

Abstract
Numerous stimuli, including oncogenic signaling, DNA damage or eroded telomeres trigger proliferative arrest, termed cellular senescence. Accumulating evidence suggests that cellular senescence is a potent barrier to tumorigenesis in vivo, however oncogene induced senescence can also promote cellular transformation.1,2 Several oncogenes, whose overexpression results in cellular senescence, converge on the TOR (target of rapamycin) pathway. We therefore examined whether attenuation of TOR results in delay or reversal of cellular senescence. By using primary human fibroblasts undergoing either replicative or oncogenic RAS-induced senescence, we demonstrated that senescence can be delayed, and some aspects of senescence can be reversed by inhibition of TOR, using either the TOR inhibitor rapamycin or by depletion of TORC1 (TOR Complex 1). Depletion of TORC2 fails to affect the course of replicative or RAS-induced senescence. Overexpression of REDD1 (Regulated in DNA Damage Response and Development), a negative regulator of TORC1, delays the onset of replicative senescence. These results indicate that TORC1 is an integral component of the signaling pathway that mediates cellular senescence.
Keywords: TOR, TORC1, rapamycin, senescence
Introduction
Cellular senescence is a collective term that has been applied to several related but distinct processes. These have as their common endpoint the cessation of cell proliferation tied to characteristic changes in cellular morphology, increase in lysosomal mass, secretion of inflammatory cytokines and increased expression of pro-senescence proteins.2-5
This report addresses two well-characterized types of senescence: RAS-induced and replicative senescence. Proliferative arrest triggered by the overexpression of oncogenic RAS in primary cell culture is a well-studied example of oncogene-induced senescence.6,7 In primary human fibroblasts, RAS-induced senescence begins with a period of increased proliferation, followed, after approximately six days, by proliferative arrest.6 This type of senescence depends on the activation of the p38MAPK (p38 Mitogen-Activated Protein Kinase) pathway, a pathway activated by stress stimuli, including inflammatory cytokines, UV irradiation and heat shock.8-11 Activation of p38MAPK, in turn, phosphorylates and activates the tumor suppressor p53, whose expression is necessary for induction of RAS-induced senescence.6,12 Overexpression of oncogenic RAS also leads to activation of the DNA damage response.13,14
Unlike RAS-induced senescence, replicative senescence develops slowly and occurs after approximately 50 cell divisions ex vivo in human cells.15 Most normal somatic cells do not express hTERT (human Telomerase Reverse Transcriptase), which is required for maintenance of telomeres and, as a result, gradually lose the ends of their telomeres with every duplication.16 Critically short telomeres trigger a DNA damage response that is sufficient to maintain the senescence-associated proliferative arrest.17-19
Both replicative and RAS-induced senescence are characterized by a common set of senescence-associated markers: secretion of cytokines, including IL8 (Interleukin 8), activation of the p38MAPK pathway, induction of SA-β-gal (Senescence-Associated β-galactosidase) activity and increased expression of the pro-senescent proteins, tumor suppressor p53 and p21 (cyclin-dependent kinase inhibitor 1A).19-24 Cellular senescence is multifaceted: on one hand, it is an important barrier to transformation and cancer;5 on the other, it is implicated in inflammation and could promote aging.2 Several mechanisms exist that can delay, or in some cases bypass, cellular senescence: depletion of senescence-promoting proteins like the tumor suppressors p53 or RB (RetinoBlastoma protein) can reverse both replicative and RAS-induced senescence,25 and overexpression of hTERT in replicative senescent cells reverses the senescence and leads to immortalization.26 In addition, recent reports indicate that senescence induced by DNA damaging agents can be delayed by chemical inhibitors of TOR, implying that TOR contributes to the establishment of senescence.27 Attenuation of TOR signaling through serum withdrawal or through treatment with rapamycin results in cellular quiescence: induction of DNA damage or p53 does not lead to senescence in these cells.28 Under these conditions, inhibition of TOR suppresses geroconversion, i.e., transition of quiescence into senescence.29
Signaling of several prominent oncogenes, including RAS, PI3K (Phosphoinositide-3-Kinase), AKT (v-Akt Thymoma viral oncogene homolog 1) and RHEB (RAS-Homolog Enriched in Brain), targets TOR and is deregulated in most cancers.30-35 As a result, TOR has become one of the most actively pursued drug targets.36-38 The effects of the TOR inhibitors are, however, complex, because these compounds also interfere with important TOR-dependent negative feedback loops that affect the PI3K signaling pathway.39-43
TOR belongs to PIKK (Phosphatidylinositol-3-Kinase-related kinase) family of serine/threonine kinases that includes DNA-PK (DNA-dependent Protein Kinase catalytic subunit), ATM (Ataxia-Telangiectasia Mutated) and ATR (Ataxia- and Rad3-related). The latter three kinases are activated in response to DNA damage and can stall cell cycle progression.44 TOR exists in two distinct multi-protein complexes: TORC1 and TORC2.45-47 TORC1 is a sensor of amino acids, oxygen and growth factors, controlling a variety of cellular processes that extend from cell growth and proliferation to autophagy.48,49 TORC2 recently emerged as the AKT kinase; however, its functions are not well understood.50,51
TORC1 phosphorylates S6K1 and 4E-BP1 (eukaryotic translation initiation factor 4E-binding Protein 1).52,53 S6K1 phosphorylates ribosomal protein S6 and governs cell size and glucose homeostasis.54 Phosphorylation of 4E-BP1 releases and thereby activates eIF4E (eukaryotic translation Initiation Factor 4E) and allows translation of eIF4E-sensitive RNAs, many of which regulate cell proliferation.55,56 4E-BP1 also negatively regulates p53 and fibroblasts lacking 4E-BP1 undergo premature senescence.57
In order to characterize the role of TOR in replicative and RAS-induced senescence, we attenuated TOR activity with small-molecule inhibitors and by biological intervention. We found that inhibition of TORC1 retards, and to some extent reverses, phenotypic indicators of cellular senescence. In contrast, selective downregulation of TORC2 has no detectable effect on the process of cellular senescence. These findings thus identify TORC1 as an important regulator of senescence.
Results
Inhibition of TORC1 by rapamycin partly reverses senescence-associated signaling in BJ fibroblasts.
Cellular senescence is marked by secretion of inflammatory cytokines, upregulation of pro-senescent protein p21 and phosphorylation of p38 kinase. Inflammatory cytokines including IL8 reinforce the senescence-associated phenotype in an autocrine loop.58,59 We confirmed by western blot the induction of IL8 in late-passage BJ human primary fibroblasts and in BJ fibroblasts overexpressing the RAS oncogene, as compared with early passage controls and to cells transfected with empty vector, respectively. We also detected phosphorylated p38 and higher levels of senescence-associated p21 in both RAS-induced senescent cells and in late-passage BJ fibroblasts (Fig. 1A). Although no significant induction was observed in the phosphorylation of the TORC1 targets S6K1 and 4E-BP1 in senescent cells as compared with pre-senescent cells, levels of total 4E-BP1 were lower in both RAS-induced and replicative senescent cells (Fig. 1A).

Figure 1. Attenuation of TORC1 reverses senescence-associated signaling. (A) SDS-PAGE western blot of BJ fibroblasts treated once over three days with either rapamycin (10 nM) or PP242 (200 nM). Left panel: early passage (senescent –) or late passage (senescent +) during replicative senescence examined for levels of senescence-associated proteins and for levels of the downstream targets of TORC1 and TORC2. Right panel: RAS-induced senescent cells (senescent +) or control cells with empty vector WH (senescent –) examined for levels of senescence-associated proteins and for levels of the downstream targets of TORC1 and TORC2. (B) SDS-PAGE western blot of a time course of cells passaged in the presence of rapamycin (10 nM) for 2, 8, 48 and 200 h (8 d), examined for levels of the downstream targets of TORC1. (C) Staining for senescence-associated β-galactosidase (SA-β-gal) of replicative senescent cells treated with rapamycin (10 nM) or PP242 (200 nM) for 72 h.
