

Summary
Discovering the unintended “off-targets” that predict adverse drug reactions (ADRs) is daunting by empirical methods alone. Drugs can act on multiple protein targets, some of which can be unrelated by traditional molecular metrics, and hundreds of proteins have been implicated in side effects. We therefore explored a computational strategy to predict the activity of 656 marketed drugs on 73 unintended “side effect” targets. Approximately half of the predictions were confirmed, either from proprietary databases unknown to the method or by new experimental assays. Affinities for these new off-targets ranged from 1 nM to 30 μM. To explore relevance, we developed an association metric to prioritize those new off-targets that explained side effects better than any known target of a given drug, creating a Drug-Target-ADR network. Among these new associations was the prediction that the abdominal pain side effect of the synthetic estrogen chlorotrianisene was mediated through its newly discovered inhibition of the enzyme COX-1. The clinical relevance of this inhibition was borne-out in whole human blood platelet aggregation assays. This approach may have wide application to de-risking toxicological liabilities in drug discovery.
Adverse drug reactions (ADRs) can limit the use of otherwise effective drugs. Next to lack of efficacy, they are the leading cause for attrition in clinical trials of new drugs1–3 and are more prominent still in the failure of molecules to advance from pre-clinical research into human trials.4 Some ADRs are caused by modulation of a drug’s primary target,5 others result from non-specific interactions of reactive metabolites.6 In many cases, however, ADRs are caused by unintended activity at off-targets. Notorious examples of off-target toxicity include that of the appetite suppressant Fen-Phen, withdrawn from the market after numerous patient deaths. These owed to the activation of the 5-HT2B receptor by one of its metabolites, norfenfluramine, leading to proliferative valvular heart disease.7 Similarly, well-known drugs, such as the antihistamine terfenadine, have been withdrawn because of they caused arrhythmias and death, which have been attributed to their off-target inhibition of the hERG potassium channel.8,9 Prediction of unknown off-target drug interactions might prevent such disastrous drug toxicities, which are often detected only after fatalities in the clinic, and might allow safer molecules to be prioritized for pre-clinical development. Methods to systematically predict off-targets, and associate these with side effects, have thus attracted intense interest,10–16 frequently in the form of either chemical genomics17,18 or informatics19–26 approaches.
Whereas the informatics methods have never been tested systematically on a large scale, in principle they can be deployed against thousands of targets. Here we present a large-scale, prospective evaluation of safety target prediction using one such method, the Similarity Ensemble Approach (SEA).25–27 SEA calculates whether a molecule will bind to a target based on the chemical features it shares with those of known ligands, using a statistical model to control for random similarity. Because SEA relies only on chemical similarity, it can be applied systematically and, for those targets that have known ligands, comprehensively. For 656 drugs approved for human use (Supplementary Table S1), targets were predicted from among 73 proteins (Supplementary Table S2, Supplementary Methods) with established association of ADRs,22,28 for which assays were available at Novartis. Encouragingly, many of the predictions were confirmed, often at pharmacologically relevant concentrations. This motivated us to develop a guilt-by-association metric that linked the new targets to the ADRs of those drugs for which they are the primary or well-known off-targets, creating a Drug-Target-ADR network. The applicability and the limitations of this approach will be considered.
Testing the predictions
The 656 drugs were computationally screened for their likelihood to bind to 73 targets (Supplementary Table S2) using SEA.25–27 The targets belong to the Novartis in vitro safety panels based on their association with ADRs.22,28 Here we insisted that they also be described in ChEMBL,29 enabling correspondence with SEA predictions (Supplementary Table S2). ChEMBL annotates over 285,000 ligands modulating over 1,500 different human targets with affinities better than 30 μM. SEA calculated the similarity of each drug versus each set of ligands for the 73 targets, comparing the overall set similarity to a model of such expected at random. For instance, the sodium channel blocker aprindine loosely resembled the set of histamine H1 ligands; though no single H1 ligand was strongly similar to the drug (Table 1), the overall similarity of the set was much above that expected at random, leading to a highly significant SEA expectation value (E-value) of 5×10−26 between aprinidine and H1 receptor ligands. Only 1,644 of the over 47,000 possible drug-target pairs had significant E-values. Of these, 403 were already known in ChEMBL, and so were trivially confirmed; we do not consider these further. Of the remaining 1,241 predictions, 348 (28%) were unknown to ChEMBL, but could be found in proprietary ligand-target databases that were unavailable to SEA (Methods). The remaining 893 predictions represented previously unexplored drug-target associations.
