

Abstract
The Plasmodium falciparum parasite is an obligate intracellular pathogen whose invasion and remodeling of the human erythrocyte results in the clinical manifestations of malarial disease. The functional analysis of erythrocyte determinants of invasion and growth is a relatively unexplored frontier in malaria research, encompassing studies of natural variation of the erythrocyte, as well as genomic, biochemical and chemical biological and transgenic approaches. These studies have allowed the functional analysis of the erythrocyte in vitro, resulting in the discovery of critical erythrocyte determinants of Plasmodium infection. Here, we will focus on the varied approaches used for the study of the erythrocyte in Plasmodium infection, with a particular emphasis on erythrocyte invasion.
Keywords: Malaria, Erythrocyte, Plasmodium, Invasion, Functional analysis, Lentivirus, Genetics, Polymorphism
1. Plasmodium and the erythrocyte
Over one-third of the world’s population is at risk of malaria infection, and malaria accounts for between 655,000 and 1.2 million deaths each year, according to recent reports (WHO, 2010; Murray et al., 2012). Malaria is caused by infection with the protozoan parasite of the genus Plasmodium. Erythrocyte invasion and growth by Plasmodium spp. parasites is an obligatory step in the life-cycle of the parasite and central to the virulence of malarial disease. There are many host and parasite protein interactions that contribute to the process of invasion and growth (Fig. 1).
Fig. 1.
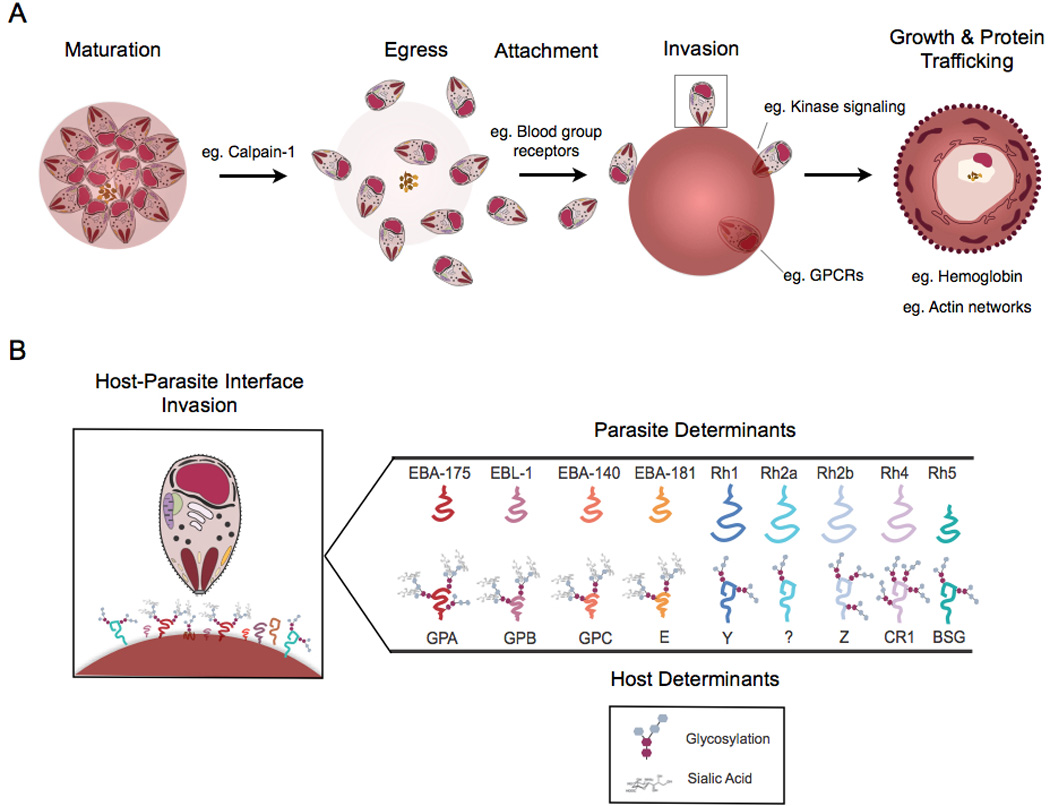
Host-parasite interactions that facilitate erythrocyte invasion and erythrocyte remodeling. (A) The erythrocytic stage of the Plasmodium life cycle depends on host proteins at a number of stages: schizont maturation, merozoite egress, merozoite attachment, merozoite invasion, and intracellular growth and parasite protein trafficking to the surface of the infected erythrocyte. Throughout this process, host Calpain-1 has been determined to be critical for parasite escape from the parasitophorous vacuole and erythrocyte egress, binding to erythrocyte blood group receptors has been shown to be required for invasion, signaling via G-protein coupled receptors (GPCRs) and host kinases have been shown to be necessary for intracellular establishment, and normal hemoglobin has been shown to be required for proper actin network formation and parasite adhesive protein export to the surface of the erythrocyte. (B) The host-parasite interface is shown in detail with respect to the invading merozoite. Parasite invasion ligands (Parasite Determinants) and human erythrocyte receptors (Host Determinants) which have been identified to date are shown. Host receptor modifications (glycosylation and terminal sialic acid residues) are shown. GPA, glycophorin A; GPB, glycophorin B; GPC, glycophorin C; CR1, complement receptor 1; BSG, Basigin; EBA, erythrocyte binding antigen; Rh, reticulocyte binding protein homolog.
Most recent studies have focused on identifying the parasite determinants for invasion into and growth within erythrocytes. Associative studies have uncovered intriguing associations between host polymorphisms in erythrocyte proteins, susceptibility to invasion by the Plasmodium parasite and virulence. Such studies have prompted the need for the direct functional analysis of erythrocyte determinants, focused on natural variation of the erythrocyte, genomic approaches, biochemical approaches, chemical biological approaches or recent transgenic approaches to either deplete, block, inhibit or transgenically alter the erythrocyte surface (Fig. 2). Here we review the different approaches that are being developed for the in vitro functional analysis of the erythrocyte (Table 1), with a special emphasis on invasion that has been the focus of considerable study.
Fig. 2.