To determine whether rapamycin reverses the senescence-associated phenotype in both replicative-senescent and RAS-induced senescent cells, we treated BJ fibroblasts for three days with either 10 nM rapamycin or with 200 nM of the ATP-competitive inhibitor of TORC1 and TORC2, PP242. In late passage fibroblasts, attenuation of TORC1 by rapamycin and, to a lesser extent, by PP242 resulted in a drop of IL8 levels, reducing the inflammatory signaling to pre-senescent levels (Fig. 1A). In RAS-induced senescent cells, a single treatment with rapamycin or PP242 did not result in significant reduction of IL8, nor in detectable dephosphorylation of p38. However, in both RAS-induced and replicative senescent cells, treatment with rapamycin or PP242 resulted in a strong reduction in the total levels of p21 and a significant reduction in the total levels of p53 (Fig. 1A). We did not see a significant upregulation of the total p53 levels in replicative senescent cells; this result is in accord with previous studies showing that total levels of p53 do not rise in late passage fibroblasts, but rather its affinity for DNA increases in senescent cells.60,61 RAS-induced senescence, on the other hand, is associated with accumulation of p53.6 In response to rapamycin, we observed a drop in total levels of p53 in both RAS-induced and replicative senescent cells (Fig. 1A). We further noticed a drop in total levels of AKT, Rictor, Raptor and Cyclin D1 after three days of treatment with rapamycin (Fig. S1). Expression of Cyclin D1 is dependent on cellular levels of eIF4E;62 other proteins could be affected due to global decrease in translation related to S6-dependent ribosome biogenesis.
We confirmed that in both RAS-induced and replicative senescent cells, treatment with rapamycin resulted in attenuation of TORC1, as evidenced by reduced phosphorylation of the ribosomal subunit S6 and of 4E-BP1 S65. Treatment with rapamycin, and, to a lesser extent, with PP242 led to an overexpression of the hypophosphorylated 4E-BP1, evidenced by more rapidly migrating bands on SDS-PAGE gel (Fig. 1A). We showed that the accumulated 4E-BP1 was dephosphorylated on both T70 and S65 and that the dephosphorylation at these sites was sustained for several days (Fig. 1B). We also conducted a time-course experiment in which rapamycin-treated cells were harvested at 2, 8, 48 and 200 h after addition of the drug. For the last time point, the medium was changed, and the inhibitor was replenished every 48 h. This experiment showed that total levels of 4E-BP1 were upregulated and peaked at 8 h of exposure to the drug. Phosphorylation of S6 remained low, even at later time points (Fig. 1B).
Senescent cells increase in size, become flattened and display elevated activity of SA-β-gal.2 To determine whether inhibition of TORC1 can reverse these senescence-associated changes, 1 nM or 10 nM rapamycin was added to replicative senescent BJ cells at passage 55 and to control cells at passage 30 three days prior to staining for SA-β-gal. Treatment with rapamycin had no effect on cellular morphology or on β-gal-staining in the cells that already entered replicative senescence (Fig. 1C). The treatment also failed to relieve replicative arrest, as evidenced by lack of proliferation. These data imply that although short-term attenuation of TORC1 can reduce senescence-associated inflammatory signaling and other molecular signatures, it fails to completely reverse cellular senescence.
Inhibition of TORC1 leads to the activation of TORC2 and phosphorylation of its downstream target, AKT S473.50 Treatment of BJ fibroblasts with rapamycin over 24 h led to dephosphorylation of S6 and concomitant phosphorylation of AKT S473. Even at high doses of rapamycin, AKT S473 remained phosphorylated, suggesting that rapamycin had no effect on the TORC2 kinase (Fig. S2). Activation of negative feedback loops resulting from inhibition of TORC1 has been previously characterized and includes both activation of the AKT and the MAPK pathways.43 We detected increased phosphorylation on both AKT S473 and ERK (Extracellular signal-Regulated Kinase) T202/ Y204 in response to 10 nM rapamycin treatment, lasting three days in both pre-senescent and senescent fibroblasts (Fig. 1A). To avoid off-target effects due to high concentration of ATP-competitive inhibitors on related kinases63 and to determine whether they can specifically confer long-term inhibition of TORC2, we used low concentrations of Torin1 and INK128 (Fig. S2). Short-term exposure (5 h) to 5 nM Torin1 or to 10 nM INK128 resulted in dephosphorylation of S6 and of AKT S473. However, cells treated for 24 h with single dose of 5 nM Torin1 showed rephosphorylation of both S6 and AKT S473 (Fig. S2). To determine whether this rephosphorylation was due to the short half-life of the inhibitor or activation of negative feedback loops, we replenished media and inhibitor every 5 h over a 24 h period. Surprisingly, such replenishment of Torin1 or INK128 did not prevent rephosphorylation of AKT at S473 but blocked rephosphorylation of S6 (Fig. S2). Phosphorylation of S6 reflects the activity of its upstream kinase, S6K, whose isoforms p70S6K1 and p85S6K1 are directly phosphorylated by TORC1. In contrast, the TORC2 target AKT S473 became rephosphorylated despite sustained activity of the inhibitor. These data show that continuous exposure to ATP-competitive inhibitors suppresses specific functions of TORC1 but appears to have only a transitory effect on the activity of TORC2. An alternative explanation for these observations is to ascribe the rephosphorylation of AKT S473 to a different kinase. Because of this uncertainty about the effect of ATP-competitive inhibitors on the long-term functions of TORC2, we used rapamycin for all future experiments.
Continuous attenuation of TORC1 postpones the onset of replicative and disrupts RAS-induced senescence.
Commitment to cellular senescence includes remodeling of the cytoskeleton and expression of senescence-associated proteins as well as of cytokines that reinforce the phenotype.22 Since rapamycin can interfere with the expression of some of these parameters, we considered it possible that continuous attenuation of TORC1 could postpone the process of senescence. Pre-treatment with rapamycin was shown to prevent the loss of proliferative potential in cells treated with DNA damaging agents.28 We therefore cultured BJ primary human skin fibroblasts starting with the pre-senescent passage 30 in the presence of 1 nM and 10 nM rapamycin. After an additional 30 passages, the untreated fibroblasts assumed the senescence-associated phenotype: flat morphology and proliferative arrest (Fig. 2A–C). In contrast, cells treated with rapamycin continued to divide past passage 60, despite the initial drop in their rate of duplication (Fig. 2A). At passage 60, the treated cells not only displayed a higher duplication rate, but also a higher cumulative number of duplications. These rapamycin-treated fibroblasts showed a significant dose-dependent drop in SA-β-gal staining, and only 71% and 42% of cells treated with 1 nM and 10 nM rapamycin, respectively, retained either senescent morphology or staining at passage 60, whereas about 90% of untreated cells showed strong staining and senescence-associated morphology (Fig. 2B). Rapamycin-treated cells also displayed pre-senescent levels of p21, lower levels of p53 and significantly lower levels of IL8 (Fig. 2C). Even with continuous treatment with rapamycin at 10 nM for over two months, phosphorylation of S6 remained low. These observations show that attenuation of TORC1 in pre-senescent fibroblasts delays the onset of replicative senescence.
