

Abstract
The aspartate biosynthetic pathway provides essential metabolites for many important biological functions, including the production of four essential amino acids. Since this critical pathway is only present in plants and microbes any disruptions will be fatal to these organisms. An early pathway enzyme, L-aspartate-β-semialdehyde dehydrogenase (ASADH), produces a key intermediate at the first branch point of this pathway. Developing potent and selective inhibitors against several orthologs in the ASADH family can serve as lead compounds for antibiotic development. Kinetic studies of two small molecule fragment libraries have identified inhibitors that show good selectivity against ASADHs from two different bacterial species, Streptococcus pneumoniae and Vibrio cholerae, despite the presence of an identical constellation of active site amino acids in this homologous enzyme family. Structural characterization of enzyme-inhibitor complexes have elucidated different modes of binding between these structurally related enzymes. This information provides the basis for a structure guided approach to the development of more potent and more selective inhibitors.
Keywords: enzyme inhibition, enzyme inactivation, structural studies, X-ray crystallography, ASA dehydrogenase
The aspartate biosynthetic pathway is found only in plants and microbes. The commitment step to this pathway is the phosphorylation of aspartic acid catalyzed by aspartokinases (1). The next enzyme, aspartate-β-semialdehyde dehydrogenase (ASADH), catalyzes a reductive dephosphorylation to produce aspartate β-semialdehyde. At this point the pathway can branch to produce either homoserine as the common precursor for the synthesis of methionine, threonine and isoleucine, or go through dihydrodipicolinate and diaminopimelate to synthesize lysine (2). Thus, one-quarter of the amino acids required for protein synthesis in these organisms are linked through and synthesized by this pathway. In addition to these essential amino acids this pathway also produces several important metabolites that play crucial roles in microbial developmental processes, including cell wall biosynthesis, protective dormancy, and virulence factor production. These intermediates include dihydrodipicolinate, a precursor of dipicolinate which is the major component of bacterial spores (3), and diaminopimelate, required for cross-linking of the peptidoglycan polymers in bacterial cell wall synthesis (4). Another product of this pathway, S-adenosylmethionine, is an essential methyl group donor that also serves as a precursor for quorum sensing signaling molecules (5,6) with critical roles in triggering virulence factors in infectious organisms.
Selective perturbations of the asd gene that encodes for ASADH have been shown to be lethal to microorganisms (7), and numerous studies to determine the minimal set of genes needed for organism survival have identified the asd gene as being essential (8-11). Our goal is to identify selective inhibitors of this validated target that can be developed into lead compounds for antimicrobial development.
The identification of new inhibitors against a target enzyme have traditionally followed two quite different approaches, either using the known structures of substrates and products to guide the synthesis of structural analogues or by screening compound libraries to identify novel groups of inhibitory structures. Screening to identify initial hits has been driven by the search for high affinity compounds for each potential drug target. Modifications of these initial hits to further enhance target affinity and improve target selectivity are then used to develop the advanced leads that move towards clinical trials. In contrast to this traditional approach, fragment-based drug discovery is designed around the hypothesis that high affinity is not the best selection criteria with which to identify initial hits (12). Instead, ligand efficiency (L.E.) has been proposed as the selection metric, where ligand efficiency is defined as the free energy of binding (ΔG) per heavy (non-hydrogen) atom in the ligand (13). Typical fragments, developed from incorporating useful functional groups into molecular scaffolds, are in the molecular weight range of 120-250 Da and have affinities in the high micromolar to low millimolar range. These fragments are then screened to probe the essential binding sites within the active site of a target enzyme. The aim is to identify a set of minimal functional components that bind to an enzyme target with reasonable affinity and high L.E. values.
We have examined both the substrate analogue and the fragment screening approaches to identify new enzyme inhibitors that show selectivity against representative ASADH enzymes isolated from different microorganisms. Kinetic studies had been used to screen fragment molecule libraries to identify new compounds that bind to ASADHs with high ligand efficiencies. Inhibitors were identified with selectivity against either Gram-negative or Gram-positive bacterial enzyme forms, or compounds that inhibit only a fungal form of ASADH (14). Different compounds from these groups of inhibitors have now been crystallized with ASADHs from a representative Gram-positive Streptococcus pneumoniae and a representative Gram-negative Vibrio cholerae bacterial species. The structures reported herein are being used to guide the design of more potent inhibitors with enhanced selectivity towards a single microbial species or a subset of bacterial or fungal species.
Results and Discussion
Examination of substrate analogue inhibitors
Structural analogues of the amino acid substrate and product of the ASADH-catalyzed reaction have been shown to inhibit this enzyme with affinities in the low micromolar range (15,16). Complexes of a variety of analogues bound to ASADHs isolated and purified from the gram-positive bacterium S. pneumoniae (spASADH) and from the gram-negative bacterium V. cholerae (vcASADH) have now been crystallized and structurally characterized to determine the nature of the interactions that lead to binding and inhibition of enzymatic activity. spASADH has greater than 40% sequence identity to the other Gram-positive ASADHs, but only 24% identity to vcASADH (17).