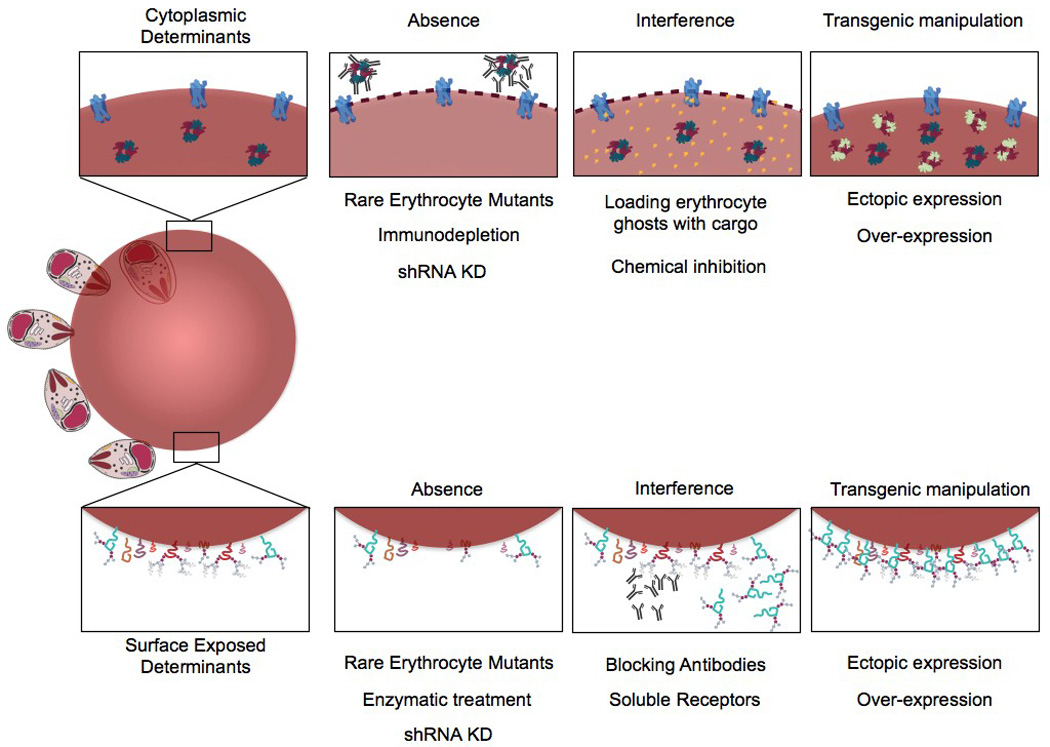
Methodologies employed for functional analysis of erythrocyte determinants of Plasmodium invasion and growth. Various experimental procedures have been employed to target both cytoplasmic and surface exposed erythrocyte determinants of invasion. Experiments employing naturally occurring mutants, biochemical approaches, chemical biological approaches and transgenic approaches have been used to identify and confirm the role of host determinants of invasion via absence, interference, ectopic expression, and over-expression. shRNA KD, small hairpin RNA knock-down.
Table 1.
Summary of approaches for analysis of erythrocyte determinants of 698 Plasmodium infection.
| Approach | Pros | Cons |
|---|---|---|
| Natural Mutants | ||
| Assays using natural erythrocyte mutants | Polymorphisms selected by nature (physiologically relevant) Viable erythrocytes naturally obtained |
Confounding polymorphisms also present Difficult to control experiments with cells from different donors Inability to complement the mutation |
|
Functional genomics of natural mutants |
Using natural selection to identify novel candidate resistance/susceptibility loci |
Weak signals due to (weak) linkage disequilibrium To date, GWAS has required further deep sequencing to uncover signals |
| Biochemical Approaches | ||
| Pull-down | Detects physiologically interacting proteins, ex vivo or in vitro, with or without cross-linkers | Possible that weak interactions will not be detected by pull-down |
| Gel-overlay, Erythrocyte binding assays (EBAs) with enzyme treated erythrocytes | Can use both antibody mediated detection or radioactive methods | Protein abundance and stability can effect the sensitivity Requires stable protein interactions |
| Invasion assays with enzyme treated erythrocytes | Determine the effect of removing sets of receptors in the same genetic background on parasite invasion | Non-specific removal of receptors. Removes groups of both known and unknown receptors. |
| Antibody mediated inhibition | Specific inhibition of a receptor by blocking access to the parasite ligands Can determine the effect of blocking a given receptor in the same genetic background High throughput |
Antibody dependent (not all antibodies are inhibitory) Agglutination Off target effects of antibodies (can impact both invasion and membrane rigidity, for example) |
| Avexis | Pentamerizing the bait or prey increases detection of transient interactions | Limited by what can be expressed (size, glycosylation, number of transmembrane domains) False positives |
| Chemical genetic Approaches | ||
| Chemical inhibition | Potent inhibition can be achieved Temporal inhibition can be achieved Depending on the inhibitor, inhibition can be reversed |
Off-target effects Specificity of inhibition can be dependent on concentration Host versus parasite specificity can be a narrow window, and difficult to achieve |
| Immunodepletion of cytoplasmic proteins | The alternative to transgenic modification with the same result – specific depletion of a given protein | Limited to cytoplasmic proteins |
| Transgenic Approaches | ||
| shRNA Knock-down | Stable and either inducible or constant Amenable to forward genetic screens Can study the effect of the absence (or decrease) of a given protein in the erythrocyte |
Level of knock-down varies depending on the gene Knock-down (rather than knock-out) can be insufficient to yield a phenotype Erythrocyte viability can be effected depending on when the knock-down has an effect May be necessary to induce knock-down later in erythropoiesis to avoid developmental effects |
| Ectopic expression | Inducible expression, varying level of expression, can be achieved with specific vectors and promoters | Matching physiologic levels of expression with appropriate promoters |
| Allelic replacement | Complementation is possible (ectopically expressing a protein in a null background) | Must codon optimize the transgenically expressed allele to avoid shRNA mediated KD of replacement as well as endogenous Matching levels of expression with appropriate promoters |
GWAS, genome-wide association study; shRNA, small hairpin RNA; KD, knock down; AVEXIS, avidity-based extracellular interaction screen.
2. Studies of natural variation of the erythrocyte
Malaria has exerted a detectably strong selective pressure on the human genome. Over the last few decades, a number of erythrocyte polymorphisms have been found to provide resistance to malaria. These polymorphisms are often spatially distributed in areas of intense malaria transmission (Williams, 2006). Polymorphic erythrocyte proteins such as hemoglobin for sickle cell trait (HbAS) and glucose-6-phosphate dehydrogenase (G6PD) have been associated with protection from malaria in vivo (Allison et al., 1961; Allison and Clyde, 1961; Gilles et al., 1967; Ruwende and Hill, 1998), and it has been postulated based on studies in mice (Min-Oo et al., 2003; Ayi et al., 2004, 2008) that pyruvate kinase deficiencies could also provide protection in humans (Daily and Sabeti, 2008); however, the mechanisms governing such protection are poorly understood. These polymorphic erythrocytes have been used in in vitro assays to determine their relevance to the processes of Plasmodium invasion, growth and development, protein trafficking and erythrocyte surface display, and rosetting or cytoadherence (Roth et al., 1983; Fairhurst et al., 2005; Ayi et al., 2008; Cholera et al., 2008; Cyrklaff et al., 2011). Importantly, these polymorphic erythrocytes provide specific candidate host proteins that are likely critical in these virulence processes.
2.1. Invasion
All of the known receptors for erythrocyte invasion belong to polymorphic human blood group antigen families. A strong body of evidence implicating specific surface receptors in Plasmodium parasite invasion and growth stems from work examining the role of naturally-occurring erythrocyte mutants. These erythrocytes completely lack a protein, contain a partial deletion or are polymorphic - either in sequence or expression level and have provided valuable clues implicating specific proteins as receptors for invasion. Naturally occurring mutants have been used both to determine the specificity of binding of different parasite ligands or to determine the impact of specific mutations on parasite invasion efficiency. These include glycophorin A null cells (Miller et al., 1977; Pasvol et al., 1982a; Facer, 1983; Pasvol and Jungery, 1983), glycophorin B null cells (S-s-u) (Pasvol et al., 1982a; Facer, 1983; Pasvol and Jungery, 1983), glycophorin C mutant and null cells Gerbich and Leach (Serjeantson, 1989; Lobo et al., 2003; Maier et al., 2003) and Duffy antigen receptor for chemokines (DARC) null cells (Horuk et al., 1993; Chitnis, 2001). Polymorphic proteins have also been analyzed, including ABO blood group variants (Fraser et al., 1966; Cserti and Dzik, 2007; Uneke, 2007; Rowe et al., 2009b), Band 3 deletion mutants (SAO) (Cortes et al., 2004), and Oka blood group variants of the Basigin protein (BSG) (Crosnier et al., 2011).