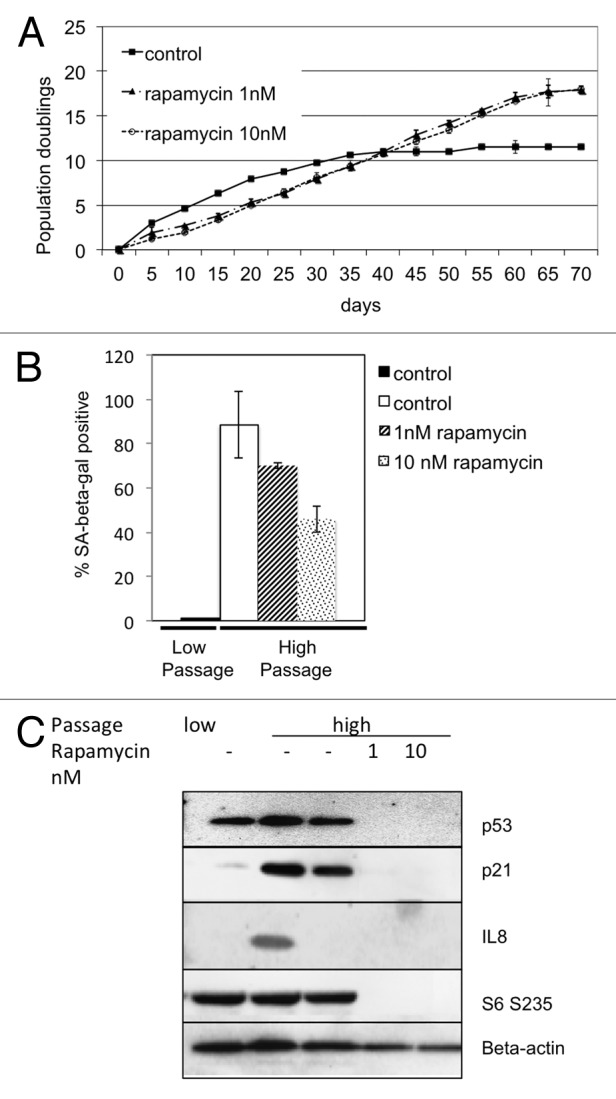
Figure 2. Attenuation of TORC1 signaling in early-passage fibroblasts delays replicative senescence. (A) Cells treated over 70 d with either 1nM or 10 nM rapamycin, and duplication rate measured every 5 d in triplicate. Cumulative duplications displayed on the Y-axis. Rate of duplications is displayed as the slope of the curve. (B) SA-β-gal staining of cells passaged in the presence of rapamycin (1 nM or 10 nM) for 70 d (high passage) or control, unpassaged (low passage). (C) Western blot of high passage and low passage cells cultured in the presence of rapamycin (1 nM or 10 nM).
Unlike replicative senescence, RAS-induced senescence proceeds more rapidly and leads to proliferative arrest within a few days.6 We treated early passage BJ primary fibroblasts with 2 nM or 5 nM rapamycin at day 2 post-transduction with either oncogenic H-RASV12 (RAS) or empty vector (WH). Overexpression of RAS resulted in transition to senescence within 10 days in untreated cells. These cells stained strongly for SA-β-gal and displayed the morphological markers of senescence. Fibroblasts treated with rapamycin, in contrast, continued proliferating and showed a significant reduction in SA-β-gal staining from near 100% to 50% at higher concentrations of the inhibitor (Fig. 3A and B). These results support the conclusion that both RAS-induced senescence and replicative senescence can be disrupted by attenuation of TORC1.
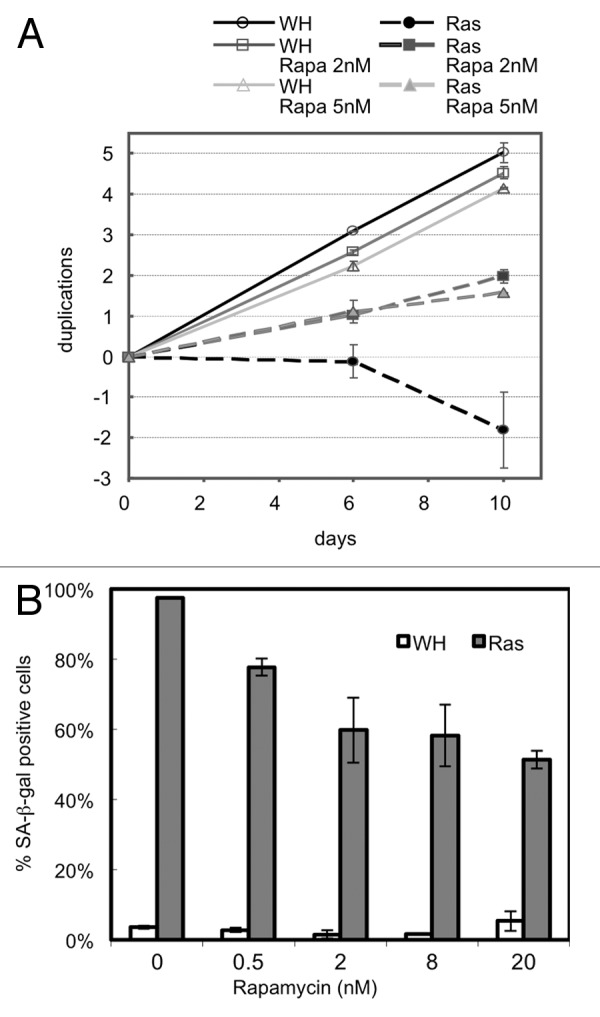
Figure 3. Attenuation of TORC1 signaling in RAS-induced cells disrupts RAS-induced senescence. (A) Growth curve of HRAS-V12-transduced cells or empty vector control (WH) in the presence and absence of rapamycin. Cells treated with rapamycin 2 nM or 5 nM, day two post infection. (B) SA-β-gal staining of cells transduced with either HRAS-V12 or empty vector control after passaging with 0.5, 2, 8 or 20 nM rapamycin from day 2 of infection.
Disruption of TORC1 but not TORC2 disrupts oncogenic RAS-induced senescence.
The categorical protein of the TORC1 complex is Raptor (Regulatory Associated Protein of TOR), a protein whose interaction with TOR is sensitive to rapamycin.64,65 The TORC2 complex contains Rictor (Raptor-Independent Companion of the TOR, complex) as the distinctive component.46,47,66 Unlike TORC1, TORC2 is mostly insensitive to rapamycin, although long-term treatment has a negative effect on TORC2 integrity.67 The rapamycin-induced delay in cellular senescence could, therefore, be caused by a disruption of TORC1, TORC2 or both complexes. In order to address this question, we depleted components of the TOR complexes with shRNA against TOR, Raptor or Rictor in cells and examined RAS-induced senescence. Two independent shRNAs were constructed that effectively reduced expression of TOR, Raptor or Rictor (Fig. 4A). Knockdown of either TOR or Raptor disrupted RAS-induced senescence in BJ cells. Whereas the control cells entered senescence upon expression of activated RAS, cells expressing TOR or Raptor shRNAs continued to proliferate and displayed a drop in SA-β-gal staining (Fig. 4B, C and E). In contrast, depletion of Rictor in these cells did not affect the process of RAS-induced cellular senescence (Fig. 4D and E). These results suggest that TORC1 is essential for oncogenic RAS-induced senescence, while inactivation of TORC2 has no effect on senescence induction. Based on this finding, we reason that the anti-senescence activity of rapamycin relies on its ability to inhibit the TORC1 rather than the TORC2 complex.

Figure 4. Depletion of TORC1, but not TORC2 subunits disrupts RAS-induced senescence. (A) BJ fibroblasts transduced with shRNA against TOR, Raptor, Rictor or with scrambled (shSC) shRNA. Relative mRNA levels determined by qRT-PCR. (C-D) TOR-depleted, Rictor-depleted, or Raptor-depleted cells infected with either HRAS-V12 or empty vector control (WH) and passaged for 12 d. Total population doublings displayed on the Y-axis. Slope of the curve indicates growth rate. Left panel: staining of SA-β-gal of RAS-induced or control cells. (E) SA-β-gal of Rictor, Raptor, TOR-depleted cells.
Overexpression of the negative regulator of TOR, REDD1, delays the onset of replicative senescence.