Table 1.
New drug-off-target predictions confirmed by in vitro experiment. Representative, confirmed predictions are shown.
| Drug | Closest ChEMBL Molecule | Tca | Target | SEA E-value | IC50 [μM] | Closest known targetb | BLAST E- valuec |
|---|---|---|---|---|---|---|---|
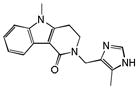 Alosetron |
|
0.25 | HTR2B | 1.6×10−17 | 0.02 | KCNH7 | 3.6e+2 |
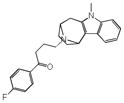
 Aprindine |
|
0.38 | HRH1 | 5.0×10−26 | 0.78 | SCN5A | 3.3e−1 |
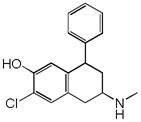
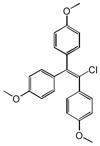 Chlorotrianisene |
|
0.31 | PTGS1 | 1.9×10−17 | 0.16 | ESR1 | 9.0e+2 |
 Clemastine |
|
0.31 | SLC6A4 | 1.1×10−14 | 0.42 | KCNH2 | 6.1e+1 |
|
Dyclonine |
|
0.36 | DRD4 | 1.5×10−17 | 4.1 | SLC6A3 | 2.3e+2 |
 Fasudil |
|
0.37 | ADRA2A | 1.1×10−7 | 4.0 | CCR2 | 1.5e−9 |
 Prenylamine |
|
0.31 | OPRM1 | 1.1×10−8 | 1.8 | CACNA1G | 3.5e+0 |
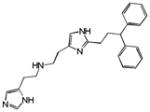
|
0.30 | HRH1 | 3.2×10−66 | 7.9 | SCN5A | 3.3e−1 | |
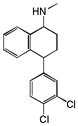 Sertraline |
|
0.33 | HRH2 | 5.1×10−45 | 1.4 | HTR4 | 6.8e−51 |
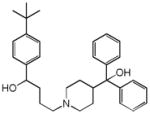 Terfenadine |
|
0.24 | SCN5A | 8.9×10−8 | 7.1 | CYP2D6 | 5.9e+1 |
Tc: The Tanimoto coefficient using ECFP_4-based molecular similarity to the closest ChEMBL reference molecule in the target set.
Closest known target: a known target of the drug that has highest sequence similarity to the predicted target.
BLAST E-value: based on the sequence identity of the predicted target to the closest known target; values greater than 10−5 represent unrelated proteins.
Of these predictions, 694 were tested at Novartis. For 478, activity was less than 25% at 30 μM; these were considered disproved. For another 65 predictions, activity was between 25 and 50% at 30 μM; these were considered ambiguous. Finally, for 151 of the new drug-target predictions IC50 values of less (better) than 30 μM were measured in concentration-response curves (Figure 1a, Supplementary Figure S1). In 125 cases, the drugs had an IC50 value better than 10 μM and in 48 activities were sub-micromolar (Table 1, Supplementary Table S3, Supplementary Figure S1). In summary, of the 1,042 predictions that were tested (694 by assay, 348 by databases), 48% were confirmed either in proprietary databases, unknown to the method and to those undertaking the SEA calculation, or in Novartis assays in full concentration response, and just under 46% were disproved (Figure 1a).
Figure 1. Predicting off-targets, and their novelty.