While no known null exists for Complement receptor 1 (CR1), cells with high and low erythrocyte surface expression of CR1 are common, and these expression level differences impact Plasmodium falciparum invasion (Spadafora et al., 2010; Tham et al., 2010)
2.2. Protein trafficking and display
While the observation that HbAS is associated with protection from severe malaria was made decades ago (Allison, 1964) and hemoglobin C (HbC) has also been shown to be protective (Flint et al., 1998; Agarwal et al., 2000; Modiano et al., 2001), researchers have been searching for the mechanism of this protection. Plasmodium falciparum invasion and growth appear to be decreased in HbAS cells in vitro (Friedman, 1978; Pasvol et al., 1978), but it has been suggested that other factors must be involved for protection to be observed in vivo.
Parasite adhesive proteins, such as PfEMP1, are exported to the surface of the infected eythrocyte where they adhere to host receptors resulting in sequestration of P. falciparum-infected erythrocytes. It has been found that display of PfEMP1 and cytoadherance is altered on the surface of HbC containing cells (Fairhurst et al., 2005), and HbAS and HbS (hemoglobin associated with sickle cell anemia) containing cells (Cholera et al., 2008). A precise mechanism of abnormal display in HbSC (hemoglobin associated with sickle cell – hemoglobin C disease) and HbCC (homozygous for hemoglobin C) cells has been recently proposed (Cyrklaff et al., 2011). Using cryoelectron tomography, researchers were able to observe that the parasite generates a human actin network to link the developing parasite with Maurer’s clefts, allowing proper trafficking of adhesive parasite proteins to the surface of the infected erythrocyte (Cyrklaff et al., 2011). The presence of hemoglobin C or S resulted in disordered actin networks and aberrant trafficking, possibly making these cells vulnerable to clearance by the spleen.
Rosetting is a process by which parasitized erythrocytes bind to uninfected erythrocytes, forming clusters (rosettes) that are thought to contribute to malaria pathology by blocking blood flow through small vessels (Kaul et al., 1991; Rowe et al., 2009a) and by facilitating the invasion of merozoites, resulting in increased parasitemia and anemia, as has been observed in human clinical isolates and in Saimiri monkeys (Rowe et al., 2002; Le Scanf et al., 2008). Before its role in invasion was appreciated, the CR1 protein was identified as an erythrocyte receptor important in the process of rosetting (Rowe et al., 1997). Later, ABO blood group polymorphisms, and in particular blood group O, were found to confer protection against severe malaria through a mechanism of reduced rosetting (Rowe et al., 2007).
2.3. Functional genomics to identify novel candidates
While the roles of a few candidate polymorphic proteins have been identified through associative studies and confirmed in vitro, these known factors cannot explain the entirety of human genetic resistance to malaria, implying that many genetic factors have yet to be discovered (Mackinnon et al., 2005). Functional genomics can provide a means of identifying such novel candidates. In recent years, genome-wide association studies (GWAS) have provided important insights as to the genetic determinants of diseases. It is only recently that such tools have been adapted to study genetically highly diverse populations, such as those found in Africa (Jallow et al., 2009; Teo et al., 2010). One such study examining GWA with severe malaria in 2,500 Gambian children was able to identify a single known malaria resistance loci (HbS) (Jallow et al., 2009). The authors concluded that the insufficient tagging efficiency of a 500 K gene chip array (Affymetrix, USA), in addition to low allele frequencies and other factors which decreased statistical power, were responsible for the lack of known and novel malaria resistance loci identified by the GWAS. However, when deep sequencing and multipoint imputation modeling around the HbS loci were conducted, the significance of the signal was increased and the causal single nucleotide polymorphism (SNP) was identified. Further known signals of resistance such as G6PD polymorphism and susceptibility (such as non-O ABO blood group) were confirmed upon deep sequencing, but significance levels were not sufficient to be detected by the GWAS methodology. It was postulated that the weak linkage disequilibrium present in African populations makes identifying causal variants more difficult and will increase the number of SNPs required to tag common variants in the population. Additionally, many resistance markers are present at either low frequencies or have reached fixation – decreasing the power of GWAS to detect those. Further, the population-specific sequence information is critical in designing and interpreting GWAS. In African populations resistance mutations can arise independently in loci under particularly strong selection and this results in different causal SNPs in different African populations, complicating the ability to conduct multicenter replication studies. With advances in sequencing technology and increased sequence coverage of diverse African populations, perhaps these challenges can be overcome and will increase the power of the GWAS approach for identifying determinants of malarial disease in Africa.
3. Biochemical approaches
Numerous biochemical approaches have been applied to identify the host erythrocyte receptors and the Plasmodium parasite ligands that are involved in the process of merozoite invasion. The primary challenge with biochemical approaches is stability and the temporal nature of the protein interactions. While some ligand-receptor interactions have been successfully identified biochemically (Galinski et al., 1992; Sim et al., 1994; Ogun and Holder, 1996), many have eluded characterization.
3.1. Enzymatic cleavage
In addition to naturally occurring erythrocyte mutants, which are often rare, studies have been conducted using enzymatic cleavage to specifically remove non-overlapping sets of receptors from the surface of the erythrocyte (Binks and Conway, 1999; Okoyeh et al., 1999; Thompson et al., 2001). The advantage of these studies is that comparisons can be made between cells from the same donor, minimizing the confounding effects due to genetic variation. The disadvantage is that enzymatic treatment usually removes many potential determinants simultaneously. Enzyme-treated erythrocytes have been utilized in various assays, such as erythrocyte binding assays (EBAs), gel overlay and invasion assays, to identify receptor-ligand interactions. Each assay is based on differences in protein-protein interactions between the wild type (mock treated) and enzymatically cleaved erythrocytes. Whether recombinant proteins are incubated with erythrocytes, washed and eluted, as with EBAs; native protein gels of treated erythrocytes are run and overlaid with recombinant ligand as in gel overlays; or whether invasion of viable merozoites into enzyme treated cells are compared as in standard invasion assays, each assay relies on comparing the interactions due to the absence of a given set of receptors.
Early studies were done with neuraminidase treatment of erythrocytes to determine dependence of different strains on sialic acid (Mitchell et al., 1986). Enzymatic cleavage of erythrocyte receptors with different proteases has been employed to define different receptors used by the parasite for binding and invasion of the erythrocyte, known as invasion pathways (Dolan et al., 1990; Reed et al., 2000; Duraisingh et al., 2003). Such methodologies have allowed for comparisons between invasion pathway utilization for both laboratory and field isolated parasites (Okoyeh et al., 1999; Baum et al., 2003; Lobo et al., 2004; Nery et al., 2006; Bei et al., 2007; Deans et al., 2007; Jennings et al., 2007; Lantos et al., 2009; Gomez-Escobar et al., 2010).