Because REDD1 (Regulated in DNA Damage response and Development1) negatively regulates TOR in response to DNA damage, hypoxia, glucose deprivation and glucocorticoids,68-70 we investigated its role in cellular senescence. Overexpression of REDD1 in BJ fibroblasts via lentiviral transduction led to dephosphorylation of S6 S235 (Fig. 5A). This is consistent with earlier reports showing that overexpression of REDD1 inhibits the TORC1 axis.68,71 In addition, we observed increased levels of hypophosphorylated 4E-BP1 and increased phosphorylation of AKT S473. These results indicate that REDD1 preferentially downregulates TORC1 in BJ fibroblasts. We observed a decrease in cell size and weaker SA-β-gal staining at passage 55+ in cells expressing REDD1 as compared with the wild-type control cells (Fig. 5B and C). Moreover, unlike wild-type cells that entered proliferative arrest at passage 55 and beyond, REDD1-overexpressing cells continued to proliferate at a rate similar to the pre-senescent fibroblasts (Fig. 5B and data not shown). These results demonstrate that inhibition of TORC1 by REDD1 leads to attenuation of cellular senescence. As a positive control, we overexpressed eIF4E. eIF4E overexpression results in accelerated senescence in mouse embryonic fibroblasts and B cells.72 eIF4E regulates expression of Cyclin D1,62 a protein that is elevated in senescent cells and that is downregulated in response to attenuation of TORC1 (Fig. S1). We observed an increase in levels of Cyclin D1 in response to eIF4E overexpression (Fig. 5A). BJ primary fibroblasts reached senescence within one week of stable overexpression of eIF4E, and over 70% stained strongly with SA-β-gal (Fig. 5B and C).
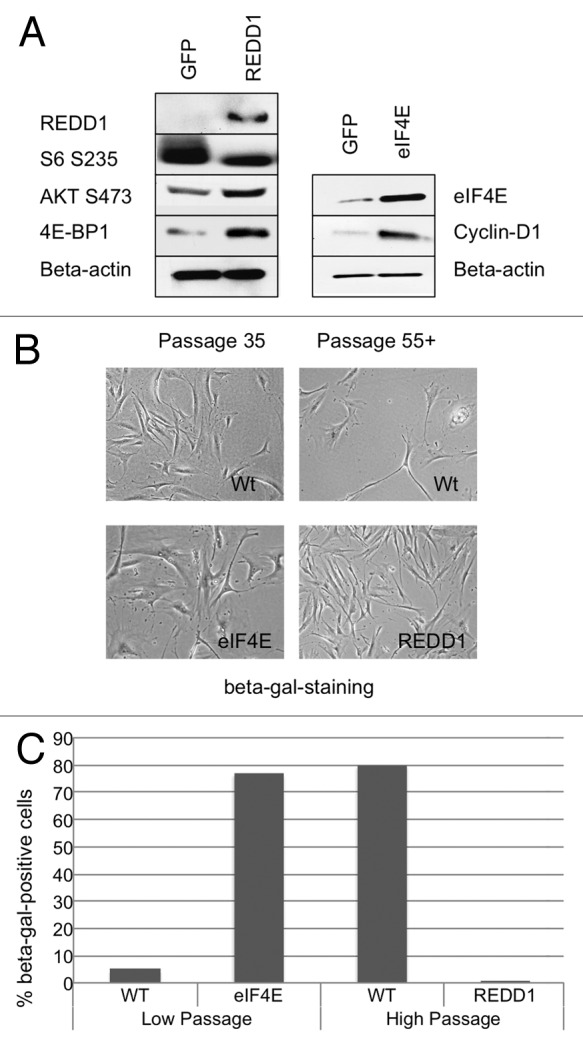
Figure 5. Stable expression of negative regulator of TOR, REDD1, delays the onset of replicative senescence and stable overexpression of eIF4E, accelerates it. (A) Cells expressing REDD1, GFP or eIF4E were assayed by SDS-PAGE western blot (right panel). SDS-PAGE western blot of cells transduced with either REDD1 lentivector (left panel) or eIF4E vector (right panel). (B) Beta-gal staining and passage number of REDD1, eIF4E or control (low passage and high passage) cells. (C) SA-β-gal quantitation of high passage REDD1 cells, eIF4E and low passage control cells.
Overexpression of 4E-BP1 leads to depletion of p53 and p21.
4E-BP1 negatively regulates eIF4E. We demonstrated that total levels of 4E-BP1 are upregulated in response to both treatment with rapamycin and overexpression of REDD1. We therefore asked whether overexpression of 4E-BP1 in primary fibroblasts would reverse some markers of senescence.
Senescence-associated proteins p53 and p21 are central to both induction and maintenance of senescence.57 Fibroblasts lacking 4E-BP1 undergo premature senescence, and 4E-BP1 plays an important role in senescence by negatively regulating pro-senescent protein p53.57 We therefore transiently overexpressed 4E-BP1 in BJ fibroblasts and studied the effect of this overexpression on TOR targets and on markers of senescence. We confirmed overexpression of 4E-BP1 by western blot (Fig. 6). Most 4E-BP1 was present in the dephosphorylated form. The level of phosphorylated 4E-BP1 S65 did not change in response to the overexpression of 4E-BP1; this indicates that most 4E-BP1 was present in the inhibitory, active form. We detected no significant change in the level of phosphorylated S6, suggesting that TORC1-S6K1 axis was not inhibited in response to overexpression of 4E-BP1 (Fig. 6). However, total levels of both p53 and p21 decreased in response to overexpression of 4E-BP1.
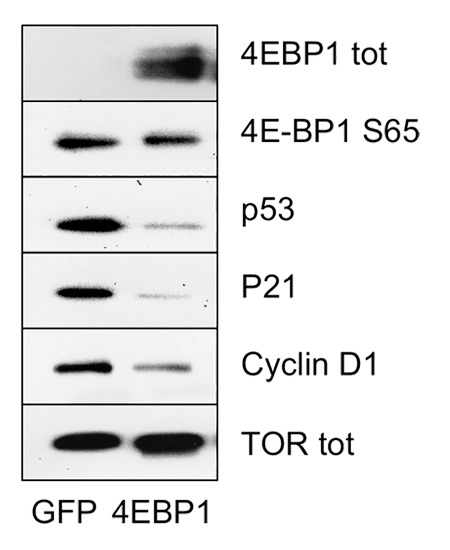
Figure 6. Overexpression of 4E-BP1 results in downregulation of p53, p21 and Cyclin D1. SDS-PAGE western blot of BJ cells at passage 35, transiently transfected with GFP or 4E-BP1.
In addition we observed decreased levels of Cyclin D1, a marker frequently associated with senescent cells,73 due to 4E-BP1 overexpression. This is in accord with the observation that cells undergoing eIF4E-induced senescence display higher levels of Cyclin D1, and that expression of Cyclin D1 can be downregulated by inhibiting TORC1 signaling. These results demonstrate that overexpression of 4E-BP1 alone can regulate key players in cellular senescence, p53 and p21.
Discussion
In this report, we demonstrate that both oncogene-induced senescence and replicative senescence can be disrupted by attenuating TORC1 signaling. Our results show that inhibition of TORC1 with the selective inhibitor rapamycin or by depletion of Raptor or TOR proteins allow cells to duplicate past the point of proliferative arrest. Both proliferation arrest and senescence-associated signaling are delayed in response to rapamycin treatment: fibroblasts passaged in the presence of rapamycin maintain low inflammatory signaling, including IL8 and phosphorylated p38. However, in RAS-dependent senescence, we did not detect a significant decrease in p38 phosphorylation or IL8 after treatment with rapamycin. This difference between replicative and RAS-induced senescence could be due to strong signaling from RAS that compensates some rapamycin-induced changes in TORC1 signaling.