(a) Success of SEA predictions. Known: predicted off-targets confirmed via proprietary databases. Confirmed: predictions tested in vitro achieving IC50 <30 μM; Ambiguous: predictions with 25–50% activity at 30 μM. Inactive: <25% activity. (b) SEA enriched for non-trivial similarity. Drugs were binned (grey) by lowest Tc yielding valid SEA predictions. Hit rates of SEA (red) and 1NN (blue) with s.d. (c) Non-intuitive (chlorotrianisene) and straightforward (medrysone) SEA predictions, with Tc to closest references. Chlorotrianisene is only marginally similar to indomethacin, but is (correctly) predicted for COX-1. (d) Sequence similarities of each confirmed drug off-target to drug’s closest known targets.
In assessing these results, one would like to compare the true- to the false-positive and to the false-negative predictions. Whereas this work offers guidance on the first question, we can only address false negatives for a few compounds (Supplementary Results). Among these was astemizole, which had affinities ranging from 0.1 to 9 μM on the 5-HT2A, 5-HT2B and 5-HT2C, H2 and D2 receptors, as measured in other projects at Novartis. These targets were missed owing to a charge post-filter, separate from SEA itself, which excluded compounds with net charge dissimilar from the reference ligands.30 Astemizole was improperly assigned31 a charge of +2, wrongly differentiating it from the known ligands; the SEA E-values linking astemizole to these targets were themselves between 10−25 to 10−27. Other failures could be attributed to SEA itself. For instance, promazine bound to the histamine H1 and H2 receptors with low to mid-nanomolar affinities, but the SEA E-values at 10−4 to 10−3 were below our significance cutoff. This work was undertaken with ChEMBL2 as a source of ligand-target association; had we used the more recent ChEMBL10, H1 would have been predicted with an E-value of 10−9 (see http://sea.bkslab.org), and had we used ChEMBL12 and a newer version of SEA both targets would have been predicted. Clearly, with its reliance on topology and on inference from known ligand-target associations, SEA will have false negatives.
A key question is whether the new predictions were in any way surprising. One way to evaluate this is to compare the similarity of drugs predicted for new targets to the closest previously known ligand for that target. We used Tanimoto coefficients (Tc), which compare the groups in common between two molecules, here represented by ECFP_4 fingerprints. Tc values between nearest molecules were small, often less than 0.432; visual inspection of these pairs confirms the dissimilarity suggested by the low Tc values (Table 1). More systematically, SEA may be compared to a method that predicts targets based only on one nearest neighbor (a 1NN model) (Figure 1b). For close analogs (Tc values > 0.7, Figure 1c), the fraction of true positives was comparable between 1NN and SEA (Figure 1b). But across most similarity thresholds, SEA substantially outperformed 1NN, and by nearly two-fold in the low similarity range. Thus, for the Rho kinase inhibitor fasudil, SEA predicted only the adrenergic α2A receptor, with an E-value of 1.1×10−7, which was experimentally confirmed (IC50 = 4 μM). This occurred despite the low similarity of the closest known α2 ligand, which had a Tc value of 0.37 to fasudil. Conversely, at this similarity threshold the 1NN model predicted nine targets, only three of which were confirmed (Supplementary Table S4). For chlorotrianisene, two of the three targets predicted by SEA were confirmed; conversely, at its 0.31 Tc for cyclooxygenase-1 (COX-1) the 1NN model predicted ten targets, only two of which were confirmed.
We also investigated how often the new off-target would have been obvious based on sequence similarity of the targets.25,26,33 We calculated the BLAST sequence similarity of predicted targets to any known target of a drug (Table 1, Supplementary Table S3). Of the 151 new off-target predictions, 39 (26%) had BLAST E-values greater (worse) than 10−5, suggesting the previously known targets shared no sequence similarity with the new off-targets (Table 1, Supplementary Table S3, Figure 1d). For example, the anesthetic dyclonine was shown to bind the histamine H2 receptor (HRH2), while the closest known target was the Nav1.8 channel (SCN10A), which has no significant sequence similarity (BLAST E-value > 1) and is functionally unrelated to H2. Similarly, the anti-nausea drug alosetron antagonized the 5-HT2B receptor with an IC50 of 18 nM, though 5-HT2B has no sequence similarity to the ion channel targets of this drug (Table 1). Chlorotrianisene potently inhibits the enzyme COX-1, which is unrelated by sequence to the primary nuclear hormone receptor of this drug, the estrogen receptor (Table 1).
