of tumor angiogenesis
6 μm tumor frozen sections were fixed with 4% paraformaldehyde in PBS for 10 min at 4°C and washed 3x with PBS. A three-step biotin-streptavidin-horseradish peroxidase method was used for antibody detection. After fixation, the sections were blocked with 10% rabbit serum and incubated overnight at 4°C with rat monoclonal CD31 antibody (550274, BD Biosciences). The sections were then incubated with biotinylated rabbit anti-rat IgG (Vector Labs) and streptavidin-HRP (Dako). Immunoreactivity was revealed with 3, 3′ diaminobenzidine. For quantitation of vessels, CD31-positive vessels were counted in 4–6 fields within the central area of each tumor using a 20x objective lens and an ocular grid (0.25 mm2 per field). The total numbers of vessel per unit area was calculated, and the data was represented graphically.
Immunohistochemistry
Formalin-fixed paraffin-embedded tumor sections were de-paraffinized, rehydrated and washed in PBS. Antigen retrieval was performed in 10 mM sodium citrate, pH 6.0 for 10 min using a pressure cooker. After blocking with 3% hydrogen peroxide for 10 min, sections were incubated with 10% goat serum for 1 h. Then, sections were incubated with primary antibodies (anti-β-galactosidase: Abcam, #ab-96239, at a dilution of 1:1,000) overnight at 4°C. Antibody binding was detected using a biotinylated secondary (Vector Labs) followed by strepavidin-HRP (Dako). Immunoreactivity was revealed using 3, 3′ diaminobenzidine. Finally, samples were counter-stained with hematoxylin, dehydrated, mounted and observed under the microscope.
Bioinformatics analysis
The transcriptional profiles of tumor stroma isolated by laser-capture from human primary breast cancers,48 were retrieved and re-analyzed, essentially as we previously described.49
Supplementary Material
Acknowledgments
F.S. and her laboratory were supported by grants from the Breast Cancer Alliance (BCA) and the American Cancer Society (ACS). U.E.M. was supported by a Young Investigator Award from the Margaret Q. Landenberger Research Foundation. M.P.L. was supported by grants from the NIH/NCI (R01-CA-080250; R01-CA-098779; R01-CA-120876; R01-AR-055660), and the Susan G. Komen Breast Cancer Foundation. R.G.P. was supported by grants from the NIH/NCI (R01-CA-70896, R01-CA-75503, R01-CA-86072, and R01-CA-107382) and the Dr. Ralph and Marian C. Falk Medical Research Trust. The Kimmel Cancer Center was supported by the NIH/NCI Cancer Center Core grant P30-CA-56036 (to R.G.P.). Funds were also contributed by the Margaret Q. Landenberger Research Foundation (to M.P.L.). This project is funded, in part, under a grant with the Pennsylvania Department of Health (to M.P.L. and F.S.). The Department specifically disclaims responsibility for any analyses, interpretations or conclusions. This work was also supported, in part, by a Centre grant in Manchester from Breakthrough Breast Cancer in the UK (to A.H.) and an Advanced ERC Grant from the European Research Council.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/20718
References
- 1.Sotgia F, Martinez-Outschoorn UE, Howell A, Pestell RG, Pavlides S, Lisanti MP. Caveolin-1 and cancer metabolism in the tumor microenvironment: markers, models, and mechanisms. Annu Rev Pathol. 2012;7:423–67. doi: 10.1146/annurev-pathol-011811-120856. [DOI] [PubMed] [Google Scholar]