The instability of β-aspartyl phosphate has prevented determination of the specific binding interactions between this substrate and the enzyme. Aspartyl β-difluorophosphonate (β-AFP) is a structural analogue of this substrate in which the high energy acyl phosphate group has been replaced by a more stable fluorine-substituted phosphonate group (16) (Figure 1). The structure of a ternary complex with NADP and β-AFP bound to spASADH was determined to assess the mode of binding of the phosphorylated amino acid substrate (Table 1). An Fo-Fc omit map calculated after initial refinement cycles shows evidence of β-AFP binding in the active site of both subunits in the functional dimer of spASADH (Figure 2). However, breaks in the electron density of the ligand indicate some disorder in inhibitor binding. The best refined portion of the density is tetragonal in shape and corresponds to the phosphonate moiety of the inhibitor. This group is bound in the site where the phosphate group required for phosphorylation of the substrate (ASA) in the reverse reaction had previously been shown to bind (18). This anion site is composed of a “recognition dyad” where the phosphonate group is oriented similarly to phosphate through electrostatic interactions with the positively-charged side chains of Arg99 and Lys223. Hydrogen-bonds between the phosphonate oxygens and the amide side chain of Asn127 provide additional stability. There is also density proximal to the guanidinium group of Arg245 (Figure 2) which has been modeled as the disordered carboxylate of β-AFP that is bound in the carboxylate site of the ASA substrate (19). The significantly weaker density corresponding to carbon backbone of the inhibitor is more difficult to model and is likely a consequence of several different orientations of these interior carbons. The α-amino group of the bound inhibitor cannot be oriented towards the active site side chain (Glu220) that is normally responsible for binding the corresponding substrate α-amino group (20) without causing significant geometric distortion. The inability to form this stabilizing interaction is the likely explanation for the lack of interpretable electron density for this functional group and for the central carbon atoms in this structure.
Figure 1.

Structures of the β-aspartyl phosphate substrate and substrate analogue inhibitors.
Table 1.
Diffraction data and refinement statistics for ASADH-inhibitor complexes
| Enzyme form |
Ligands | Space group, Unit cell (Å, °) |
Resolution (Å) a |
Rsymm a |
I/σI a | Completeness (%) a |
Rw/Rfree | rmsd (bonds,angles) |
Ramachandran plot (%) b |
PDB ID |
|---|---|---|---|---|---|---|---|---|---|---|
| spASADH | aspartyl β-difluorophosphonate + NADP |
P21 60, 98.7, 64.5 β=100.8° |
1.80 (1.86-1.80) |
0.064 (0.34) |
6.8 (2.2) |
89 (89) | 0.20/0.24 | 0.007, 1.11 | 94.5/3.1/1.4 | 3Q11 |
| vcASADH | S-allyl-L-cysteine sulfoxide | P43 212 107.6, 107.6, 154 |
1.80 (1.86-1.80) |
0.057 (0.32) |
22.6 (2.2) |
95 (64) | 0.19/0.21 | 0.006, 0.94 | 96.5/2.4/1.1 | 3Q0E |
| spASADH | cysteamine | P212121 72.6, 78.8, 244.6 |
2.26 (2.34-2.26) |
0.057 (0.30) |
13.6 (2.2) |
83 (44) | 0.19/0.24 | 0.008, 1.14 | 94.5/3.7/1.8 | 3Q1L |
| vcASADH | S-carbamoyl-L-cysteine + NADP |
P4212 155, 155, 69.2 |
1.75 (1.81-1.75) |
0.068 (0.35) |
14.9 (3.2) |
85 (78) | 0.19/0.21 | 0.006, 1.03 | 96.3/2.9/0.8 | 3PZR |
| spASADH | D-2,3-diaminopropionate | P212121 73.1, 78.2, 246.1 |
2.20 (2.28-2.20) |
0.067 (0.19) |
18.5 (5) |
90 (80) | 0.20/0.25 | 0.008, 1.09 | 94.1/4.1/1.8 | 3PYL |
| spASADH | D-2,3-diaminopropionate + NADP |
P21 60, 100.1, 64.5 β=100.8° |
2.00 (2.03-2.00) |
0.090 (0.22) |
10.5 (4.2) |
93 (70) | 0.18/0.21 | 0.006, 1.00 | 95.8/2.8/1.4 | 3PZB |
| spASADH | D-2-aminoadipate + 2′,5′-ADP |
P21 60.1, 99.4, 64.6 β=101.1° |
2.00 (2.07-2.00) |
0.060 (0.13) |
12.3 (5.7) |
82 (65) | 0.20/0.23 | 0.007, 1.01 | 95.6/3.1/1.3 | 3PWS |
| spASADH | 2-aminoterephthalate + NADP |
P21 60.1, 99.3, 64.5 β=101° |
1.60 (1.66-1.60) |
0.055 (0.30) |
17.5 (3.5) |
93 (67) | 0.16/0.18 | 0.012, 1.42 | 95.9/2.7/1.4 | 3PYX |
| spASADH | cyclohexane-1,4-dicarboxylate + 2′,5′-ADP |
P21 59.9, 99.7, 64.4 β=100.9° |
1.50 (1.55-1.50) |
0.034 (0.18) |
22.4 (5.1) |
87 (44) | 0.13/0.16 | 0.011, 1.41 | 96.0/2.6/1.4 | 3PWK |
statistics for the highest resolution shell are in parentheses
Residues in preferred, allowed and generously allowed regions of the Ramachandran plot as reported by Coot (25).