Associating in vitro targets with ADRs
To systematically assess the potential clinical relevance of the discovered targets, we developed a quantitative score that associated in vitro activity with patient adverse drug reactions. We enumerated all possible target-ADR pairs for 2,760 drugs with available adverse event annotations, expressing as an enrichment score the co-occurrence of pairs that were more common than expected by chance. (Supplementary Table S5). For example, “abdominal pain upper” has been reported for 45 drugs that interact with COX-1. The ADR “abdominal pain upper” was linked with 6,046 drug-target pairs, while COX-1 was linked with 2,188 drug-ADR pairs; there were a total of 681,797 target-ADR pairs overall. Thus the pair “abdominal pain upper” – “COX-1” was enriched 2.3-fold above random (Methods), with a χ2 p-value of 9.9×10−9. A total of 3,257 significant target-ADR associations were identified (Supplementary Table S5).
Having identified new off-targets for the drugs, and linked these with observed ADRs, we sought Drug-Target-ADR connections that illuminate the clinical relevance of the predictions. Of the 151 confirmed new drug-target associations tested at Novartis, 82 were significantly associated with one or more ADRs, resulting in a total of 247 Drug-Target-ADR links. In 116 cases, the enrichment factor of the new Drug-Target-ADR link was stronger than that for any previously known target (Table 2, Supplementary Table S6). For example, prenylamine was found to bind the histamine H1 receptor (HRH1), which we associate with a sedation ADR (ef = 4.9). By contrast, none of prenylamine’s known targets was associated with this side effect. For other cases, known targets represented an alternative explanation for an ADR. For instance, we found that diphenhydramine binds to the dopamine transporter (SLC6A3, Table 2), which is associated with tremor.34 Though tremor was also associated with one of diphenhydramine’s known targets, sodium channel SCN10A (ef = 1.9),35 its association with the dopamine transporter was higher (ef = 2.02), indicating a possible mechanistic link with the new off-target. Conversely, diphenhydramine’s “dry mouth” side effect was better explained by its known antagonism of the M3 muscarinic receptor (CHRM3, ef = 2.45, Supplementary Table S5).
Table 2. Characteristic new, confirmed targets that are associated with adverse drug reactions of the drugs.
Adverse drug reactions are listed that are more strongly associated with the predicted target than any known target together with pharmacokinetic data (AUC and Cmax), where available. Where pharmacokinetics (PK) and pharmacodynamic activity (PD) was available, drugs have been identified that behave comparably to the predicted drug and also cause the adverse event.
| Drug name | Target | Activity (μM) (median) | AUC [μM*h] | Cmax [μM] | Adverse event | EF ratioa | Alternative target | Comparable drugb |
|---|---|---|---|---|---|---|---|---|
| Chlorotrianisene | PTGS1 | 0.16 | NA | NA | Abdominal pain upper | 2.32 | ||
| Rash | 1.79 | |||||||
| Clemastine | SLC6A4 | 0.42 | NA | NA | Sleep disorder | 2.15 | ||
| Cyclobenzaprine | HRH1 | 0.02 | 0.16–4.10 (0.69) | 0.01–0.13 (0.06) | Ataxia | 1.73 | Desipramine | |
| Somnolence | 1.49 | Aripiprazole | ||||||
| Diphenhydramine | SLC6A3 | 4.33 | 2.57–3.42 (3.00) | 0.26–0.26 (0.26) | Tremor | 2.02/1.90 | SCN10A | Citalopram |
| Loxapine | CHRM2 | 1.12 | 0.03–0.43 (0.21) | 0.02–0.41 (0.14) | Tachycardia | 2.08/1.97 | CHRM1 | Sibutramine |
| Methylprednisolone | PGR | 1.30 | 0.09–10.76 (1.28) | 0.06–2.11 (0.31) | Depression | 3.87/2.49 | NR3C1 | Flutamide |
| Prenylamine | HRH1 | 7.87 | 0.12 | 1.2 | Sedation | 4.94 | ||
| Ranitidine | CHRM2 | 5.56 | 5.66–121.90 (9.67) | 1.14–9.11 (2.12) | Constipation | 1.63 | Haloperidol | |
| Ritodrine | OPRM1 | 9.18 | 0.03–0.32 (0.11) | 0.01–0.15 (0.04) | Hyperhidrosis | 3.21 | Oxycodone |
EF ratio: ADR-Target enrichment for predicted/best alternative known target.