- 2.Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Power surge: supporting cells “fuel” cancer cell mitochondria. Cell Metab. 2012;15:4–5. doi: 10.1016/j.cmet.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 3.Lisanti MP, Martinez-Outschoorn UE, Lin Z, Pavlides S, Whitaker-Menezes D, Pestell RG, et al. Hydrogen peroxide fuels aging, inflammation, cancer metabolism and metastasis: the seed and soil also needs “fertilizer”. Cell Cycle. 2011;10:2440–9. doi: 10.4161/cc.10.15.16870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lisanti MP, Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Pestell RG, Howell A, et al. Accelerated aging in the tumor microenvironment: connecting aging, inflammation and cancer metabolism with personalized medicine. Cell Cycle. 2011;10:2059–63. doi: 10.4161/cc.10.13.16233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, et al. Oxidative stress in cancer-associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle. 2010;9:3256–76. doi: 10.4161/cc.9.16.12553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, et al. Autophagy in cancer-associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFκB activation in the tumor stromal microenvironment. Cell Cycle. 2010;9:3515–33. doi: 10.4161/cc.9.17.12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martinez-Outschoorn UE, Pestell RG, Howell A, Tykocinski ML, Nagajyothi F, Machado FS, et al. Energy transfer in “parasitic” cancer metabolism: mitochondria are the powerhouse and Achilles’ heel of tumor cells. Cell Cycle. 2011;10:4208–16. doi: 10.4161/cc.10.24.18487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sotgia F, Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Lisanti MP. Understanding the Warburg effect and the prognostic value of stromal caveolin-1 as a marker of a lethal tumor microenvironment. Breast Cancer Res. 2011;13:213. doi: 10.1186/bcr2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, Howell A, et al. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle. 2011;10:4047–64. doi: 10.4161/cc.10.23.18151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sotgia F, Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, et al. Mitochondrial metabolism in cancer metastasis: visualizing tumor cell mitochondria and the “reverse Warburg effect” in positive lymph node tissue. Cell Cycle. 2012;11:1445–54. doi: 10.4161/cc.19841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, Chiavarina B, Flomenberg N, et al. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle. 2010;9:3485–505. doi: 10.4161/cc.9.17.12721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinez-Outschoorn UE, Whitaker-Menezes D, Pavlides S, Chiavarina B, Bonuccelli G, Casey T, et al. The autophagic tumor stroma model of cancer or “battery-operated tumor growth”: A simple solution to the autophagy paradox. Cell Cycle. 2010;9:4297–306. doi: 10.4161/cc.9.21.13817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Tanowitz HB, Sotgia F, et al. Stromal-epithelial metabolic coupling in cancer: integrating autophagy and metabolism in the tumor microenvironment. Int J Biochem Cell Biol. 2011;43:1045–51. doi: 10.1016/j.biocel.2011.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ertel A, Tsirigos A, Whitaker-Menezes D, Birbe RC, Pavlides S, Martinez-Outschoorn UE, et al. Is cancer a metabolic rebellion against host aging? In the quest for immortality, tumor cells try to save themselves by boosting mitochondrial metabolism. Cell Cycle. 2012;11:253–63. doi: 10.4161/cc.11.2.19006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Witkiewicz AK, Casimiro MC, Dasgupta A, Mercier I, Wang C, Bonuccelli G, et al. Towards a new “stromal-based” classification system for human breast cancer prognosis and therapy. Cell Cycle. 2009;8:1654–8. doi: 10.4161/cc.8.11.8544. [DOI] [PubMed] [Google Scholar]
- 16.Witkiewicz AK, Dasgupta A, Nguyen KH, Liu C, Kovatich AJ, Schwartz GF, et al. Stromal caveolin-1 levels predict early DCIS progression to invasive breast cancer. Cancer Biol Ther. 2009;8:1071–9. doi: 10.4161/cbt.8.11.8874. [DOI] [PubMed] [Google Scholar]