Figure 2.

Binding of the substrate analogue aspartyl β-difluorophosphonate to the spASADH/ NADP complex. A 1.8 Å 2Fo-Fc electron density map (blue, 1.5σ level) shows the active site groups and an Fo-Fc omit-electron density map (orange, 2.5σ level) is superimposed with the model of the substrate analogue inhibitor. The phosphonate group makes electrostatic interactions with Arg99 and Lys223 and hydrogen bonds to Asn127, while the carboxyl group of the inhibitor interacts with Arg245.
S-allyl-L-cysteine sulfoxide is another analogue of the substrate aspartyl β-phosphate in which the phosphate moiety has been replaced by an allyl group (Figure 1). This substrate analogue was found to be a potent inhibitor of ASADH with a measured Ki of 8 μM and a very high ligand efficiency value of 0.7 kcal/mol/heavy atom (13). The structure of this inhibitor bound in a ternary complex with vcASADH and NADP contains additional electron density adjacent to the active site Cys134 side chain that is consistent with inactivation of vcASADH through disulfide bond formation (Figure 3). This active site-directed inactivator is oriented through an electrostatic interaction between its carboxyl group and Arg101, the functional group that is part of the phosphate binding site. In addition, attack of the cysteine nucleophile to form this disulfide bond causes reductive displacement of the allyl group, leading to a covalently bound cysteinyl moiety.
Figure 3.
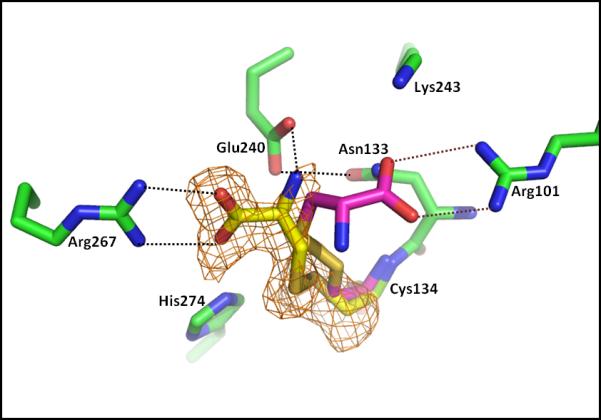
Binding of active site-directed inactivators to the cysteine nucleophile of vcASADH. S-carbamoyl-L-cysteine and S-allyl-L-cysteine are covalently bound to the catalytic Cys134 of the vcASADH/NADP complex. In each case formation of the disulfide bond leads to displacement of the S-moiety of the inhibitors. However, the cysteinyl moiety produced from the S-carbamoyl cysteine inhibitor (yellow structure) is oriented towards Arg267 (orange Fo-Fc omit map, 2.5σ level calculated to 1.75 Å), while the same moiety produced from S-allyl-L-cysteine (red structure) is oriented towards Arg101, with the interactions shown in red dotted lines.
Identification of Inhibitors by fragment library screening
Now that a picture has emerged showing how β-aspartyl phosphate binds in the active site of ASADH, a screening approach was initiated to identify new chemical entities than could potentially interact with this target enzyme family with greater conformational diversity, to increase the likelihood of binding, and improved selectivity against different members of this homogeneous enzyme family. Fragment library screening experiments were initially conducted by soaking either apo-ASADH crystals or crystals of the ASADH-cofactor complexes into four-compound cocktails of these small fragments as described in Methods. As inhibitor-containing cocktails were identified, soaking with the individual compounds and co-crystallization studies were conducted with these inhibitors. Several thiol-containing amino acid analogues showed high potency inhibition of the ASADHs (14), and were subsequently identified to be active site-directed inactivators yielding similar complexes to those observed from the substrate analogue studies described above. However, other amino acid analogues were found to selectively inhibit only a single ASADH enzyme form. Structural characterization of these enzyme-inhibitor complexes provides additional insights into the different modes of inhibitor binding and will provide the basis for the development of additional inhibitors with improved selectivity for ASADHs from specific microorganisms.