Treatment with rapamycin led to decrease in key regulators of senescence, p21 and p53. Overexpression of p21 alone is sufficient to drive cells to senescence.74 Similarly, fibroblasts lacking p53 escape from oncogene-induced senescence.6,57 p53, however, can also inhibit the TOR pathway and suppress p21 senescence by promoting quiescence.75 We demonstrated that attenuation of TORC1 results in downregulation of both proteins, and low levels of p21 are sustained over several weeks of treatment.
In addition to a decrease in p21 and p53 levels, both types of senescent cells displayed increased levels of 4E-BP1, present in the hypophosphorylated form. Transient overexpression of 4E-BP1 alone led to a drop in both p21 and p53 levels. It is therefore possible that inhibition of TORC1 delays senescence through upregulation of hypophosphorylated 4E-BP1. Further experiments with inducible expression of 4E-BP1 in RAS-induced cells answer this question.
We were not able to document sustained inhibition of TORC2 with small-molecule inhibitors. Although ATP-competitive inhibitors led to dephosphorylation of AKT S473 within 5 h, treatment lasting longer than 5 hours using inhibitor concentrations that do not affect DNA-PK resulted in rephosphorylation of this site. Phosphorylation of Akt S473 is the most commonly used indicator of TORC2 activity, yet this site could conceivably also be phosphorylated by other kinases, which would explain the observed rephosphorylation in the presence of TOR inhibitors. We are currently investigating this possibility.
Previous studies by Demidenko et al. demonstrated that senescence induced by expression of p21 is attenuated in a similar manner by either rapamycin or LY294002 (inhibitor of PI3K). The authors reasoned that the attenuation is therefore due to inhibition of TORC1 rather than TORC2.76 Our knockdown experiments with shRNA allowed us to discriminate between the functions of TORC1 and TORC2 in senescence. Depletion of TOR or of Raptor disrupted the course of senescence, whereas the depletion of Rictor was without effect. Assuming that the knockdowns affected the senescence-relevant activities of TORC1 and TORC2, these observations suggest that TORC1 is an essential mediator of senescence, but TORC2 is dispensable. However, these data do not rule out the possibility that the extent of Rictor depletion via shRNA was insufficient to interfere with a putative function of TORC2 in RAS-induced senescence.
Cellular senescence is an important barrier to oncogenic transformation. Recent studies demonstrate the presence of oncogene-induced senescence in vivo; abrogation of the senescence response leads to development of hepatocellular carcinoma in mice.1 Mechanisms that disrupt senescence in either damaged cells or pre-malignant cells could lead to oncogenic transformation. TORC1 positively regulates cell growth and proliferation. However, its attenuation activates several negative feedback loops, ultimately resulting in the phosphorylation and activation of AKT, with a possible positive effect on cell proliferation.41,43,77 AKT is activated in response to rapamycin and in response to long-term treatment with ATP-competitive inhibitors. In addition to activation of AKT, both rapamycin and ATP-competitive inhibitors lead to an increased phosphorylation of ERK, a protein whose activation protects the cell against apoptosis.78 Activated AKT and ERK have been demonstrated to occur in vivo in solid tumors from patients undergoing treatment with rapalogs.39,43 Attenuation of TORC1 could be the driving mechanism that leads to escape from senescence in these cells.
Potent inducers of senescence, p53 and p21, are downregulated in response to inhibition of TORC1. It is possible that inactivation of TORC1 postpones senescence via at least two different mechanisms: downregulation of p53 and p21 and upregulation of AKT phosphorylation at S473. Activated AKT promotes survival by multiple means: it phosphorylates MDM2, leading to nuclear translocation and degradation of p53. It also induces degradation of the cyclin-dependent kinase inhibitor p27Kip1, facilitating cell cycle progression, and inhibits apoptosis through inactivation of pro-apoptotic factors, including caspase-9, the BH3-only protein Bad and pro-apoptotic FOXO (forkhead box O) transcription factors.79-86
Three of the PIKK family members have been implicated in phosphorylating AKT S473: DNA-PK,87 ATM88 and TORC2.50,51,89 DNA-PK and ATM are activated in response to DNA damage. TORC1 is attenuated when growth conditions are suboptimal: in response to lack of nutrients, osmotic stress, hypoxia and low energy signals. Negative feedback loops resulting from inhibition of TORC1 lead to AKT S473 phosphorylation via activation of TORC2. It is therefore intriguing to speculate that phosphorylation of this site by the abovementioned PIKK family members serves to stall apoptosis and allow cells to recover. Supporting this notion, a recent study demonstrated that suppression of AKT S473 phosphorylation sensitizes cancer cells to apoptosis induced by TORC1 inhibition. Likewise, insulin-induced phosphorylation of AKT at S473 leads to avoidance of apoptosis in cancer cells with inhibited TORC1.90
We observed an increase in AKT S473 phosphorylation in both replicative and RAS-induced senescent cells and a further increase in phosphorylation of this site with rapamycin treatment and prolonged treatment with ATP-competitive inhibitors. It is possible that AKT S473 allows senescent cells to evade apoptosis despite accumulated DNA damage signaling and promotes proliferation in cells with inhibited TORC1. TBK1 (TANK-Binding Kinase 1) has been implicated in AKT activation91,92 and may contribute to S473 phosphorylation in the presence of Torin1.
A recent study by Kennedy et al. demonstrated that pancreatic cancer cells bearing activated RAS and inactivated tumor suppressor PTEN (phosphatase and tensin homolog) reenter senescence upon rapamycin treatment as judged by decreased BrdU staining and drop in immunohistochemical staining for p21 and p53.93 These observations are in contrast to our data and to the findings of Astle et al., with BJ primary human fibroblasts documenting downregulation of p53 and p21 as a consequence of inhibiting the AKT/TORC1 pathway.94 This discrepancy could be due to inactivation of PTEN in the pancreatic cancer cells.
In addition to phosphorylation and activation of AKT, treatment with rapamycin leads to an increase in hypophosphorylated 4E-BP1. We saw lower levels of 4E-BP1 in senescent fibroblasts and an increase in the total levels of the protein in response to rapamycin treatment. Therefore, in addition to negatively regulating translation via dephosphorylation of the S6 and dephosphorylation and activation of 4E-BP1, rapamycin also leads to accumulation of inhibitory 4E-BP1, further repressing eIF4E-dependent translation. Although only a subpopulation of mRNAs are eIF4E-dependent,95 long-term inhibition of TORC1 would have global effect on translation and lead to a drop in protein levels. We noticed this pattern with several proteins after long-term treatment with rapamycin (Fig. S1). If downregulation of protein translation delays senescence by reducing the amount of total protein in the cell, then inhibition of protein-degrading machinery should have the opposite outcome and lead to accelerated senescence. It has been shown by Kang et al. that disruption of autophagy leads to premature senescence in primary fibroblasts.96 Our data imply that translation via TORC1 contributes to cellular senescence, and attenuation of translation postpones it. Inhibition of TORC1 relieves the translational load in the cells and activates pro-proliferative AKT, allowing cells to postpone senescence.
Rapamycin and its derivatives had limited success in cancer therapy.97 Inhibition of senescence may at least in part explain this lack of effectiveness, as it involves downregulation of the potent tumor suppressors p53 and p21. Inhibition of TORC1 also leads to phosphorylation of AKT S473, which has been linked to pro-proliferative functions of AKT.51,90,98 Senescence serves as a barrier to oncogenic transformation; disruption of oncogene-induced senescence by inhibition of TORC1 could lift this important barrier.
Materials and Methods
Cell culture.
BJ cells (human foreskin fibroblasts) were cultured in MEM (Minimum Essential Medium Eagle), supplemented with 10% FBS (fetal bovine serum), 100 units/ml penicillin and 100 ∝g/ml streptomycin, 2 mM L-glutamine, 1% non-essential amino acids. Cell culture medium was changed every three days during passaging.