- 17.Witkiewicz AK, Dasgupta A, Sammons S, Er O, Potoczek MB, Guiles F, et al. Loss of stromal caveolin-1 expression predicts poor clinical outcome in triple negative and basal-like breast cancers. Cancer Biol Ther. 2010;10:135–43. doi: 10.4161/cbt.10.2.11983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Witkiewicz AK, Dasgupta A, Sotgia F, Mercier I, Pestell RG, Sabel M, et al. An absence of stromal caveolin-1 expression predicts early tumor recurrence and poor clinical outcome in human breast cancers. Am J Pathol. 2009;174:2023–34. doi: 10.2353/ajpath.2009.080873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Witkiewicz AK, Kline J, Queenan M, Brody JR, Tsirigos A, Bilal E, et al. Molecular profiling of a lethal tumor microenvironment, as defined by stromal caveolin-1 status in breast cancers. Cell Cycle. 2011;10:1794–809. doi: 10.4161/cc.10.11.15675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Witkiewicz AK, Whitaker-Menezes D, Dasgupta A, Philp NJ, Lin Z, Gandara R, et al. Using the “reverse Warburg effect” to identify high-risk breast cancer patients: stromal MCT4 predicts poor clinical outcome in triple-negative breast cancers. Cell Cycle. 2012;11:1108–17. doi: 10.4161/cc.11.6.19530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Di Vizio D, Morello M, Sotgia F, Pestell RG, Freeman MR, Lisanti MP. An absence of stromal caveolin-1 is associated with advanced prostate cancer, metastatic disease and epithelial Akt activation. Cell Cycle. 2009;8:2420–4. doi: 10.4161/cc.8.15.9116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu KN, Queenan M, Brody JR, Potoczek M, Sotgia F, Lisanti MP, et al. Loss of stromal caveolin-1 expression in malignant melanoma metastases predicts poor survival. Cell Cycle. 2011;10:4250–5. doi: 10.4161/cc.10.24.18551. [DOI] [PubMed] [Google Scholar]
- 23.Chiavarina B, Whitaker-Menezes D, Migneco G, Martinez-Outschoorn UE, Pavlides S, Howell A, et al. HIF1-alpha functions as a tumor promoter in cancer-associated fibroblasts, and as a tumor suppressor in breast cancer cells: Autophagy drives compartment-specific oncogenesis. Cell Cycle. 2010;9:3534–51. doi: 10.4161/cc.9.17.12908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A. 2001;98:12072–7. doi: 10.1073/pnas.211053698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krtolica A, Campisi J. Cancer and aging: a model for the cancer promoting effects of the aging stroma. Int J Biochem Cell Biol. 2002;34:1401–14. doi: 10.1016/S1357-2725(02)00053-5. [DOI] [PubMed] [Google Scholar]
- 26.Coppé JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem. 2011;286:36396–403. doi: 10.1074/jbc.M111.257071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Young AR, Narita M. Connecting autophagy to senescence in pathophysiology. Curr Opin Cell Biol. 2010;22:234–40. doi: 10.1016/j.ceb.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 28.Narita M, Young AR, Narita M. Autophagy facilitates oncogene-induced senescence. Autophagy. 2009;5:1046–7. doi: 10.4161/auto.5.7.9444. [DOI] [PubMed] [Google Scholar]
- 29.Tian LM, Xie HF, Xiao X, Yang T, Hu YH, Wang WZ, et al. Study on the roles of β-catenin in hydrogen peroxide-induced senescence in human skin fibroblasts. Exp Dermatol. 2011;20:836–8. doi: 10.1111/j.1600-0625.2011.01324.x. [DOI] [PubMed] [Google Scholar]
- 30.Liu DH, Chen YM, Liu Y, Hao BS, Zhou B, Wu L, et al. Rb1 protects endothelial cells from hydrogen peroxide-induced cell senescence by modulating redox status. Biol Pharm Bull. 2011;34:1072–7. doi: 10.1248/bpb.34.1072. [DOI] [PubMed] [Google Scholar]
- 31.Waghray M, Cui Z, Horowitz JC, Subramanian IM, Martinez FJ, Toews GB, et al. Hydrogen peroxide is a diffusible paracrine signal for the induction of epithelial cell death by activated myofibroblasts. FASEB J. 2005;19:854–6. doi: 10.1096/fj.04-2882fje. [DOI] [PubMed] [Google Scholar]
- 32.Kurz DJ, Decary S, Hong Y, Erusalimsky JD. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci. 2000;113:3613–22. doi: 10.1242/jcs.113.20.3613. [DOI] [PubMed] [Google Scholar]