3.2. Antibody mediated blocking and inhibition
For erythrocyte determinants of invasion that are surface exposed, interfering with the protein’s accessibility to the parasite through blocking antibodies, Fab fragments, or soluble receptor competition assays, have provided useful clues as to the mechanism of action of these determinants. High concentrations of antibodies required to inhibit parasite invasion can cause agglutination of the erythrocytes and changes in membrane rigidity. The use of Fab fragments can be used to circumvent these issues. While Fab fragments can be useful in invasion neutralizing experiments, they are not applicable to studies of opsonophagocytosis via binding of IgG to Fc receptors on phagocytic cells. Antibodies against glycophorin A have been shown to inhibit parasite invasion (Pasvol et al., 1982a; Pasvol et al., 1982b; Jungery, 1985), however this inhibition may be due either to receptor blockage or to an increase in erythrocyte membrane rigidity (Pasvol et al., 1989). In the case of CR1, certain monoclonal antibodies were able to disrupt the interaction between the CR1 receptor and its parasite ligand, PfRh4, in biochemical pull-down experiments (Tham et al., 2010). Monoclonal antibodies were also shown to potently block the PfRh5-BSG interaction through binding to BSG at incredibly low inhibitory concentrations (Crosnier et al., 2011).
In addition to blocking the host-parasite interactions with antibodies, soluble erythrocyte receptors have also been shown to inhibit parasite invasion. Recently, soluble CR1 was shown to inhibit parasites that relied on PfRh4 for invasion (Tham et al., 2010). Inhibiting the PfRh5-BSG interaction using pentamerized BSG proved a potent inhibitor of all laboratory parasite isolates and recently in vitro-adapted Senegalese isolates tested to date, implying that this interaction may be essential for invasion of all strains (Crosnier et al., 2011). The fact that targeting a receptor can give such potent cross-strain inhibition gives credence to the concept of host targeted therapies for malaria. While targeting the parasite ligand could give rise to rapidly evolving allelic forms, targeting a conserved receptor in the human host might actually circumvent the issue of polymorphism.
3.3. Identification of novel receptors: AVEXIS
One major challenge has been identifying receptor-ligand interactions due to their often weak and transient nature. To address this problem, an AVEXIS (avidity-based extracellular interaction screen) was developed to allow for high throughput screening of low-affinity protein-protein interactions (Bushell et al., 2008). By multimerizing either the bait or the prey, the method allows for detection of low affinity interactions that are intractable by standard biochemical techniques. The AVEXIS system was applied to Plasmodium ligand-receptor interactions involved in invasion and resulted in the identification of BSG as the receptor for PfRh5 (Crosnier et al., 2011). While this system successfully identified a major receptor-ligand interaction, one of the challenges is that only single pass transmembrane proteins (or proteins with large ectodomains predicted to be soluble) were included in the 40 erythrocyte protein preys. Therefore while some interactions may be identified, others remain to be discovered.
4. Chemical biological approaches
Besides biochemical approaches, chemical genetic approaches have been useful in validating existing players in the process of parasite invasion and egress and in identifying novel candidates. An important role for erythrocyte G-Protein signaling in P. falciparum invasion and growth was identified through hypotonic erythrocyte lysis, followed by loading erythrocytes with membrane insoluble protein cargo to inhibit the erythrocyte protein activity (Murphy et al., 2006). Such experiments with re-sealed modified erythrocytes were able to establish a requirement of host G-Protein Coupled Receptors (GPCRs) for Plasmodium invasion. Targeting GPCRs has subsequently been suggested as a novel anti-malarial strategy, either singly or in combination with other anti-malarial drugs (Murphy et al., 2006).
In addition to identifying signaling pathways involved in parasite invasion and growth, chemical genetic approaches have been applied to investigate the role of host proteases in parasite egress. By treating infected erythrocytes with DCG04—a papain family protease inhibitor—a specific block in schizont egress was identified (Chandramohanadas et al., 2009). The protease target for DCG04 was found to be human Calpain-1 through biochemical approaches (pull-downs with tagged DCG04 followed by mass spectrometry), and confirmed through hypotonic erythrocyte lysis followed by immunodepletion of the protease (Chandramohanadas et al., 2009). Further, small interfering RNA (siRNA) knock-downs in nucleated human U2OS cells and mouse fibroblast cell lines lacking both calpain-1 and calpain-2 were used in invasion and egress assays for the related apicomplexan parasite Toxoplasma gondii, and both showed the same egress phenotype. However, while the method of immunodepletion followed by re-sealing erythrocyte ghosts is suited to the study of certain cytoplasmic proteins, unfortunately it cannot be applied to the study of transmembrane proteins such as erythrocyte surface receptors.
Chemical genetic screening to identify Plasmodium Mitogen-activated protein (MAP) kinase inhibitors of parasite growth unexpectedly identified inhibitors of two mammalian kinases—PAK1 and MEK1, with no clear orthologs in the Plasmodium genome—that inhibited blood stage growth of P. falciparum, and both blood and liver stages of Plasmodium berghei (Sicard et al., 2011). Such studies emphasize the importance of chemical genetic methods to identify novel host targets, especially in the erythrocyte.
5. Transgenic approaches
While studies using naturally occurring erythrocyte polymorphisms have been useful in determining the genetic basis of resistance to infection with Plasmodium, as with all studies of naturally occurring mutants, the paired control by definition is blood from another donor that is deemed to be wild-type for that polymorphism. These “controls” can be (and often are) polymorphic at other loci that are not under direct observation, can be of different ages, and potentially have been handled and stored differently, all of which can significantly confound the results. One way to truly observe the impact of absence on P. falciparum invasion and growth is to transgenically modify erythrocytes from the same donor, or modify them biochemically as with enzymatic treatment.
The in vitro generation of erythrocytes from stem cells has been explored both in terms of genetic targeting of the erythrocyte to understand function, as well as for targeted stem cell therapies. Previously, complete differentiation into fully mature erythrocytes required implantation into non-obese diabetic/severe combined immunodeficient (NOD/SCID) mice (Miyoshi et al., 1999; Neildez-Nguyen et al., 2002; Puthenveetil et al., 2004). In 2005, a protocol was developed for the generation of synchronous reticulocytes and fully mature, enucleated erythrocytes from hematopoietic stem cells completely in vitro and with marked expansion in numbers from the progenitor cells (Giarratana et al., 2005). The advances in this methodology made it technically possible to create genetically modified human erythrocytes in vitro. By targeting the hematopoietic stem cell precursors with lentiviral constructs designed to stably knock-down or transgenically express proteins, the terminally differentiated erythrocyte will contain mutations in isogenic backgrounds. The ability to knock-down protein expression with short hairpin RNAs (shRNAs), ectopically express or over-express proteins using expression vectors, and perform allelic replacement experiments has dramatically increased the tool kit available to researchers studying host-parasite interactions in Plasmodium invasion.
To date, there have been few studies that have utilized this erythrocyte culture system to genetically modify erythroid cells and erythrocytes in vitro. One study controlled expression of the surface exposed human blood group antigen Jka (Bagnis et al., 2009) while another studied the role of hemoglobin switching through regulation by BCL11A (Sankaran et al., 2009). Additionally, we combined in vitro culture of hematopoietic stem cells to terminally differentiated enucleated erythrocytes, with lentivirus-delivered shRNA-mediated knockdown of erythrocyte receptors. Specifically, we targeted glycophorin A (Bei et al., 2010), a receptor that interacts with PfEBA-175, demonstrating strain-specific differences in invasion efficiency. We used this approach to deplete erythrocytes of BSG (Crosnier et al., 2011), the recently identified receptor for PfRh5, demonstrating that it appears to be essential for efficient invasion of all strains.