Comparable drug: drugs that are known to bind the predicted target (bold: predicted target is the primary target), share the ADR and behave similarly in terms of PK and PD (see Supporting Methods).
We asked whether the drug’s affinity for its predicted ADR-target was relevant given its pharmacology, comparing the predictions against other drugs with similar pharmacodynamics and pharmacokinetics (Table 2). This was possible for 36 Drug-Target-ADR links (Supplementary Table S6). For instance, cyclobenzaprine was shown to bind to the histamine H1 receptor at 21 nM, while its median Cmax in plasma was 61 nM; nine other drugs binding H1 in the nanomolar range with comparable Cmax values were found (Table 2). Though some of the measured drug-target affinities were modest, the PK often confirmed that they were nevertheless relevant. For instance, the affinity of ranitidine (Zantac) for the M2 muscarinic receptor, which we associate with its constipation ADR, is only 5.6 μM. Nevertheless, with an AUC of 5.7 to 122 μM*h in plasma, this association seems plausible. Similarly, diphenhydramine has an IC50 of only 4.3 μM against the dopamine transporter, a target that we associate with the drug’s tremor side effect. Nevertheless, diphenhydramine’s AUC of 2.6 to 3.4 μM*h supports the relevance of its modest IC50.
Drug-Target-ADR networks
Network graphs help visualize the new and known drug-target links, and the adverse events with which they are associated (Figure 2a–c). For example, the estrogen receptor (ESR1) modulator chlorotrianisene was found to inhibit PTGS1 (COX-1), indeed with an affinity substantially better than its affinity for ESR1. Drugs that modulate the two proteins can share two of chlorotrianisene’s adverse reactions, “erythema multiforme” and “oedema”, but “rash” and “abdominal pain upper” link only to drugs inhibiting COX-1, and both of these are associated with chlorotrianisene almost uniquely among the estrogen receptor modulators (Figure 2a, Supplementary TableS5). For prenylamine, a new G protein-coupled receptor (GPCR) cluster (HRH1, OPRM1, ADRB2) emerges that is unrelated to the drugs primary ion channel activity but uniquely link to its sedative and myocardial infarction ADRs (Figure 2a). For domperidone, its known activity at dopamine receptors is associated with a Parkinsonism-like phenotype (“hyperprolactinaemia” and “extrapyramidal disorder”), while “somnolence” only associates with the newly discovered opioid activity (Figure 2b).
Figure 2. Off-target networks.

(a–c) Off-target networks for three drugs. Known targets of the drugs are grey while newly predicted targets are blue; the adverse events associated with each are orange and red, respectively. Red adverse events are significantly (ef > 1, q-Value < 0.05) associated with the new off-targets. Targets related by sequence are connected by grey edges. (d) Chlorotrianisene inhibits platelet aggregation. Two independent experiments (red and blue) shown for chlorotrianisene and indomethacin. Vehicle: negative control; ASA: Acetylsalicylic acid (positive control). Asterisks indicate significant (*: paired student t-test p-Value < 0.05) and highly significant (**: p-Value < 0.01) differences to vehicle control with s.d.