- 33.Gerland LM, Peyrol S, Lallemand C, Branche R, Magaud JP, Ffrench M. Association of increased autophagic inclusions labeled for beta-galactosidase with fibroblastic aging. Exp Gerontol. 2003;38:887–95. doi: 10.1016/S0531-5565(03)00132-3. [DOI] [PubMed] [Google Scholar]
- 34.Robbins E, Levine EM, Eagle H. Morphologic changes accompanying senescence of cultured human diploid cells. J Exp Med. 1970;131:1211–22. doi: 10.1084/jem.131.6.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stöckl P, Hütter E, Zwerschke W, Jansen-Dürr P. Sustained inhibition of oxidative phosphorylation impairs cell proliferation and induces premature senescence in human fibroblasts. Exp Gerontol. 2006;41:674–82. doi: 10.1016/j.exger.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 36.Goldstein S, Ballantyne SR, Robson AL, Moerman EJ. Energy metabolism in cultured human fibroblasts during aging in vitro. J Cell Physiol. 1982;112:419–24. doi: 10.1002/jcp.1041120316. [DOI] [PubMed] [Google Scholar]
- 37.Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, et al. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell. 2006;5:187–95. doi: 10.1111/j.1474-9726.2006.00199.x. [DOI] [PubMed] [Google Scholar]
- 38.White E, Lowe SW. Eating to exit: autophagy-enabled senescence revealed. Genes Dev. 2009;23:784–7. doi: 10.1101/gad.1795309. [DOI] [PubMed] [Google Scholar]
- 39.Chen J, Goligorsky MS. Premature senescence of endothelial cells: Methusaleh’s dilemma. Am J Physiol Heart Circ Physiol. 2006;290:H1729–39. doi: 10.1152/ajpheart.01103.2005. [DOI] [PubMed] [Google Scholar]
- 40.Sasaki M, Ikeda H, Yamaguchi J, Miyakoshi M, Sato Y, Nakanuma Y. Bile ductular cells undergoing cellular senescence increase in chronic liver diseases along with fibrous progression. Am J Clin Pathol. 2010;133:212–23. doi: 10.1309/AJCPWMX47TREYWZG. [DOI] [PubMed] [Google Scholar]
- 41.Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y. Autophagy mediates the process of cellular senescence characterizing bile duct damages in primary biliary cirrhosis. Lab Invest. 2010;90:835–43. doi: 10.1038/labinvest.2010.56. [DOI] [PubMed] [Google Scholar]
- 42.Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y. Modulation of the microenvironment by senescent biliary epithelial cells may be involved in the pathogenesis of primary biliary cirrhosis. J Hepatol. 2010;53:318–25. doi: 10.1016/j.jhep.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 43.Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y. Autophagy may precede cellular senescence of bile ductular cells in ductular reaction in primary biliary cirrhosis. Dig Dis Sci. 2012;57:660–6. doi: 10.1007/s10620-011-1929-y. [DOI] [PubMed] [Google Scholar]
- 44.Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y. A possible involvement of p62/sequestosome-1 in the process of biliary epithelial autophagy and senescence in primary biliary cirrhosis. Liver Int. 2012;32:487–99. doi: 10.1111/j.1478-3231.2011.02656.x. [DOI] [PubMed] [Google Scholar]
- 45.Chatzistamou I, Dioufa N, Trimis G, Sklavounou A, Kittas C, Kiaris H, et al. p21/waf1 and smooth-muscle actin α expression in stromal fibroblasts of oral cancers. Cell Oncol (Dordr) 2011;34:483–8. doi: 10.1007/s13402-011-0044-6. [DOI] [PubMed] [Google Scholar]
- 46.Luo Y, Zou P, Zou J, Wang J, Zhou D, Liu L. Autophagy regulates ROS-induced cellular senescence via p21 in a p38 MAPKα dependent manner. Exp Gerontol. 2011;46:860–7. doi: 10.1016/j.exger.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Demidenko ZN, Blagosklonny MV. Quantifying pharmacologic suppression of cellular senescence: prevention of cellular hypertrophy versus preservation of proliferative potential. Aging (Albany NY) 2009;1:1008–16. doi: 10.18632/aging.100115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14:518–27. doi: 10.1038/nm1764. [DOI] [PubMed] [Google Scholar]