In addition to applying reverse genetic methodologies to the surface receptors of the erythrocyte utilized by Plasmodium for invasion, transgenic erythrocytes could be generated to determine the role of depletion of cytoplasmic and cytoskeletal erythrocyte proteins on pathogenesis related processes such as erythrocyte deformability, parasite protein trafficking and export, intracellular signaling, species-specific tropism and gametocytogenesis. The ability to genetically modify the erythrocyte through targeted knock-down, ectopic expression and/or over-expression of transgenes, allelic replacement, and temporal expression or depletion of target proteins, lends an additional level of sophistication and specificity to experiments aimed at identifying and characterizing host-parasite interactions.
Transgenic approaches have been applied to correct erythrocyte disorders such as murine beta-thalassemia (Imren et al., 2002) and sickle cell disease (Pawliuk et al., 2001), the latter being corrected in human hematopoietic stem cells transplanted into mice. Recently, the same authors that pioneered the ex vivo culture of erythrocytes have piloted the use of such cells for transplant into humans (Giarratana et al., 2011), again broadening the horizon for how these genetically modified cells might be used in gene therapy. Additionally, with recent advances in induced pluripotent stem (iPS) cells, there has been increased interest in the use of gene therapy to repair erythrocyte disorders using autologous cells from the patient (Lengerke and Daley, 2010).
6. The prospect for future technologies
As the weapons in the arsenal become more advanced and diverse, we are able to target the host-parasite interface with increasing precision and specificity. Advances in the methodologies of both human genomics and erythrocyte genetics have identified novel determinants of Plasmodium infection and have given us the tools with which to interrogate those. GWASs are being adapted to highly diverse African populations by correcting for underlying population structure, making studies aimed at determining novel human determinants of Plasmodium infection more feasible (Teo et al., 2010). While current reverse genetic methodologies for the erythrocyte have been performed using adult hematopoietic stem cells (Bagnis et al., 2009; Sankaran et al., 2009; Bei et al., 2010; Crosnier et al., 2011), the potential to apply such methodologies to continuously self-renewing iPS cells is the next goal. For instance, gene knockouts might be possible using iPS cells. As reverse genetic methodologies are being adapted and applied to the erythrocyte to identify and confirm suspected determinants of Plasmodium infection, forward genetic screens to identify novel determinants are becoming a reality.
As these methodologies develop and are refined, one can envision applications not only in the field of Plasmodium biology, but also in the fields of erythrocyte biology, hematology and gene therapy. While these methods enable the identification and validation of erythrocyte determinants of Plasmodium infection, it is our hope that they will also contribute to the development of potential host-targeted therapies for malaria and other diseases.
Highlights.
-
►
Approaches for the functional analysis of erythrocyte determinants of malaria infection are described.
-
►
Methods to identify critical host surface molecules required for erythrocyte invasion by Plasmodium parasites are emphasised.
-
►
Potential for transgenic manipulation of erythrocytes in vitro is described.
-
►
Pros and cons of the different technologies used for molecular analysis of the erythrocyte are identified.
Acknowledgements
We acknowledge the following support: Centers for Disease Control and Prevention, USA (grant R36 CK000119-01 to AKB); Burroughs Welcome Fund New Investigator in the Pathogenesis of Infectious Diseases, USA (award to MTD); National Institutes of Health, USA (grant R01AI057919 and R01AI091787 to MTD); Bill & Melinda Gates Foundation Grand Challenges Explorations Grant (MTD).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agarwal A, Guindo A, Cissoko Y, Taylor JG, Coulibaly D, Kone A, Kayentao K, Djimde A, Plowe CV, Doumbo O, Wellems TE, Diallo D. Hemoglobin C associated with protection from severe malaria in the Dogon of Mali, a West African population with a low prevalence of hemoglobin S. Blood. 2000;96:2358–2363. [PubMed] [Google Scholar]
- Allison AC, Charles LJ, Mc GI. Erythrocyte glucose-6-phosphate dehydrogenase deficiency in West Africa. Nature. 1961;190:1198–1199. doi: 10.1038/1901198a0. [DOI] [PubMed] [Google Scholar]
- Allison AC, Clyde DF. Malaria in African children with deficient erythrocyte glucose-6-phosphate dehydrogenase. Br Med J. 1961;1:1346–1349. doi: 10.1136/bmj.1.5236.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison AC. Polymorphism and Natural Selection in Human Populations. Cold Spring Harb Symp Quant Biol. 1964;29:137–149. doi: 10.1101/sqb.1964.029.01.018. [DOI] [PubMed] [Google Scholar]
- Ayi K, Turrini F, Piga A, Arese P. Enhanced phagocytosis of ring-parasitized mutant erythrocytes: a common mechanism that may explain protection against falciparum malaria in sickle trait and beta-thalassemia trait. Blood. 2004;104:3364–3371. doi: 10.1182/blood-2003-11-3820. [DOI] [PubMed] [Google Scholar]
- Ayi K, Min-Oo G, Serghides L, Crockett M, Kirby-Allen M, Quirt I, Gros P, Kain KC. Pyruvate kinase deficiency and malaria. N Engl J Med. 2008;358:1805–1810. doi: 10.1056/NEJMoa072464. [DOI] [PubMed] [Google Scholar]
- Bagnis C, Chapel S, Chiaroni J, Bailly P. A genetic strategy to control expression of human blood group antigens in red blood cells generated in vitro. Transfusion. 2009;49:967–976. doi: 10.1111/j.1537-2995.2008.02078.x. [DOI] [PubMed] [Google Scholar]
- Baum J, Pinder M, Conway DJ. Erythrocyte invasion phenotypes of Plasmodium falciparum in The Gambia. Infect Immun. 2003;71:1856–1863. doi: 10.1128/IAI.71.4.1856-1863.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bei AK, Membi CD, Rayner JC, Mubi M, Ngasala B, Sultan AA, Premji Z, Duraisingh MT. Variant merozoite protein expression is associated with erythrocyte invasion phenotypes in Plasmodium falciparum isolates from Tanzania. Mol Biochem Parasitol. 2007;153:66–71. doi: 10.1016/j.molbiopara.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Bei AK, Brugnara C, Duraisingh MT. In vitro genetic analysis of an erythrocyte determinant of malaria infection. J Infect Dis. 2010;202:1722–1727. doi: 10.1086/657157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binks RH, Conway DJ. The major allelic dimorphisms in four Plasmodium falciparum merozoite proteins are not associated with alternative pathways of erythrocyte invasion. Molecular and biochemical parasitology. 1999;103:123–127. doi: 10.1016/s0166-6851(99)00115-2. [DOI] [PubMed] [Google Scholar]