Active site-directed inactivators from fragment libraries
Crystal soaking experiments with compound cocktails from a soluble fragment library (SFL) produced some enzyme-complex structures with additional electron density present in the active site region, demonstrating the binding of one or more cocktail components. Analysis of the difference electron density map obtained from one of these structures is consistent with formation of a covalent disulfide bond between one of the cocktail components and the active site nucleophile Cys128 of spASADH, similar to that observed for the binding of S-allyl-L-cysteine sulfoxide. The shape of the unassigned density and subsequent kinetic studies with the individual cocktail components (14) identified this inactivator as cysteamine. Two alternative conformations of this bound inactivator were required to explain the broadened electron density seen at the amine end of the inactivator (data not shown). In each conformation the amine is located within hydrogen-bonding distance from one of the two terminal nitrogens in the guanidinium group of Arg245. The amine group of the inactivator has to be deprotonated and neutral to make a favorable binding interaction with this positively charged side chain, thereby causing this inactivator to be oriented in the opposite direction in the active site from the disulfide adduct produced by reaction with S-allyl-L-cysteine sulfoxide.
Another example of a covalently-bound fragment was identified in the ternary complex of vcASADH with its cofactor NADP and S-carbamoyl-L-cysteine from the SFL. In this structure contiguous electron density from the active site cysteine (Cys134) supports the formation of a disulfide bond with L-cysteine portion of S-carbamoyl-L-cysteine (Figure 3). As was seen with the modification of this cysteine side chain by S-allyl-L-cysteine sulfoxide, formation of this disulfide adduct causes displacement of the carbamoyl group, with no evidence seen for this moiety in the electron density map. A precedent for this reaction was previously observed in the reductive demethylation of another active site-directed inactivator, S-methyl-L-cysteine sulfoxide upon binding to vcASADH (19). These results show that even protected thiol-containing inhibitors will react with the cysteine nucleophile of this enzyme family, leading to covalent inactivation through disulfide bond formation.
Despite producing the same Cys134-cysteinyl adduct that was obtained through the reaction with S-allyl-L-cysteine sulfoxide, the adduct obtained upon modification with S-carbamoyl-L-cysteine is also oriented differently in the active site. In this case the terminal carboxyl group forms an electrostatic interaction with the other active site arginine (Arg267) and this orientation is stabilized by additional electrostatic and hydrogen-bonding interactions between the amino group of the inactivator and the side chain functional groups of Glu240 and Asn133 (Figure 3). The presence of two different orientations of the identical structure in the active site is unusual, and indicates similar binding energies between these different orientations thereby highlighting the important role played by each of these active site arginines in ligand binding. These structures suggest the possibility of designing inhibitors with appropriately spaced functional groups that could simultaneously access both active site arginines and lead to improved affinity and selectivity.
These four different inhibitor complexes of ASADH were each found to crystallize in different space groups (Table 1) that are indicative of either an open or a closed conformation of the enzyme. The V. cholerae form of ASADH typically crystallizes in the P43212 space group in the absence of the nucleotide cofactor and in P4212 in a more closed conformation with bound cofactor (19). Similarly, the S. pneumoniae ASADH structures crystallize in P212121 in the absence of cofactor and in P21 with a bound cofactor (17). However, in each case the orientation of the substrate binding groups are not affected by these conformational changes.
Non-covalent fragment library inhibitors
A non-covalently bound compound from library screening was detected while analyzing electron density maps of a complex of spASADH obtained by soaking enzyme crystals with a SFL cocktail containing D,L-2,3-diaminopropionate. Electron density in the catalytic sites of both dimers (this complex crystallize with two dimers in the asymmetric unit) of spASADH indicated ligand binding, and a model of this inhibitor was fit into the density and refined (Table 1). Unfortunately, the data quality of this structure was not sufficient to unambiguously determine if a single enantiomer was bound from this racemic mixture. A ternary complex of this inhibitor in the presence of NADP was subsequently co-crystallized, and this complex produced better diffraction quality crystals with data extending to 2 Å resolution (Table 1). At this resolution the choice of the correct enantiomer was obvious, with the D-isomer of 2,3-diaminopropionate bound in the active site of spASADH (Figure 4). This unexpected result is in contrast to the absolute requirement of L-stereochemistry for the amino acid substrate for the ASADHs. The binding of this inhibitor is stabilized through ion pair formation between the carboxylate group of the ligand and the guanidinium group of Arg245. An additional hydrogen-bond is formed between the 2-amino group and the carboxylate of Glu220, an interaction that does not occur with the corresponding amino group of the β-AFP inhibitor. The binding of D-2,3-diaminopropionate in the spASADH active site also induces a change in the conformation of the Asn127 side chain (Figure 4) similar to that observed during the formation of the tetrahedral intermediate complex with ASADH from H. influenza (21). This amide group moves into position to form an additional hydrogen-bond between the 2-amino group of the ligand and Oδ of Asn127, thereby alleviating a potential clash between the inhibitor amino group and this amide side chain. Surprisingly, the 3-amino group of D-2,3-diaminopropionate does not make any productive interactions with either the enzyme or the cofactor. The lack of binding interactions is reflected in the weaker electron density observed for this amino group and suggests the likelihood of designing additional amino acid analogues through derivatization at the 3-position that could produce more potent and selective ASADH inhibitors.
Figure 4.