Several of these Drug-Target-ADR associations that emerged were surprising. Among them were the association of the muscle relaxant cyclobenzaprine with somnolence, the H2 antagonist ranitidine with constipation, and chlorotrianisene with upper abdominal pain (Table 2). Cyclobenzaprine caught our attention because even its mechanism of action target for muscle relaxation has not been characterized, and its association with the off-target discovered here, the H1 receptor (IC50 20 nM), precedes the identification of its primary target. The central nervous system H1 receptor is strongly associated with somnolence, consistent with the drug’s ADR, and supported by its pharmacokinetics. Similarly, the constipation effect of ranitidine is consistent with its activity on the M2 muscarinic receptor. Though its affinity for M2 is modest at 5.5 μM, its pharmacokinetics make this affinity relevant to this ADR.
Perhaps the most compelling demonstration of a Drug-Target-ADR association is one in vivo, or in an accepted in vivo biomarker. The observation that chlorotrianisene was a potent COX-1 (PTGS1) inhibitor seemed a reasonable explanation for the upper abdominal pain (epigastralgia) side effect provoked by the drug, and one that lent itself to direct testing in an accepted biomarker. Epigastralgia is a well-known ADR of non-steroidal anti-inflammatory drugs (NSAIDs), which inhibit the cyclooxygenase enzymes COX-1 and COX-2. COX-1 has housekeeping effects in the gastric mucosa,36 and its inhibition can lead to mucosal thinning and to gastroduodenal ulceration, leading to upper gastric pain and indeed the thousands of annual hospitalizations that associated with NSAID use.37 NSAIDs also inhibit platelet aggregation by direct inhibition of their endogenous COX-1 enzyme.38 Intriguingly, this effect is unreported for other synthetic estrogens, which, to the contrary, are more likely to promote platelet aggregation.39,40 A widely accepted model for platelet aggregation may be run ex-vivo, in whole blood, allowing one to test for target engagement of COX-1 in this effect.
Accordingly, collagen-induced platelet aggregation was measured in freshly drawn human blood from six healthy volunteer donors. Acetylsalicylic acid, the active ingredient in aspirin, inhibited platelet aggregation by 42 to 48% at 250 μM. The more potent NSAID indomethacin inhibited platelet aggregation by 50% at 50 μM. Chlorotrianisene inhibited platelet aggregation in whole blood with a potency almost indistinguishable from that of indomethacin, and more potently than acetylsalicylic acid (Figure 2d). These results are consistent with an in vivo inhibitory activity of chlorotrianisene on COX-1, and with the epigastralgia that is among its common side effects.
Drug and target promiscuity
To investigate overall patterns of drug and target promiscuity, we integrated the experimental results from this and other Novartis studies. The most promiscuous target was the voltage-gated sodium channel (SCN5A), to which 70 of 126 tested drugs bound (56%) (Figure 3a). From a target family standpoint, however, this was an exception, as most other promiscuous targets were small molecule-recognizing GPCRs; of non-GPCRs, only SCN5A and the ion channel hERG (KCNH2) were targeted more than average (>13% of drugs tested). Transporters had mid-range promiscuity; enzymes, nuclear receptors, and ligand-gated ion channels were less promiscuous, while peptide-recognizing receptors were hit least of all (Supplementary Tables S2, S7).
Figure 3. Target and drug promiscuity.

(a) Target promiscuity. Targets are sorted based on the percentage of drugs hitting the target below 30 μM (indicated by numbers next to target names). Colors code for seven distinct target classes. (b) Promiscuous drugs are often hydrophobic and cationic. Each point represents one drug. Ionization at pH 7.4 and ALogP values were calculated from drug structures. Hit rate: percentage of targets the drug binds below 30 μM.