Cellular senescence was analyzed as described previously.9 For growth curves, 104 cells were plated into each well in 12-well plates in triplicates. Every 3–5 d, cells were trypsinized from plates, and cell numbers were counted with Beckman Coulter Z1 Particle Counter. At each split, 104 cells were reseeded to each well in fresh plates and allowed to grow until the next split. Population doublings (PD) were calculated with the formula PD = log(N2/N1)/log2, where N1 is the number of cells seeded and N2 is the number of cells recovered. Cellular senescence was confirmed by SA-b-gal staining. Cells were washed with 1x PBS, fixed with 0.5% gluteraldehyde solution in 1x PBS for 15 min at room temperature, washed again with 1x PBS containing 1 mM magnesium chloride, incubated in X-Gal staining solution (35 mM potassium ferricyanide, 35 mM potassium ferrocyanide trihydrate, 2 mM magnesium chloride in 1x PBS and 1 mg/ml X-Gal) overnight and washed with H2O. At least 200 cells were counted under the microscope in randomly chosen fields from each culture well.
Short-term inhibition of TORC1.
Rapamycin (10 nM) or PP242 (200 nM) was added once and cells harvested after 72 h for SDS-PAGE western blot.
Long-term inhibition of TORC1: Rapamycin (1 nM or 10 nM) was added every three days with change of media and cells harvested after passage 60 for SDS-PAGE western blot.
Transfections.
Cells were grown to 95% confluence and transfected with Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s protocol.
For lentiviral infections, cDNA of REDD1 (OriGene) was cloned into pLV-EF1alpha-MCS-IRES-Puro (Biosettia Inc.) between BamHI and EcoRI restriction sites. The PCR primers used to amplify REDD1 cDNA were REDD1-F: GCC CGG GTT GGA TCC GCC ACC ATG CCT AGC CTT TGG GAC CGC TTC T and REDD1-R: GGG AGA GGG GCT AGC TCA ACA CTC CTC AAT GAG CAG CTG T.
4E-BP1 cDNA (Origene) was cloned into pEN_tmcs (Invitrogen) vector between SpeI and XhoI sites. The PCR primers used to amplify 4E-BP1 cDNA were FORspeI4ebp1: GTT ATA CTA GTG CCA CCA TGT CCG GGG GCA GC and REVxhoI4ebp1: CGT AGC TCG AGC TAA GGA AGG GGT GGT TGC. Recombination reaction was performed with pSLIK-hygro vector (Invitrogen) according to the manufacturer’s Gateway cloning protocol. Recombinant lentiviruses were produced by transfection into HEK293T cells in the presence of packaging plasmids (pMDL-G, pRSV-REV and pVSV-G) using Lipofectamine 2000 as described in ViraPowerTM Lentiviral Expression System protocol (Invitrogen).
shRNA.
shRNAs for TOR, Raptor and Rictor were designed and cloned into pLV-H1-E2F1-puro based on the single-oligonucleotide RNA interference technology (Biosettia) using oligonucleotides with the following sequences:
sh-mTOR-2627-AAA AGC TAT GTA GTA GAG CCC TAT TGG ATC CAA TAG GGC TCT ACT ACA TAG C,
sh-mTOR-3097-AAA AGG TCA TGC CCA CGT TCC TTT TGG ATC CAA AAG GAA CGT GGG CAT GAC C, sh-RAPTOR-2116-AAA AGC AGG TGC TGT TAA GCC AAT TGG ATC CAA TTG GCT TAA CAG CAC CTG C, sh-RAPTOR-2400-AAA AGG ACC ATG ACG GCT TTC ATT TGG ATC CAA ATG AAA GCC GTC ATG GTC C, sh-RICTOR-3439-AAA AGG AAC AAC TCT GTA ATG AAT TGG ATC CAA TTC ATT ACA GAG TTG TTC C, sh-RICTOR-333-AAA AGC TAC GAG CGC TTC GAT ATT TGG ATC CAA ATA TCG AAG CGC TCG TAG C, sh-RICTOR-1201-AAA AGC ACT GAT ACT CTC TGC ATT TGG ATC CAA ATG CAG AGA GTA TCA GTG C.
BJ cells were grown to 70% confluence in a 90 mm plate and infected with the shRNA-encoding lentiviruses in fresh medium containing 8 ∝g/mL polybrene. Transduced cells were selected with 1.2 ∝g/mL of puromycin or 120 ∝g/mL hygromycin two days post-transduction.
Western blot analysis.
Cells were harvested in RIPA buffer (50 mM Tris-Cl, pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate and 0.1% SDS) supplemented with complete protease inhibitor cocktail mix (Roche), 1 mM sodium orthovanadate and 200 nM PMSF. Cell lysates were centrifuged at 13,000 rpm at 4°C for 10 minutes and supernatants were collected. Protein concentration was determined using BCA protein assay reagent (Bio-RAD) according to manufacturer’s protocol. Twenty ∝g of protein was separated by SDS-PAGE western blot and transferred to a PVDV membrane (Millipore). The blots were incubated with antibodies in 5% BSA and 1x TBST (Tris-Buffered Saline and Tween 20) and secondary antibodies in 5% milk and 1x TBST. Primary antibodies for IL8 and p53 were purchased from Santa Cruz Biotechnology and all the remaining primary antibodies and the secondary antibodies were from Cell Signaling Technology. The proteins bands were visualized using West Pico substrate (Thermo Scientific Pierce) using Bio-RAD Chemi-DOC XRS (Bio-RAD) according to the manufacturer’s protocol.
Real-time RT-PCR.
RNA was isolated from cells using Trizol (Invitrogen) according to manufacturer’s protocol. 200 ng of RNA was converted to cDNA with oligo(dT) primer and iScriptTM Reverse Transcription Supermix (Bio-rad). Quantitative real-time PCR was performed in triplicates with gene-specific primers and SsoAdvancedTM SYBR® Green Supermix (Bio-rad) in a BioRad CFX96 REAL TIME SYSTEM following manufecturer’s protocols. Primer sequences for RT-PCR:
mTOR-rt-F4 CAG GGT TCG AGA TAA GCT CACmTOR-rt-R4 TTA CCA GAA AGG GCA CCA GRaptor-rt-F2 TGA GCG TCA ATG GAG ATG TGRaptor-rt-R2 AGA TGG CGG TGA ACT GAT TGRictor-rt-F3 ACT AAA TGT CAT GAG ACT GGG CRictor-rt-R3 TTG TAT GAA CCT CCG ACA CGThe mRNA level for each gene was normalized to that of GAPDH.