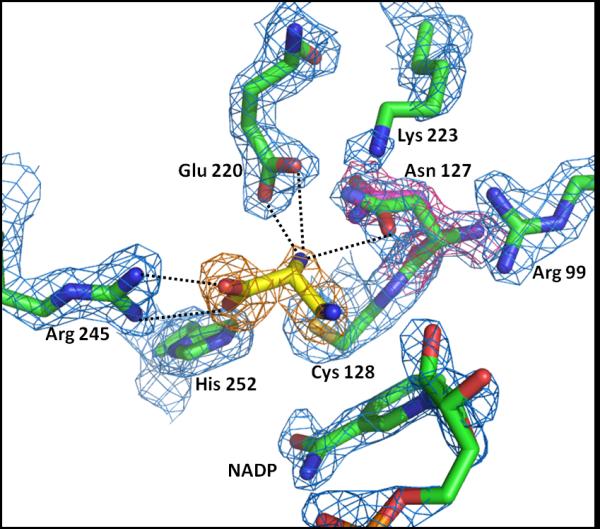
Binding of D-2,3-diaminopropionate to the spASADH/NADP complex. A 2.0 Å resolution 2Fo-Fc map (blue, 1.5σ level) shows bound NADP and the active site amino acids, while an Fo-Fc omit map (orange, 2.7σ level) shows the orientation and stereochemistry of the bound inhibitor. The side chain of Asn127 has shifted from its original position (2Fo-Fc map, red electron density, 1.7σ level) in response to inhibitor binding.
Design of improved ASA dehydrogenase inhibitors
Some compounds that are structurally related to these new inhibitors were modeled into the active site of the ASADHs using the binding interactions observed in these initial structures as a guide. These compounds were also examined kinetically as possible inhibitors, with the selection based on the potential for favorable interactions from these docking studies, and several were found to be inhibitors with good binding affinity (14). The interactions of these newly identified inhibitors of spASADH were next examined structurally to assess any differences between the modeled binding interactions and the actual interactions that are made when these compounds bind to the enzyme.
Extending the carbon skeleton of D-2,3-diaminopropionate allows the introduction of an additional carboxyl group to potentially interact within the phosphate binding pocket that contains the positively-charged side chains of Arg99 and Lys223. A linear, six-carbon dicarboxylic acid was found to make the optimal docking interactions with both arginyl residues in the active site (Figure 5). Several amino dicarboxylic acids were tested, and DL-2-aminoadipate was found to have a low micromolar Ki against vcASADH (0.23 mM when adjusted for the binding of a single enantiomer), but showed little inhibitory activity against spASADH (14). Co-crystallization of 2-aminoadipate in a complex with vcASADH and NADP resulted in observed electron density for the carboxylates of the inhibitor (data not shown), but did not show well-ordered binding of this inhibitor in the active site. Co-crystallization of the same inhibitor in a complex with spASADH and 2′,5′-ADP (a mononucleotide analogue of NADP) lead to more ordered electron density in the inhibitor binding pocket, confirming the interactions predicted from docking studies, despite the lack of strong inhibition of this enzyme form by 2-aminoadipate. The D-isomer of 2-aminoadipate selectively binds in this active site through two sets of bidentate interactions between the carboxylate groups of the inhibitor and the guanidinium groups of Arg99 and Arg245 (Figure 5). The position of the amino group of this inhibitor is also stabilized by interactions with side chain carbonyl oxygen of Asn127 and with the carboxylate oxygens of Glu220.
Figure 5.

Binding of D-2-aminoadipate to the spASADH/2′,5′-ADP complex. The orientation of this inhibitor (magenta structure) is superimposed with the refined model of 2-aminoadipate (2.0 Å Fo-Fc omit map, orange density, 2.5σ level).
While the design of inhibitors that can make the desired interactions with the two active site arginyl groups has now been accomplished, only a modest enhancement in binding affinity was achieved with these inhibitors (14). It is likely that the measured binding constant is somewhat compromised by requiring the enzyme to trap and bind a single rotomer of this conformationally flexible, linear dicarboxylic acid. The possibility of several bound orientations and multiple conformers can still lead to overall strong inhibition, but will result in diffuse and disordered electron density as was observed with the binding of 2-aminoadipate. A conformationally constrained dicarboxylic acid could potentially overcome this barrier, and several cyclic analogues were examined to test this idea. Terephthalic acid (benzene-1,4-dicarboxylic acid) was found to be only a modest inhibitor of spASADH (Ki ~ 5 mM), but the addition of an amino group (2-aminoterephthalate) lead to a greater than 6-fold enhanced affinity (Ki = 0.78 mM). The structure of a ternary complex with this inhibitor and NADP bound to spASADH shows the expected binding of 2-aminoterephthalate in the active site (Figure 6). Each carboxyl group makes electrostatic interactions with the active site arginyl residues, but even at this higher resolution (1.6 Å) weaker electron density does not allow accurate modeling of the aromatic ring into a single orientation. Although kinetic studies showed an enhanced binding contribution made by incorporating an amino group into this inhibitor, no obvious interactions were found between the amino group and any enzyme active site functional groups. Instead the amino group is oriented through an internal hydrogen-bond with the carboxyl group at C1 that could help position this group to make an improved bidentate interaction with the side chain of Arg245 (Figure 6). This stabilizing hydrogen bond would not be available in terephthalic acid and may explain the weaker affinity of this inhibitor towards spASADH.