Inverting this analysis, the most promiscuous drug, chlorhexidine, hit 34 (64%) out of 54 targets against which it was tested, and another nine drugs were active on over 50% of their tested targets (Figure 3b, Supplementary Table S8). Twenty-five drugs bound to proteins from among all major target classes. Highly promiscuous drugs often were lipophilic and cationic at physiological pH (Figure 3b).41,42
Predicting off-targets & adverse events
This study begins to quantify drug polypharmacology at scale: the 656 drugs considered here each modulated an average of seven safety targets, sometimes across multiple classes, and more than 10% of drugs acted on nearly half (45%) of the 73 targets (Figure 3b). It is sobering that this promiscuity is observed for approved human drugs, which have typically already been optimized to minimize toxicity. For lead molecules that are progressing toward the clinic, this level of off-target promiscuity might be higher still.28 Anticipating these off-targets is difficult, as they can be unrelated in sequence and structure to the primary targets of a drug, and even known target-ADR associations are not always straightforward. Two results of this study speak to these challenges. First, of the 1,042 predicted drug-target associations that were tested, 48% were confirmed (Figure 1a). With 46% of the predictions disproved, the method remains imperfect, but this rate nevertheless may be high enough to prioritize compound classes and targets for testing. Second, a guilt-by-association metric can link off-targets with ADRs. A three-way association between drugs, molecular targets, and ADRs may be systematically calculated and interpreted (Figures 2a–c).
Surprisingly, drugs often modulated off-targets unrelated to their primary target. Of the 151 off-targets that were confirmed by new experiment, 39 were unrelated by sequence to any of the drug’s known targets (Figure 1d). For example, the antitussive clemastine and the antihistamine diphenhydramine (an active ingredient in Tylenol PM and several other products), both of which act on the histamine H1 GPCR, also modulate the serotonin transporter (5-HTT, SLC6A4), to which the primary target is unrelated by sequence or structure. Conversely, the SERT inhibitor sertraline acts on the histamine H2 GPCR. The activity of drugs on targets that are unrelated by sequence or structure to their primary targets can seem capricious and certainly makes prioritization of likely targets more difficult. A ligand-based approach offers an orthogonal view of target relationships and so can illuminate similarities that are opaque from a molecular biology perspective. The converse is also true, and the two views will often be complementary.
The association between chlorotrianisene, COX-1, and epigastralgia illustrates the potential of the approach. The therapeutic target of chlorotrianisene, the estrogen nuclear hormone receptor, bears no sequence or structural similarity to the COX-1 enzyme, but the likelihood of cross-activity between the two targets is articulated by ligand similarity (Table 1). Correspondingly, the linking of abdominal pain and COX-1 only emerges when one quantitatively compares the ADRs of known COX-1 inhibitors to what one would expect at random. The potent inhibition of platelet aggregation by chlorotrianisene in whole blood (Figure 2d) is consistent with the systemic, in vivo activity of this drug at relevant concentrations on COX-1.
Certain shortcomings of the method should not escape the reader’s attention. Almost 46% of the predicted drug-target associations were disproved, and the method, which is inference-based, undoubtedly has important false negatives. As such, SEA cannot replace compound-target testing. What it can do is identify compounds early in development for possible liabilities that would ordinarily be identified only much later in drug progression. Similarly, the guilt-by-association method is inference-based and mechanism-naïve, and so will miss some target-ADR associations and make others that are invalid. Also, only some side effects fall into the remit of this approach, which assumes an off-target mechanism. Side effects, like other pharmacological events, have a strong exposure component, and can result from complex interactions with regulatory networks.43 Thus, topical drugs like econazole or chlorhexidine, though promiscuous in vitro, have fewer ADRs than expected because they never achieve sufficient systemic exposure in vivo. Conversely, a drug like tacrolimus might be relatively selective in vitro, but is associated with multiple side effects owing to its broad immunosuppressant effects.
These caveats should not obscure the potential of this approach to predict and understand drug side effects. The method was deployed automatically at scale, without human intervention. Whereas its predictions were sometimes disproved, they were just as often confirmed. If some of the targets it suggested required no imaginative leap—as when a steroid was predicted for a new nuclear hormone receptor—a quarter of the confirmed targets were unrelated by sequence or structure to any of the known targets of the drugs (Figures 1c, 1d, Tables 1 and S2). Pragmatically, the ability to calculate Drug-Target-ADR networks provides a tool to anticipate liabilities among candidate drugs being advanced toward the clinic, or yet earlier, for prioritization of chemotypes in preclinical series. If such networks cannot replace direct experimentation, they can usefully prioritize off-targets for consideration. As we struggle to develop new therapeutics, this and related approaches11,19,21–24,44,45 can identify molecules with liabilities early in their development, and so focus effort on those candidates that are least subject to them.