- 49.Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Transcriptional evidence for the “Reverse Warburg Effect” in human breast cancer tumor stroma and metastasis: similarities with oxidative stress, inflammation, Alzheimer’s disease, and “Neuron-Glia Metabolic Coupling”. Aging (Albany NY) 2010;2:185–99. doi: 10.18632/aging.100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chinnadurai G, Vijayalingam S, Gibson SB. BNIP3 subfamily BH3-only proteins: mitochondrial stress sensors in normal and pathological functions. Oncogene. 2008;27(Suppl 1):S114–27. doi: 10.1038/onc.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Daumer KM, Milliman JN, Chiavarina B, et al. Tumor cells induce the cancer-associated fibroblast phenotype via caveolin-1 degradation: implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle. 2010;9:2423–33. doi: 10.4161/cc.9.12.12048. [DOI] [PubMed] [Google Scholar]
- 52.Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29:2570–81. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mazure NM, Pouysségur J. Atypical BH3-domains of BNIP3 and BNIP3L lead to autophagy in hypoxia. Autophagy. 2009;5:868–9. doi: 10.4161/auto.9042. [DOI] [PubMed] [Google Scholar]
- 54.Ha SD, Ham B, Mogridge J, Saftig P, Lin S, Kim SO. Cathepsin B-mediated autophagy flux facilitates the anthrax toxin receptor 2-mediated delivery of anthrax lethal factor into the cytoplasm. J Biol Chem. 2010;285:2120–9. doi: 10.1074/jbc.M109.065813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, et al. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci. 2003;116:1679–88. doi: 10.1242/jcs.00381. [DOI] [PubMed] [Google Scholar]
- 56.Ravikumar B, Moreau K, Rubinsztein DC. Plasma membrane helps autophagosomes grow. Autophagy. 2010;6:1184–6. doi: 10.4161/auto.6.8.13428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol. 2010;12:747–57. doi: 10.1038/ncb2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goldman SJ, Taylor R, Zhang Y, Jin S. Autophagy and the degradation of mitochondria. Mitochondrion. 2010;10:309–15. doi: 10.1016/j.mito.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yamashita M, Ogawa T, Zhang X, Hanamura N, Kashikura Y, Takamura M, et al. Role of stromal myofibroblasts in invasive breast cancer: stromal expression of alpha-smooth muscle actin correlates with worse clinical outcome. Breast Cancer. 2012;19:170–6. doi: 10.1007/s12282-010-0234-5. [DOI] [PubMed] [Google Scholar]
- 60.Nomura H, Uzawa K, Yamano Y, Fushimi K, Ishigami T, Kouzu Y, et al. Overexpression and altered subcellular localization of autophagy-related 16-like 1 in human oral squamous-cell carcinoma: correlation with lymphovascular invasion and lymph-node metastasis. Hum Pathol. 2009;40:83–91. doi: 10.1016/j.humpath.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 61.Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Narita M, Young AR, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science. 2011;332:966–70. doi: 10.1126/science.1205407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zoncu R, Sabatini DM. Cell biology. The TASCC of secretion. Science. 2011;332:923–5. doi: 10.1126/science.1207552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aita VM, Liang XH, Murty VV, Pincus DL, Yu W, Cayanis E, et al. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics. 1999;59:59–65. doi: 10.1006/geno.1999.5851. [DOI] [PubMed] [Google Scholar]
- 65.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 66.Mariño G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, López-Otín C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J Biol Chem. 2007;282:18573–83. doi: 10.1074/jbc.M701194200. [DOI] [PubMed] [Google Scholar]
- 67.Scherz-Shouval R, Elazar Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007;17:422–7. doi: 10.1016/j.tcb.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 68.Maiuri MC, Tasdemir E, Criollo A, Morselli E, Vicencio JM, Carnuccio R, et al. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ. 2009;16:87–93. doi: 10.1038/cdd.2008.131. [DOI] [PubMed] [Google Scholar]