Figure 6.

Structure of two conformationally constrained dicarboxylic acid inhibitors bound in the active site of spASADH. A 1.5 Å Fo-Fc omit map (orange, 3.5σ level) shows complete electron density for cyclohexane-1,4-dicarboxylate (green structure), with the side chain of Cys128 occupying two different orientations in this structure. In contrast, 2-aminoterephthalic acid (red structure) adopts a different orientation to maintain contact with the active site arginines, with the amino group of this inhibitor forming an internal hydrogen-bond (red dotted line) to help orient the carboxyl group.
The constraints imposed by the planar aromatic ring may be limiting the flexibility needed to allow optimal bidentate interactions between the carboxyl groups of terephthalate and the active site guanidinium groups. To relieve this constraint but still avoid rotational flexibility cyclohexane-1,4-dicarboxylate was examined as potential inhibitor and found to bind to spASADH with 2-fold higher affinity (Ki = 0.45 mM) than that measured for 2-aminoterephthalate. The structure of this inhibitor bound to the enzyme was determined at high resolution (1.5 Å) in the presence of the cofactor analogue 2′,5′-ADP (Table 1) and, in contrast to the broken density seen for 2-aminoterephthalate, this new inhibitor shows reasonably complete electron density for the cyclohexyl ring (Figure 6). However, this improved density was not observed with the initial ternary complex structure determined with cyclohexane-1,4-dicarboxylate in the presence of the physiological cofactor, NADP. In that structure partial occupancy was observed for this inhibitor, as well as for the nicotinamide and ribose moieties of the cofactor. A potential clash between the edge of the cyclohexane and one of the ribose hydroxyl groups seems to preclude the simultaneous binding of both compounds in their preferred orientations (data not shown). In the complex with the cofactor analogue (2′,5′-ADP) that is missing the nicotinamide monophosphate moiety, cyclohexane-1,4-dicarboxylate is bound with each carboxyl group making symmetrical, bidentate interactions with the respective guanidinium groups in the active site. Additional electron density observed near His252 was interpreted as a partially-occupied water molecule (Figure 6).
The interactions with the corresponding carboxyl groups in 2-aminoterephthalate are somewhat less symmetrical than those of cyclohexane-1,4-dicarboxylate because of the planarity imposed by the aromatic ring. To compensate for these less than ideal interactions the orientation of the aromatic ring and the position of the carboxyl carbons are shifted relative to those seen for the cyclohexyl ring. The respective cyclic structures are oriented at nearly 90 degree angles to each other, while the carboxyl carbon at C1 has shifted by about 0.3 Å and that at C6 by nearly 0.7 Å to allow improved interactions for 2-aminoterephthalate with the two active site arginyl residues (Figure 6).
Conclusions
Through a comparison of these various enzyme-inhibitor structures the critical active site binding groups in the ASADH family have been identified. Based on these initial structures compounds have been designed and tested that make productive binding interactions with these functional groups. The next challenge will be to build additional binding interactions into these core structures that will capitalize on the differences in inhibitor binding to enhance the selectivity between the homologous enzymes in this family.
Experimental Procedures
Fragment libraries
Two fragment libraries were assembled by including a range of different low molecular weight compounds to probe binding at various sites on the target enzyme. The water-soluble fragment library (SFL) is composed of 384 compounds, equally divided among four different compound classes: amino acids and derivatives, metabolites and analogues, carbohydrates and bases, and water soluble organics and aromatics. An organic fragment library (OFL) of 384 compounds dissolved in DMSO was also assembled from four different classes of compounds: benzene derivatives, 5-membered heterocycles, 6-membered heterocycles, and fused/multiple ring systems (14). Stock solutions of each compound were prepared either in water (200 mM) or in DMSO (400 mM), with some pH adjustment as needed to solubilize acids or bases in the water soluble library. For screening purposes fragment cocktails were assembled by mixing one compound from each class, resulting in 96 fragment cocktails containing four compounds for each library.
Enzyme crystallization
Different crystallized enzyme forms were used for soaking either in cocktails or with the individual compounds: apo-enzyme or two different binary complexes with either NADP or 2′,5′-ADP. Crystallization was induced by the vapor diffusion method with the experiments conducted in 24-well Linbro plates. The enzyme solution (2 μl of 12-20 mg/ml) was mixed with the well solution and set up in hanging drops against 500 μl of the well solution. Crystals of apo spASADH were grown in 12-20% PEG 3350, 0.1 M MES buffer (pH 6.5), 0.05 M Ca-acetate and 10 mM DTT. Crystals of the binary complexes with 2′,5′-ADP or NADP were grown from 14-24% PEG MME 5K (or PEG 3350), 0.1 M sodium citrate (pH 7), 0.05-0.1 M sodium acetate and 10 mM DTT.