- Bushell KM, Sollner C, Schuster-Boeckler B, Bateman A, Wright GJ. Large-scale screening for novel low-affinity extracellular protein interactions. Genome Res. 2008;18:622–630. doi: 10.1101/gr.7187808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandramohanadas R, Davis PH, Beiting DP, Harbut MB, Darling C, Velmourougane G, Lee MY, Greer PA, Roos DS, Greenbaum DC. Apicomplexan parasites co-opt host calpains to facilitate their escape from infected cells. Science. 2009;324:794–797. doi: 10.1126/science.1171085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitnis CE. Molecular insights into receptors used by malaria parasites for erythrocyte invasion. Curr Opin Hematol. 2001;8:85–91. doi: 10.1097/00062752-200103000-00005. [DOI] [PubMed] [Google Scholar]
- Cholera R, Brittain NJ, Gillrie MR, Lopera-Mesa TM, Diakite SA, Arie T, Krause MA, Guindo A, Tubman A, Fujioka H, Diallo DA, Doumbo OK, Ho M, Wellems TE, Fairhurst RM. Impaired cytoadherence of Plasmodium falciparum-infected erythrocytes containing sickle hemoglobin. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:991–996. doi: 10.1073/pnas.0711401105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes A, Benet A, Cooke BM, Barnwell JW, Reeder JC. Ability of Plasmodium falciparum to invade Southeast Asian ovalocytes varies between parasite lines. Blood. 2004;104:2961–2966. doi: 10.1182/blood-2004-06-2136. [DOI] [PubMed] [Google Scholar]
- Crosnier C, Bustamante LY, Bartholdson SJ, Bei AK, Theron M, Uchikawa M, Mboup S, Ndir O, Kwiatkowski DP, Duraisingh MT, Rayner JC, Wright GJ. Basigin is a receptor essential for erythrocyte invasion by Plasmodium falciparum. Nature. 2011;480:534–537. doi: 10.1038/nature10606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cserti CM, Dzik WH. The ABO blood group system and Plasmodium falciparum malaria. Blood. 2007;110:2250–2258. doi: 10.1182/blood-2007-03-077602. [DOI] [PubMed] [Google Scholar]
- Cyrklaff M, Sanchez CP, Kilian N, Bisseye C, Simpore J, Frischknecht F, Lanzer M. Hemoglobins S and C Interfere with Actin Remodeling in Plasmodium falciparum-Infected Erythrocytes. Science. 2011;334:1283–1286. doi: 10.1126/science.1213775. [DOI] [PubMed] [Google Scholar]
- Daily JP, Sabeti P. A malaria fingerprint in the human genome? N Engl J Med. 2008;358:1855–1856. doi: 10.1056/NEJMe0801414. [DOI] [PubMed] [Google Scholar]
- Deans A-M, Nery S, Conway DJ, Kai O, Marsh K, Rowe JA. Invasion Pathways and Malaria Severity in Kenyan Plasmodium falciparum Clinical Isolates. Infect Immun. 2007;75:3014–3020. doi: 10.1128/IAI.00249-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan SA, Miller LH, Wellems TE. Evidence for a switching mechanism in the invasion of erythrocytes by Plasmodium falciparum. J Clin Invest. 1990;86:618–624. doi: 10.1172/JCI114753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duraisingh MT, Triglia T, Ralph SA, Rayner JC, Barnwell JW, McFadden GI, Cowman AF. Phenotypic variation of Plasmodium falciparum merozoite proteins directs receptor targeting for invasion of human erythrocytes. EMBO J. 2003;22:1047–1057. doi: 10.1093/emboj/cdg096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facer CA. Merozoites of P. falciparum require glycophorin for invasion into red cells. Bull Soc Pathol Exot Filiales. 1983;76:463–469. [PubMed] [Google Scholar]
- Fairhurst RM, Baruch DI, Brittain NJ, Ostera GR, Wallach JS, Hoang HL, Hayton K, Guindo A, Makobongo MO, Schwartz OM, Tounkara A, Doumbo OK, Diallo DA, Fujioka H, Ho M, Wellems TE. Abnormal display of PfEMP-1 on erythrocytes carrying haemoglobin C may protect against malaria. Nature. 2005;435:1117–1121. doi: 10.1038/nature03631. [DOI] [PubMed] [Google Scholar]
- Flint J, Harding RM, Boyce AJ, Clegg JB. The population genetics of the haemoglobinopathies. Baillieres Clin Haematol. 1998;11:1–51. doi: 10.1016/s0950-3536(98)80069-3. [DOI] [PubMed] [Google Scholar]
- Fraser GR, Giblett ER, Motulsky AG. Population genetic studies in the Congo. 3. Blood groups (ABO, MNSs, Rh, Jsa) Am J Hum Genet. 1966;18:546–552. [PMC free article] [PubMed] [Google Scholar]
- Friedman MJ. Erythrocytic mechanism of sickle cell resistance to malaria. Proc Natl Acad Sci U S A. 1978;75:1994–1997. doi: 10.1073/pnas.75.4.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galinski MR, Medina CC, Ingravallo P, Barnwell JW. A reticulocyte-binding protein complex of Plasmodium vivax merozoites. Cell. 1992;69:1213–1226. doi: 10.1016/0092-8674(92)90642-p. [DOI] [PubMed] [Google Scholar]
- Giarratana M-C, Kobari L, Lapillonne H, Chalmers D, Kiger L, Cynober T, Marden MC, Wajcman H, Douay L. Ex vivo generation of fully mature human red blood cells from hematopoietic stem cells. Nat Biotechnol. 2005;23:69–74. doi: 10.1038/nbt1047. [DOI] [PubMed] [Google Scholar]
- Giarratana MC, Rouard H, Dumont A, Kiger L, Safeukui I, Le Pennec PY, Francois S, Trugnan G, Peyrard T, Marie T, Jolly S, Hebert N, Mazurier C, Mario N, Harmand L, Lapillonne H, Devaux JY, Douay L. Proof of principle for transfusion of in vitro-generated red blood cells. Blood. 2011;118:5071–5079. doi: 10.1182/blood-2011-06-362038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilles HM, Fletcher KA, Hendrickse RG, Lindner R, Reddy S, Allan N. Glucose-6-phosphate-dehydrogenase deficiency, sickling, and malaria in African children in South Western Nigeria. Lancet. 1967;1:138–140. doi: 10.1016/s0140-6736(67)91037-9. [DOI] [PubMed] [Google Scholar]
- Gomez-Escobar N, Amambua-Ngwa A, Walther M, Okebe J, Ebonyi A, Conway DJ. Erythrocyte invasion and merozoite ligand gene expression in severe and mild Plasmodium falciparum malaria. J Infect Dis. 2010;201:444–452. doi: 10.1086/649902. [DOI] [PubMed] [Google Scholar]
- Horuk R, Chitnis CE, Darbonne WC, Colby TJ, Rybicki A, Hadley TJ, Miller LH. A receptor for the malarial parasite Plasmodium vivax: the erythrocyte chemokine receptor. Science. 1993;261:1182–1184. doi: 10.1126/science.7689250. [DOI] [PubMed] [Google Scholar]