Supplementary Material
Acknowledgements
This is manuscript 21,501 of The Scripps Research Institute. Work of the authors is funded by NIH grants CA078230, CA151574 and CA153124. M.K. is supported by a Skaggs Oxford-Scripps Predoctoral Fellowship. The authors would like to thank the Cravatt lab for antibodies and useful discussions, Vogt lab for discussions and critical reading, Stephanie Lu for technical assistance and David Sabatini and Nathanael Gray for Torin1 inhibitor.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Note
Supplemental materials can be found at: http://www.landesbioscience.com/journals/cc/article/20683
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/20683
References
- 1.Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547–51. doi: 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- 2.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–56. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blagosklonny MV. Cell cycle arrest is not senescence. Aging . 2011;3:94–101. doi: 10.18632/aging.100281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–33. doi: 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 5.Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24:2463–79. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/S0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 7.DeNicola GM, Tuveson DA. RAS in cellular transformation and senescence. Eur J Cancer. 2009;45(Suppl 1):211–6. doi: 10.1016/S0959-8049(09)70036-X. [DOI] [PubMed] [Google Scholar]
- 8.Bulavin DV, Demidov ON, Saito S, Kauraniemi P, Phillips C, Amundson SA, et al. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat Genet. 2002;31:210–5. doi: 10.1038/ng894. [DOI] [PubMed] [Google Scholar]
- 9.Wang W, Chen JX, Liao R, Deng Q, Zhou JJ, Huang S, et al. Sequential activation of the MEK-extracellular signal-regulated kinase and MKK3/6-p38 mitogen-activated protein kinase pathways mediates oncogenic ras-induced premature senescence. Mol Cell Biol. 2002;22:3389–403. doi: 10.1128/MCB.22.10.3389-3403.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bulavin DV, Kovalsky O, Hollander MC, Fornace AJ., Jr. Loss of oncogenic H-ras-induced cell cycle arrest and p38 mitogen-activated protein kinase activation by disruption of Gadd45a. Mol Cell Biol. 2003;23:3859–71. doi: 10.1128/MCB.23.11.3859-3871.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han J, Sun P. The pathways to tumor suppression via route p38. Trends Biochem Sci. 2007;32:364–71. doi: 10.1016/j.tibs.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 12.Sun P, Yoshizuka N, New L, Moser BA, Li Y, Liao R, et al. PRAK is essential for ras-induced senescence and tumor suppression. Cell. 2007;128:295–308. doi: 10.1016/j.cell.2006.11.050. [DOI] [PubMed] [Google Scholar]
- 13.Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–42. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 14.Mallette FA, Gaumont-Leclerc MF, Ferbeyre G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev. 2007;21:43–8. doi: 10.1101/gad.1487307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 16.Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43:405–13. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- 17.d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–8. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 18.Gire V, Roux P, Wynford-Thomas D, Brondello JM, Dulic V. DNA damage checkpoint kinase Chk2 triggers replicative senescence. EMBO J. 2004;23:2554–63. doi: 10.1038/sj.emboj.7600259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shay JW, Pereira-Smith OM, Wright WE. A role for both RB and p53 in the regulation of human cellular senescence. Exp Cell Res. 1991;196:33–9. doi: 10.1016/0014-4827(91)90453-2. [DOI] [PubMed] [Google Scholar]
- 20.Deng Q, Liao R, Wu BL, Sun P. High intensity ras signaling induces premature senescence by activating p38 pathway in primary human fibroblasts. J Biol Chem. 2004;279:1050–9. doi: 10.1074/jbc.M308644200. [DOI] [PubMed] [Google Scholar]
- 21.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011;30:1536–48. doi: 10.1038/emboj.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iwasa H, Han J, Ishikawa F. Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes Cells. 2003;8:131–44. doi: 10.1046/j.1365-2443.2003.00620.x. [DOI] [PubMed] [Google Scholar]
- 24.Noda A, Ning Y, Venable SF, Pereira-Smith OM, Smith JR. Cloning of senescent cell-derived inhibitors of DNA synthesis using an expression screen. Exp Cell Res. 1994;211:90–8. doi: 10.1006/excr.1994.1063. [DOI] [PubMed] [Google Scholar]
- 25.Beauséjour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003;22:4212–22. doi: 10.1093/emboj/cdg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–52. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 27.Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle. 2009;8:1888–95. doi: 10.4161/cc.8.12.8606. [DOI] [PubMed] [Google Scholar]
- 28.Leontieva OV, Blagosklonny MV. DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging . 2010;2:924–35. doi: 10.18632/aging.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blagosklonny MV. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging. 2012;4:159–65. doi: 10.18632/aging.100443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang H, Vogt PK. Constitutively active Rheb induces oncogenic transformation. Oncogene. 2008;27:5729–40. doi: 10.1038/onc.2008.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang HW, Aoki M, Fruman D, Auger KR, Bellacosa A, Tsichlis PN, et al. Transformation of chicken cells by the gene encoding the catalytic subunit of PI 3-kinase. Science. 1997;276:1848–50. doi: 10.1126/science.276.5320.1848. [DOI] [PubMed] [Google Scholar]
- 32.Staal SP. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: amplification of AKT1 in a primary human gastric adenocarcinoma. Proc Natl Acad Sci USA. 1987;84:5034–7. doi: 10.1073/pnas.84.14.5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vogt PK, Hart JR, Gymnopoulos M, Jiang H, Kang S, Bader AG, et al. Phosphatidylinositol 3-kinase: the oncoprotein. Curr Top Microbiol Immunol. 2010;347:79–104. doi: 10.1007/82_2010_80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aoki M, Batista O, Bellacosa A, Tsichlis P, Vogt PK. The akt kinase: molecular determinants of oncogenicity. Proc Natl Acad Sci USA. 1998;95:14950–5. doi: 10.1073/pnas.95.25.14950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao L, Vogt PK. Class I PI3K in oncogenic cellular transformation. Oncogene. 2008;27:5486–96. doi: 10.1038/onc.2008.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ciraolo E, Morello F, Hirsch E. Present and future of PI3K pathway inhibition in cancer: perspectives and limitations. Curr Med Chem. 2011;18:2674–85. doi: 10.2174/092986711796011193. [DOI] [PubMed] [Google Scholar]
- 37.Dancey J. mTOR signaling and drug development in cancer. Nature reviews. Clin Oncol. 2010;7:209–19. doi: 10.1038/nrclinonc.2010.21. [DOI] [PubMed] [Google Scholar]
- 38.Vanhaesebroeck B, Vogt PK, Rommel C. PI3K: from the bench to the clinic and back. Curr Top Microbiol Immunol. 2010;347:1–19. doi: 10.1007/82_2010_65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–8. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Facchinetti V, Ouyang W, Wei H, Soto N, Lazorchak A, Gould C, et al. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 2008;27:1932–43. doi: 10.1038/emboj.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dibble CC, Asara JM, Manning BD. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol Cell Biol. 2009;29:5657–70. doi: 10.1128/MCB.00735-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Efeyan A, Sabatini DM. mTOR and cancer: many loops in one pathway. Curr Opin Cell Biol. 2010;22:169–76. doi: 10.1016/j.ceb.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–74. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lempiäinen H, Halazonetis TD. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J. 2009;28:3067–73. doi: 10.1038/emboj.2009.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:457–68. doi: 10.1016/S1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- 46.Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 47.Jacinto E, Loewith R, Schmidt A, Lin S, Rüegg MA, Hall A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–8. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 48.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–45. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 49.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 51.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–37. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 52.Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc Natl Acad Sci USA. 1998;95:1432–7. doi: 10.1073/pnas.95.4.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gingras AC, Kennedy SG, O’Leary MA, Sonenberg N, Hay N. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev. 1998;12:502–13. doi: 10.1101/gad.12.4.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ruvinsky I, Sharon N, Lerer T, Cohen H, Stolovich-Rain M, Nir T, et al. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev. 2005;19:2199–211. doi: 10.1101/gad.351605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gingras AC, Raught B, Gygi SP, Niedzwiecka A, Miron M, Burley SK, et al. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 2001;15:2852–64. doi: 10.1101/gad.912401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haghighat A, Mader S, Pause A, Sonenberg N. Repression of cap-dependent translation by 4E-binding protein 1: competition with p220 for binding to eukaryotic initiation factor-4E. EMBO J. 1995;14:5701–9. doi: 10.1002/j.1460-2075.1995.tb00257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Petroulakis E, Parsyan A, Dowling RJ, LeBacquer O, Martineau Y, Bidinosti M, et al. p53-dependent translational control of senescence and transformation via 4E-BPs. Cancer Cell. 2009;16:439–46. doi: 10.1016/j.ccr.2009.09.025. [DOI] [PubMed] [Google Scholar]