Binary (enzyme plus ligand) and ternary (enzyme plus NADP/2′,5′-ADP plus ligand) complexes were obtained by co-crystallization using hanging-drop vapor-diffusion method. 2 μl drops of protein (15-25 mg/ml) or protein-nucleotide complex (5 mM nucleotide) were mixed with equal amounts of a well solution and equilibrated against the well solution containing: a) for spASADH 16-26% PEG 3350 (or PEG 8000), 0.1 M MES buffer (pH 6.5), 0.1 M sodium acetate and 10 mM DTT; b) for vcASADH the same concentrations of protein and nucleotide, with 20-25% PEG 3350, 0.1 M sodium citrate buffer (pH 7.0), 0.1 M sodium acetate and 10 mM DTT.
Complex formation
The final concentration of each cocktail component was set at 20-40 mM to ensure saturation of the binding site under the crystallization conditions. Co-crystallization of each enzyme form was carried out either as binary inhibitor complexes or as ternary complexes in the presence of NADP or 2′,5′-ADP as a cofactor analogue. For the soaking experiments pre-grown crystals of apo spASADH and vcASADH were soaked for between 1 to 30 minutes with inhibitors that were previously identified from both the SFL and OFL. After soaking each crystal was dipped into a cryo-protectant solution (containing crystallization buffer, cofactor and inhibitors mixed with 20-25% ethylene glycol) prior to freezing in liquid nitrogen.
Diffraction data collection
Crystal quality was initially assessed on an in-house R-AXIS IV image plate area detector mounted on a Rigaku FR-E rotating anode X-ray generator. Crystals of spASADH obtained by soaking in OFL cocktails were screened and diffraction data sets were collected for each crystal that produced reasonable initial diffraction patterns on the SER-CAT beam-lines (Sectors 22A and 22B) at the Advanced Photon Source (Argonne National Laboratory). X-ray diffraction data of binary and ternary co-crystallized complexes of spASADH and vcASADH with the individual inhibitors were collected both in-house and on the GM/CA-CAT beam-line Sector 23B at the APS. All diffraction data were processed using HKL2000 and scaled with SCALEPACK (22). The diffraction data obtained from each enzyme complex were processed as described below.
Crystallographic refinement and ligand binding assessment
All crystallographic calculations were carried out within the CCP4 program suite (23). The enzyme was first refined as a rigid body using diffraction data limited to 3.5 Å resolution, followed by restrained refinement with data to the highest available resolution in REFMAC5 (24). Weighting of the X-ray and geometric components in the minimization function in REFMAC5 was done by choosing w = 0.03-0.10, except for the last two data sets (Table 1) with resolutions greater than 1.7 Å, where the weighting was set to 0.4 and 0.8, respectively. Because of the high data to parameters ratio the cyclohexane-1,4-dicarboxylate complex structure was refined with anisotropic B-factors, while all others were refined using isotropic B-factors. Difference electron density maps were calculated and inspected using COOT (25) for every structure to identify possible inhibitor binding sites. If there was significant unoccupied electron density indicating possible ligand binding then a model of the inhibitor structure was created using PRODRG (26) (coordinates in PDB format and dictionary description in REFMAC5 CIF format) and fitted into the unassigned density in COOT followed by several cycles of refinement.
Acknowledgments
The authors thank DeMarco Camper and Buenafe Arachea for providing purified enzyme samples, and Geng Gao for kinetic screening to identify fragment library inhibitors. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. We thank the staff members at the GM/CA and SER-CAT beamlines for their assistance with data collection. This work was supported by a grant from the National Institutes of Health (AI077720).