- 69.Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011;17:1498–503. doi: 10.1038/nm.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Martinez-Outschoorn UE, Lin Z, Trimmer C, Flomenberg N, Wang C, Pavlides S, et al. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect: implications for PET imaging of human tumors. Cell Cycle. 2011;10:2504–20. doi: 10.4161/cc.10.15.16585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Loss of stromal caveolin-1 leads to oxidative stress, mimics hypoxia and drives inflammation in the tumor microenvironment, conferring the “reverse Warburg effect”: a transcriptional informatics analysis with validation. Cell Cycle. 2010;9:2201–19. doi: 10.4161/cc.9.11.11848. [DOI] [PubMed] [Google Scholar]
- 72.Pavlides S, Vera I, Gandara R, Sneddon S, Pestell RG, Mercier I, et al. Warburg Meets Autophagy: Cancer-Associated Fibroblasts Accelerate Tumor Growth and Metastasis via Oxidative Stress, Mitophagy, and Aerobic Glycolysis. Antioxid Redox Signal. 2011 doi: 10.1089/ars.2011.4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer-associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8:3984–4001. doi: 10.4161/cc.8.23.10238. [DOI] [PubMed] [Google Scholar]
- 74.Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle. 2010;9:3506–14. doi: 10.4161/cc.9.17.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Martinez-Outschoorn UE, Prisco M, Ertel A, Tsirigos A, Lin Z, Pavlides S, et al. Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer: achieving personalized medicine via Metabolo-Genomics. Cell Cycle. 2011;10:1271–86. doi: 10.4161/cc.10.8.15330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK, et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle. 2011;10:1772–83. doi: 10.4161/cc.10.11.15659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cahill GF, Jr., Veech RL. Ketoacids? Good medicine? Trans Am Clin Climatol Assoc. 2003;114:149–61, discussion 162-3. [PMC free article] [PubMed] [Google Scholar]
- 78.Veech RL, Chance B, Kashiwaya Y, Lardy HA, Cahill GF., Jr. Ketone bodies, potential therapeutic uses. IUBMB Life. 2001;51:241–7. doi: 10.1080/152165401753311780. [DOI] [PubMed] [Google Scholar]
- 79.McPherson PA, McEneny J. The biochemistry of ketogenesis and its role in weight management, neurological disease and oxidative stress. J Physiol Biochem. 2012;68:141–51. doi: 10.1007/s13105-011-0112-4. [DOI] [PubMed] [Google Scholar]
- 80.Lipscombe LL, Goodwin PJ, Zinman B, McLaughlin JR, Hux JE. The impact of diabetes on survival following breast cancer. Breast Cancer Res Treat. 2008;109:389–95. doi: 10.1007/s10549-007-9654-0. [DOI] [PubMed] [Google Scholar]
- 81.Barone BB, Yeh HC, Snyder CF, Peairs KS, Stein KB, Derr RL, et al. Long-term all-cause mortality in cancer patients with preexisting diabetes mellitus: a systematic review and meta-analysis. JAMA. 2008;300:2754–64. doi: 10.1001/jama.2008.824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Solomayer EF, Diel IJ, Meyberg GC, Gollan C, Bastert G. Metastatic breast cancer: clinical course, prognosis and therapy related to the first site of metastasis. Breast Cancer Res Treat. 2000;59:271–8. doi: 10.1023/A:1006308619659. [DOI] [PubMed] [Google Scholar]
- 83.Chen J, Xavier S, Moskowitz-Kassai E, Chen R, Lu CY, Sanduski K, et al. Cathepsin cleavage of sirtuin 1 in endothelial progenitor cells mediates stress-induced premature senescence. Am J Pathol. 2012;180:973–83. doi: 10.1016/j.ajpath.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Blagosklonny MV, Campisi J, Sinclair DA. Aging: past, present and future. Aging (Albany NY) 2009;1:1–5. doi: 10.18632/aging.100009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.