- 58.Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–31. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 59.Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci USA. 2009;106:17031–6. doi: 10.1073/pnas.0905299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Atadja P, Wong H, Garkavtsev I, Veillette C, Riabowol K. Increased activity of p53 in senescing fibroblasts. Proc Natl Acad Sci USA. 1995;92:8348–52. doi: 10.1073/pnas.92.18.8348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Webley K, Bond JA, Jones CJ, Blaydes JP, Craig A, Hupp T, et al. Posttranslational modifications of p53 in replicative senescence overlapping but distinct from those induced by DNA damage. Mol Cell Biol. 2000;20:2803–8. doi: 10.1128/MCB.20.8.2803-2808.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rousseau D, Kaspar R, Rosenwald I, Gehrke L, Sonenberg N. Translation initiation of ornithine decarboxylase and nucleocytoplasmic transport of cyclin D1 mRNA are increased in cells overexpressing eukaryotic initiation factor 4E. Proc Natl Acad Sci USA. 1996;93:1065–70. doi: 10.1073/pnas.93.3.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu Q, Kirubakaran S, Hur W, Niepel M, Westover K, Thoreen CC, et al. Kinome-wide selectivity profiling of ATP-competitive mammalian target of rapamycin (mTOR) inhibitors and characterization of their binding kinetics. J Biol Chem. 2012;287:9742–52. doi: 10.1074/jbc.M111.304485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110:177–89. doi: 10.1016/S0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- 65.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–75. doi: 10.1016/S0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 66.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–84. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 67.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–68. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 68.Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18:2893–904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang H, Kubica N, Ellisen LW, Jefferson LS, Kimball SR. Dexamethasone represses signaling through the mammalian target of rapamycin in muscle cells by enhancing expression of REDD1. J Biol Chem. 2006;281:39128–34. doi: 10.1074/jbc.M610023200. [DOI] [PubMed] [Google Scholar]
- 70.Molitoris JK, McColl KS, Swerdlow S, Matsuyama M, Lam M, Finkel TH, et al. Glucocorticoid elevation of dexamethasone-induced gene 2 (Dig2/RTP801/REDD1) protein mediates autophagy in lymphocytes. J Biol Chem. 2011;286:30181–9. doi: 10.1074/jbc.M111.245423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Corradetti MN, Inoki K, Guan KL. The stress-inducted proteins RTP801 and RTP801L are negative regulators of the mammalian target of rapamycin pathway. J Biol Chem. 2005;280:9769–72. doi: 10.1074/jbc.C400557200. [DOI] [PubMed] [Google Scholar]
- 72.Ruggero D, Montanaro L, Ma L, Xu W, Londei P, Cordon-Cardo C, et al. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat Med. 2004;10:484–6. doi: 10.1038/nm1042. [DOI] [PubMed] [Google Scholar]
- 73.Dulić V, Drullinger LF, Lees E, Reed SI, Stein GH. Altered regulation of G1 cyclins in senescent human diploid fibroblasts: accumulation of inactive cyclin E-Cdk2 and cyclin D1-Cdk2 complexes. Proc Natl Acad Sci USA. 1993;90:11034–8. doi: 10.1073/pnas.90.23.11034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fang L, Igarashi M, Leung J, Sugrue MM, Lee SW, Aaronson SA. p21Waf1/Cip1/Sdi1 induces permanent growth arrest with markers of replicative senescence in human tumor cells lacking functional p53. Oncogene. 1999;18:2789–97. doi: 10.1038/sj.onc.1202615. [DOI] [PubMed] [Google Scholar]
- 75.Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci USA. 2010;107:9660–4. doi: 10.1073/pnas.1002298107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Demidenko ZN, Shtutman M, Blagosklonny MV. Pharmacologic inhibition of MEK and PI-3K converges on the mTOR/S6 pathway to decelerate cellular senescence. Cell Cycle. 2009;8:1896–900. doi: 10.4161/cc.8.12.8809. [DOI] [PubMed] [Google Scholar]
- 77.Harrington LS, Findlay GM, Lamb RF. Restraining PI3K: mTOR signalling goes back to the membrane. Trends Biochem Sci. 2005;30:35–42. doi: 10.1016/j.tibs.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 78.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–31. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 79.Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, et al. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–21. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 80.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–41. doi: 10.1016/S0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 81.del Peso L, González-García M, Page C, Herrera R, Nuñez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–9. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 82.Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8:1153–60. doi: 10.1038/nm761. [DOI] [PubMed] [Google Scholar]
- 83.Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci USA. 2001;98:11598–603. doi: 10.1073/pnas.181181198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, et al. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med. 2002;8:1145–52. doi: 10.1038/nm759. [DOI] [PubMed] [Google Scholar]
- 85.Tran H, Brunet A, Griffith EC, Greenberg ME. The many forks in FOXO’s road. Sci STKE. 2003;2003:RE5. doi: 10.1126/stke.2003.172.re5. [DOI] [PubMed] [Google Scholar]
- 86.Viglietto G, Motti ML, Bruni P, Melillo RM, D’Alessio A, Califano D, et al. Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27(Kip1) by PKB/Akt-mediated phosphorylation in breast cancer. Nat Med. 2002;8:1136–44. doi: 10.1038/nm762. [DOI] [PubMed] [Google Scholar]
- 87.Feng J, Park J, Cron P, Hess D, Hemmings BA. Identification of a PKB/Akt hydrophobic motif Ser-473 kinase as DNA-dependent protein kinase. J Biol Chem. 2004;279:41189–96. doi: 10.1074/jbc.M406731200. [DOI] [PubMed] [Google Scholar]
- 88.Viniegra JG, Martínez N, Modirassari P, Hernández Losa J, Parada Cobo C, Sánchez-Arévalo Lobo VJ, et al. Full activation of PKB/Akt in response to insulin or ionizing radiation is mediated through ATM. J Biol Chem. 2005;280:4029–36. doi: 10.1074/jbc.M410344200. [DOI] [PubMed] [Google Scholar]
- 89.Hresko RC, Mueckler M. mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J Biol Chem. 2005;280:40406–16. doi: 10.1074/jbc.M508361200. [DOI] [PubMed] [Google Scholar]
- 90.Yellen P, Saqcena M, Salloum D, Feng J, Preda A, Xu L, et al. High-dose rapamycin induces apoptosis in human cancer cells by dissociating mTOR complex 1 and suppressing phosphorylation of 4E-BP1. Cell Cycle. 2011;10:3948–56. doi: 10.4161/cc.10.22.18124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xie X, Zhang D, Zhao B, Lu MK, You M, Condorelli G, et al. IkappaB kinase epsilon and TANK-binding kinase 1 activate AKT by direct phosphorylation. Proc Natl Acad Sci USA. 2011;108:6474–9. doi: 10.1073/pnas.1016132108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ou YH, Torres M, Ram R, Formstecher E, Roland C, Cheng T, et al. TBK1 directly engages Akt/PKB survival signaling to support oncogenic transformation. Mol Cell. 2011;41:458–70. doi: 10.1016/j.molcel.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kennedy AL, Morton JP, Manoharan I, Nelson DM, Jamieson NB, Pawlikowski JS, et al. Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumorigenesis. Mol Cell. 2011;42:36–49. doi: 10.1016/j.molcel.2011.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Astle MV, Hannan KM, Ng PY, Lee RS, George AJ, Hsu AK, et al. AKT induces senescence in human cells via mTORC1 and p53 in the absence of DNA damage: implications for targeting mTOR during malignancy. Oncogene. 2011 doi: 10.1038/onc.2011.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Richter JD, Sonenberg N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature. 2005;433:477–80. doi: 10.1038/nature03205. [DOI] [PubMed] [Google Scholar]
- 96.Kang HT, Lee KB, Kim SY, Choi HR, Park SC. Autophagy impairment induces premature senescence in primary human fibroblasts. PLoS ONE. 2011;6:e23367. doi: 10.1371/journal.pone.0023367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Benjamin D, Colombi M, Moroni C, Hall MN. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discov. 2011;10:868–80. doi: 10.1038/nrd3531. [DOI] [PubMed] [Google Scholar]
- 98.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.