- Imren S, Payen E, Westerman KA, Pawliuk R, Fabry ME, Eaves CJ, Cavilla B, Wadsworth LD, Beuzard Y, Bouhassira EE, Russell R, London IM, Nagel RL, Leboulch P, Humphries RK. Permanent and panerythroid correction of murine beta thalassemia by multiple lentiviral integration in hematopoietic stem cells. Proc Natl Acad Sci USA. 2002;99:14380–14385. doi: 10.1073/pnas.212507099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jallow M, Teo YY, Small KS, Rockett KA, Deloukas P, Clark TG, Kivinen K, Bojang KA, Conway DJ, Pinder M, Sirugo G, Sisay-Joof F, Usen S, Auburn S, Bumpstead SJ, Campino S, Coffey A, Dunham A, Fry AE, Green A, Gwilliam R, Hunt SE, Inouye M, Jeffreys AE, Mendy A, Palotie A, Potter S, Ragoussis J, Rogers J, Rowlands K, Somaskantharajah E, Whittaker P, Widden C, Donnelly P, Howie B, Marchini J, Morris A, SanJoaquin M, Achidi EA, Agbenyega T, Allen A, Amodu O, Corran P, Djimde A, Dolo A, Doumbo OK, Drakeley C, Dunstan S, Evans J, Farrar J, Fernando D, Hien TT, Horstmann RD, Ibrahim M, Karunaweera N, Kokwaro G, Koram KA, Lemnge M, Makani J, Marsh K, Michon P, Modiano D, Molyneux ME, Mueller I, Parker M, Peshu N, Plowe CV, Puijalon O, Reeder J, Reyburn H, Riley EM, Sakuntabhai A, Singhasivanon P, Sirima S, Tall A, Taylor TE, Thera M, Troye-Blomberg M, Williams TN, Wilson M, Kwiatkowski DP. Genome-wide and fine-resolution association analysis of malaria in West Africa. Nat Genet. 2009;41:657–665. doi: 10.1038/ng.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings CV, Ahouidi AD, Zilversmit M, Bei AK, Rayner J, Sarr O, Ndir O, Wirth DF, Mboup S, Duraisingh MT. Molecular analysis of erythrocyte invasion in Plasmodium falciparum isolates from Senegal. Infect Immun. 2007;75:3531–3538. doi: 10.1128/IAI.00122-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungery M. Studies on the biochemical basis of the interaction of the merozoites of Plasmodium falciparum and the human red cell. Trans R Soc Trop Med Hyg. 1985;79:591–597. doi: 10.1016/0035-9203(85)90164-6. [DOI] [PubMed] [Google Scholar]
- Kaul DK, Roth EF, Jr, Nagel RL, Howard RJ, Handunnetti SM. Rosetting of Plasmodium falciparum-infected red blood cells with uninfected red blood cells enhances microvascular obstruction under flow conditions. Blood. 1991;78:812–819. [PubMed] [Google Scholar]
- Lantos PM, Ahouidi AD, Bei AK, Jennings CV, Sarr O, Ndir O, Wirth DF, Mboup S, Duraisingh MT. Erythrocyte invasion profiles are associated with a common invasion ligand polymorphism in Senegalese isolates of Plasmodium falciparum. Parasitology. 2009;136:1–9. doi: 10.1017/S0031182008005167. [DOI] [PubMed] [Google Scholar]
- Le Scanf C, Vigan-Womas I, Contamin H, Guillotte M, Bischoff E, Mercereau-Puijalon O. Rosetting is associated with increased Plasmodium falciparum in vivo multiplication rate in the Saimiri sciureus monkey. Microbes Infect. 2008;10:447–451. doi: 10.1016/j.micinf.2007.12.012. [DOI] [PubMed] [Google Scholar]
- Lengerke C, Daley GQ. Autologous blood cell therapies from pluripotent stem cells. Blood Rev. 2010;24:27–37. doi: 10.1016/j.blre.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo CA, Rodriguez M, Reid M, Lustigman S. Glycophorin C is the receptor for the Plasmodium falciparum erythrocyte binding ligand PfEBP-2 (baebl) Blood. 2003;101:4628–4631. doi: 10.1182/blood-2002-10-3076. [DOI] [PubMed] [Google Scholar]
- Lobo C-A, de Frazao K, Rodriguez M, Reid M, Zalis M, Lustigman S. Invasion profiles of Brazilian field isolates of Plasmodium falciparum: phenotypic and genotypic analyses. Infect Immun. 2004;72:5886–5891. doi: 10.1128/IAI.72.10.5886-5891.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackinnon MJ, Mwangi TW, Snow RW, Marsh K, Williams TN. Heritability of malaria in Africa. PLoS Med. 2005;2:e340. doi: 10.1371/journal.pmed.0020340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier AG, Duraisingh MT, Reeder JC, Patel SS, Kazura JW, Zimmerman PA, Cowman AF. Plasmodium falciparum erythrocyte invasion through glycophorin C and selection for Gerbich negativity in human populations. Nat Med. 2003;9:87–92. doi: 10.1038/nm807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller LH, Haynes JD, McAuliffe FM, Shiroishi T, Durocher JR, McGinniss MH. Evidence for differences in erythrocyte surface receptors for the malarial parasites, Plasmodium falciparum and Plasmodium knowlesi. J Exp Med. 1977;146:277–281. doi: 10.1084/jem.146.1.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min-Oo G, Fortin A, Tam MF, Nantel A, Stevenson MM, Gros P. Pyruvate kinase deficiency in mice protects against malaria. Nat Genet. 2003;35:357–362. doi: 10.1038/ng1260. [DOI] [PubMed] [Google Scholar]
- Mitchell GH, Hadley TJ, McGinniss MH, Klotz FW, Miller LH. Invasion of erythrocytes by Plasmodium falciparum malaria parasites: evidence for receptor heterogeneity and two receptors. Blood. 1986;67:1519–1521. [PubMed] [Google Scholar]
- Miyoshi H, Smith KA, Mosier DE, Verma IM, Torbett BE. Transduction of human CD34+ cells that mediate long-term engraftment of NOD/SCID mice by HIV vectors. Science. 1999;283:682–686. doi: 10.1126/science.283.5402.682. [DOI] [PubMed] [Google Scholar]
- Modiano D, Luoni G, Sirima BS, Simpore J, Verra F, Konate A, Rastrelli E, Olivieri A, Calissano C, Paganotti GM, D'Urbano L, Sanou I, Sawadogo A, Modiano G, Coluzzi M. Haemoglobin C protects against clinical Plasmodium falciparum malaria. Nature. 2001;414:305–308. doi: 10.1038/35104556. [DOI] [PubMed] [Google Scholar]
- Murphy SC, Harrison T, Hamm HE, Lomasney JW, Mohandas N, Haldar K. Erythrocyte G protein as a novel target for malarial chemotherapy. PLoS Med. 2006;3:e528. doi: 10.1371/journal.pmed.0030528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray CJ, Rosenfeld LC, Lim SS, Andrews KG, Foreman KJ, Haring D, Fullman N, Naghavi M, Lozano R, Lopez AD. Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet. 2012;379:413–431. doi: 10.1016/S0140-6736(12)60034-8. [DOI] [PubMed] [Google Scholar]
- Neildez-Nguyen TM, Wajcman H, Marden MC, Bensidhoum M, Moncollin V, Giarratana MC, Kobari L, Thierry D, Douay L. Human erythroid cells produced ex vivo at large scale differentiate into red blood cells in vivo. Nat Biotechnol. 2002;20:467–472. doi: 10.1038/nbt0502-467. [DOI] [PubMed] [Google Scholar]
- Nery S, Deans AM, Mosobo M, Marsh K, Rowe JA, Conway DJ. Expression of Plasmodium falciparum genes involved in erythrocyte invasion varies among isolates cultured directly from patients. Mol Biochem Parasitol. 2006;149:208–215. doi: 10.1016/j.molbiopara.2006.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogun SA, Holder AA. A high molecular mass Plasmodium yoelii rhoptry protein binds to erythrocytes. Mol Biochem Parasitol. 1996;76:321–324. doi: 10.1016/0166-6851(95)02540-5. [DOI] [PubMed] [Google Scholar]
- Okoyeh JN, Pillai CR, Chitnis CE. Plasmodium falciparum field isolates commonly use erythrocyte invasion pathways that are independent of sialic acid residues of glycophorin A. Infect Immun. 1999;67:5784–5791. doi: 10.1128/iai.67.11.5784-5791.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasvol G, Weatherall DJ, Wilson RJ. Cellular mechanism for the protective effect of haemoglobin S against P. falciparum malaria. Nature. 1978;274:701–703. doi: 10.1038/274701a0. [DOI] [PubMed] [Google Scholar]