References
- 1.Cohen GN. The Common Pathway to Lysine, Methionine, and Threonine. In: Herrmann KM, Somerville RL, editors. Amino Acids: Biosynthesis and Genetic Regulation. Addison-Wesley; Reading, MA: 1983. pp. 147–171. [Google Scholar]
- 2.Viola RE. The Central Enzymes of the Aspartate Family of Amino Acid Biosynthesis. Accounts Chem Res. 34:339–349. doi: 10.1021/ar000057q. [DOI] [PubMed] [Google Scholar]
- 3.Ragkousi K, Eichenberger P, van Ooij C, Setlow P. Identification of a New Gene Essential for Germination of Bacillus subtilis Spores with Ca2+-Dipicolinate. J Bacteriol. 185:2315–2329. doi: 10.1128/JB.185.7.2315-2329.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Heijenoort J. Recent advances in the formation of the bacterial peptidoglycan monomer unit. Nat Prod Reports. 18:503–519. doi: 10.1039/a804532a. [DOI] [PubMed] [Google Scholar]
- 5.Lyon GJ, Novick RP. Peptide signaling in Staphylococcus aureus and other Gram-positive bacteria. Peptides. 25:1389–1403. doi: 10.1016/j.peptides.2003.11.026. [DOI] [PubMed] [Google Scholar]
- 6.Chen X, Schauder S, Potier N, Van Dorsselaer A, Pelczer I, Bassler BL, Highson FM. Structural identification of a bacterial quorum-sensing signal containing boron. Nature. 415:545–549. doi: 10.1038/415545a. [DOI] [PubMed] [Google Scholar]
- 7.Galan JE, Nakayama K, Curtiss R. Cloning and characterization of the asd gene of Salmonella typhimurium: use in stable maintenance of recombinant plasmids in Salmonella vaccine strains. Gene. 94:29–35. doi: 10.1016/0378-1119(90)90464-3. [DOI] [PubMed] [Google Scholar]
- 8.Gerdes SY, Scholle MD, Campbell JW, Balazsi G, Ravasz E, Daugherty MD, Somera AL, Kyrpides NC, Anderson I, Gelfand MS, Bhattacharya A, Kapatral V, D’Souza M, Baev MV, Grechkin Y, Mseeh F, Fonstein MY, Overbeek R, Barabasl AL, Oltvai ZN, Osterman AL. Experimental Determination and System Level Analysis of Essential Genes in Escherichia coli MG1655. J Bacteriol. 185:5673–5684. doi: 10.1128/JB.185.19.5673-5684.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kobayashi K, Ehrlich SD, Albertini A, et al. Essential Bacillus subtilis genes. Proc Natl Acad Sci USA. 100:4678–4683. doi: 10.1073/pnas.0730515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salama NR, Shepard B, Falkow S. Global Transposon Mutagenesis and Essential Gene Analysis of Helicobacter pylori. J Bacteriol. 186:7926–7935. doi: 10.1128/JB.186.23.7926-7935.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Becker D, Selbach M, Rollenhagen C, Ballmaier M, Meyer TF, Mann M, Bumann D. Robust Salmonella metabolism limits possibilities for new antimicrobials. Nature. 440:303–307. doi: 10.1038/nature04616. [DOI] [PubMed] [Google Scholar]
- 12.Carr R, Jhoti H. Structure-based screening of low-affinity compounds. Drug Discovery Today. 7:522–527. doi: 10.1016/s1359-6446(02)02245-6. [DOI] [PubMed] [Google Scholar]
- 13.Hopkins AL, Groom CR, Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today. 9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 14.Gao G, Liu X, Pavlovsky A, Viola RE. Identification of Selective Enzyme Inhibitors by Fragment Library Screening. J Biomolec Screen. 15:1042–1050. doi: 10.1177/1087057110381383. [DOI] [PubMed] [Google Scholar]
- 15.Cox RJ, Gibson JS, Martin MB. Aspartyl Phosphonates and Phosphoramidates: The First Synthetic Inhibitors of Bacterial Aspartate-Semialdehyde Dehydrogenase. ChemBioChem. 3:874–886. doi: 10.1002/1439-7633(20020902)3:9<874::AID-CBIC874>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 16.Han S, Moore RA, Viola RE. A facile synthesis of a difluoromethylene analog of β-aspartyl phosphate as an inhibitor of L-aspartate-β-semialdehyde dehydrogenase. Synlett. :845–846. [Google Scholar]
- 17.Faehnle CR, Liu X, Le Coq J, Viola RE. An Examination of Key Intermediates in the Catalytic Cycle of Aspartate Semialdehyde Dehydrogenase from a Gram-positive Bacteria. J Biol Chem. 281:31031–31040. doi: 10.1074/jbc.M605926200. [DOI] [PubMed] [Google Scholar]
- 18.Faehnle CR, Blanco J, Viola RE. Structural basis for discrimination between oxyanion substrate or inhibitors in aspartate-β-semialdehyde dehydrogenase. Acta Cryst. D60:2320–2324. doi: 10.1107/S0907444904026411. [DOI] [PubMed] [Google Scholar]
- 19.Blanco J, Moore RA, Kalabeeswaran V, Viola RE. A structural basis for the mechanism of aspartate-β-semialdehyde dehydrogenase from Vibrio cholerae. Protein Sci. 12:27–33. doi: 10.1110/ps.0230803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blanco J, Moore RA, Faehnle CR, Coe DM, Viola RE. The Role of Substrate-binding Groups in the Mechanism of Aspartate-β-semialdehyde Dehydrogenase. Acta Cryst. D60:1388–1395. doi: 10.1107/S0907444904012971. [DOI] [PubMed] [Google Scholar]
- 21.Blanco J, Moore RA, Viola RE. Capture of an intermediate in the catalytic cycle of L-aspartate-β-semialdehyde dehydrogenase. Proc Natl Acad Sci USA. 100:12613–12617. doi: 10.1073/pnas.1634958100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 23.Collaborative Computational Project The CCP4 suite: programs for protein crystallography. Acta Cryst. D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 24.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Cryst. D57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 25.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 26.Schuttelkopf AW, van Aalten DMF. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Cryst. D60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]