- Pasvol G, Jungery M, Weatherall DJ, Parsons SF, Anstee DJ, Tanner MJ. Glycophorin as a possible receptor for Plasmodium falciparum. Lancet. 1982a;2:947–950. doi: 10.1016/s0140-6736(82)90157-x. [DOI] [PubMed] [Google Scholar]
- Pasvol G, Wainscoat JS, Weatherall DJ. Erythrocytes deficiency in glycophorin resist invasion by the malarial parasite Plasmodium falciparum. Nature. 1982b;297:64–66. doi: 10.1038/297064a0. [DOI] [PubMed] [Google Scholar]
- Pasvol G, Jungery M. Glycophorins and red cell invasion by Plasmodium falciparum. Ciba Found Symp. 1983;94:174–195. doi: 10.1002/9780470715444.ch11. [DOI] [PubMed] [Google Scholar]
- Pasvol G, Chasis JA, Mohandas N, Anstee DJ, Tanner MJ, Merry AH. Inhibition of malarial parasite invasion by monoclonal antibodies against glycophorin A correlates with reduction in red cell membrane deformability. Blood. 1989;74:1836–1843. [PubMed] [Google Scholar]
- Pawliuk R, Westerman KA, Fabry ME, Payen E, Tighe R, Bouhassira EE, Acharya SA, Ellis J, London IM, Eaves CJ, Humphries RK, Beuzard Y, Nagel RL, Leboulch P. Correction of sickle cell disease in transgenic mouse models by gene therapy. Science. 2001;294:2368–2371. doi: 10.1126/science.1065806. [DOI] [PubMed] [Google Scholar]
- Puthenveetil G, Scholes J, Carbonell D, Qureshi N, Xia P, Zeng L, Li S, Yu Y, Hiti AL, Yee JK, Malik P. Successful correction of the human beta-thalassemia major phenotype using a lentiviral vector. Blood. 2004;104:3445–3453. doi: 10.1182/blood-2004-04-1427. [DOI] [PubMed] [Google Scholar]
- Reed MB, Caruana SR, Batchelor AH, Thompson JK, Crabb BS, Cowman AF. Targeted disruption of an erythrocyte binding antigen in Plasmodium falciparum is associated with a switch toward a sialic acid-independent pathway of invasion. Proc Natl Acad Sci U S A. 2000;97:7509–7514. doi: 10.1073/pnas.97.13.7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth EF, Jr, Raventos-Suarez C, Rinaldi A, Nagel RL. Glucose-6-phosphate dehydrogenase deficiency inhibits in vitro growth of Plasmodium falciparum. Proceedings of the National Academy of Sciences of the United States of America. 1983;80:298–299. doi: 10.1073/pnas.80.1.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe JA, Moulds JM, Newbold CI, Miller LH. P. falciparum rosetting mediated by a parasite-variant erythrocyte membrane protein and complement-receptor 1. Nature. 1997;388:292–295. doi: 10.1038/40888. [DOI] [PubMed] [Google Scholar]
- Rowe JA, Obiero J, Marsh K, Raza A. Short report: Positive correlation between rosetting and parasitemia in Plasmodium falciparum clinical isolates. The American journal of tropical medicine and hygiene. 2002;66:458–460. doi: 10.4269/ajtmh.2002.66.458. [DOI] [PubMed] [Google Scholar]
- Rowe JA, Handel IG, Thera MA, Deans A-M, Lyke KE, Koné A, Diallo DA, Raza A, Kai O, Marsh K, Plowe CV, Doumbo OK, Moulds JM. Blood group O protects against severe Plasmodium falciparum malaria through the mechanism of reduced rosetting. Proc Natl Acad Sci USA. 2007;104:17471–17476. doi: 10.1073/pnas.0705390104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe JA, Claessens A, Corrigan RA, Arman M. Adhesion of Plasmodium falciparum-infected erythrocytes to human cells: molecular mechanisms and therapeutic implications. Expert Rev Mol Med. 2009a;11:e16. doi: 10.1017/S1462399409001082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe JA, Opi DH, Williams TN. Blood groups and malaria: fresh insights into pathogenesis and identification of targets for intervention. Curr Opin Hematol. 2009b;16:480–487. doi: 10.1097/MOH.0b013e3283313de0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruwende C, Hill A. Glucose-6-phosphate dehydrogenase deficiency and malaria. J Mol Med. 1998;76:581–588. doi: 10.1007/s001090050253. [DOI] [PubMed] [Google Scholar]
- Sankaran VG, Xu J, Ragoczy T, Ippolito GC, Walkley CR, Maika SD, Fujiwara Y, Ito M, Groudine M, Bender MA, Tucker PW, Orkin SH. Developmental and species-divergent globin switching are driven by BCL11A. Nature. 2009;460:1093–1097. doi: 10.1038/nature08243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serjeantson SW. A selective advantage for the Gerbich-negative phenotype in malarious areas of Papua New Guinea. P N G Med J. 1989;32:5–9. [PubMed] [Google Scholar]
- Sicard A, Semblat JP, Doerig C, Hamelin R, Moniatte M, Dorin-Semblat D, Spicer JA, Srivastava A, Retzlaff S, Heussler V, Waters AP. Activation of a PAK-MEK signalling pathway in malaria parasite-infected erythrocytes. Cellular microbiology. 2011;13:836–845. doi: 10.1111/j.1462-5822.2011.01582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim BK, Chitnis CE, Wasniowska K, Hadley TJ, Miller LH. Receptor and ligand domains for invasion of erythrocytes by Plasmodium falciparum. Science. 1994;264:1941–1944. doi: 10.1126/science.8009226. [DOI] [PubMed] [Google Scholar]
- Spadafora C, Awandare GA, Kopydlowski KM, Czege J, Moch JK, Finberg RW, Tsokos GC, Stoute JA. Complement receptor 1 is a sialic acid-independent erythrocyte receptor of Plasmodium falciparum. PLoS Pathog. 2010;6 doi: 10.1371/journal.ppat.1000968. e1000968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo YY, Small KS, Kwiatkowski DP. Methodological challenges of genome-wide association analysis in Africa. Nat Rev Genet. 2010;11:149–160. doi: 10.1038/nrg2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tham WH, Wilson DW, Lopaticki S, Schmidt CQ, Tetteh-Quarcoo PB, Barlow PN, Richard D, Corbin JE, Beeson JG, Cowman AF. Complement receptor 1 is the host erythrocyte receptor for Plasmodium falciparum PfRh4 invasion ligand. Proc Natl Acad Sci U S A. 2010;107:17327–17332. doi: 10.1073/pnas.1008151107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JK, Triglia T, Reed MB, Cowman AF. A novel ligand from Plasmodium falciparum that binds to a sialic acid-containing receptor on the surface of human erythrocytes. Mol Microbiol. 2001;41:47–58. doi: 10.1046/j.1365-2958.2001.02484.x. [DOI] [PubMed] [Google Scholar]
- Uneke CJ. Plasmodium falciparum malaria and ABO blood group: is there any relationship? Parasitol Res. 2007;100:759–765. doi: 10.1007/s00436-006-0342-5. [DOI] [PubMed] [Google Scholar]
- WHO. Geneva, Switzerland: World Health Organizaion; 2010. World Malaria Report 2010. [Google Scholar]
- Williams TN. Human red blood cell polymorphisms and malaria. Curr Opin Microbiol. 2006;9:388–394. doi: 10.1016/j.mib.2006.06.009. [DOI] [PubMed] [Google Scholar]