Method summary
A collection of on-hand 656 FDA-approved drugs was computationally screened against a panel of 339 molecular targets representing the species-specific expansion of 73 target assays used in Novartis safety panels. Each of the 339 target proteins was represented by its set of known ligands, as extracted from the ChEMBL_2 database. The two-dimensional structural similarity of a drug to a target’s ligand set was quantified as an E-value using the SEA, then subjected to a molecular charge filter. Predictions were tested retrospectively using proprietary databases including GeneGo Metabase, Thompson Reuters Integrity, Drugbank and GVKBio; novel predictions were tested prospectively in Novartis in vitro assays. Binding assays, and, when available, functional assays were performed including scintillation proximity, fluorometric imaging, filtration, fluorescence polarization, patch clamp, time-resolved fluorescence resonance energy transfer (TR-FRET), and homogenous time-resolved fluorescence (HTRF) assays. Concentration-response curves were calculated using XLfit (v.2 or v.4, IDBS, Guildford, UK) or a corresponding in-house software. All curves were redrawn using GraphPad PRISM v.5. Adverse drug reaction data were extracted from the world drug index (WDI) and encoded using the Medicinal dictionary for regulatory affairs (MedDRA). Using target annotations from GeneGo Metabase, Integrity, Drugbank, ChEMBL, and GVKBio, target-ADR pairs for all drugs were enumerated. Disproportionality analysis in conjunction with a Chi-squared test for association was carried out for all drug-target pairs. False discovery rate was controlled using the Benjamini-Hochberg correction for multiple hypothesis testing. Pharmacokinetic data were extracted from Integrity. For target and drug promiscuity analysis, combined external and internal target annotations were used. The computational workflow apart from SEA was implemented in Pipeline Pilot version 8 and statistical analyses were performed in R. Platelet aggregometry was performed in human blood for chlorotrianisene and indomethacin with acetylsalicylic acid as a positive control.
Methods
Virtual target profiling of drugs
We assembled a set of 656 drugs (Supplementary Table S1) available for internal prospective testing together with 73 assay targets for which Novartis safety panel assays28 were available. To compare activity annotations across databases, each target was mapped to human genes using Entrez gene and ChEMBL target identifiers (Supplementary Table S2). For target prediction, the 73 targets were represented by 339 orthologous proteins from human, rat, mouse, bovine, and sheep, using ChEMBL_2 database (released 3/25/2010); ligands for these targets with affinities ≤1 μM were grouped into sets for the SEA calculation.
We computationally screened the 656 drugs against the 339-target panel, using 1024-bit folded ECFP_446 and 2048-bit Daylight47 fingerprints independently, with the Tanimoto coefficient (Tc) as the similarity metric. Tc values lie between 0 and 1, where 1 corresponds to perfect overlap of two fingerprints. Where both fingerprints yielded the same SEA prediction, we took the prediction with the lower (i.e., stronger) E-value, unless otherwise noted. The maximum pair-wise Tc value was used in the 1-nearest neighbor (1NN) model.
Predictions with E-values <10−4 were retained. As a final step we subjected the SEA predictions to a pass/no-pass charge filter, to de-prioritize those predictions where the drug’s total charge did not match the charges calculated for at least 5% of the predicted target’s known ligands.30,31 This resulted in 4,195 drug-ChEMBL target pairs that were subsequently mapped to the 73 target panel, resulting in 1,644 unique predictions (the difference reflects the orthologous redundancies).
Testing predictions
Many SEA predictions could be confirmed by interrogation of proprietary databases, available at Novartis but unavailable in San Francisco where the calculations were performed. These included the Thompson Reuters Integrity (accessed January 2011, http://thomsonreuters.com/products_services/science/science_products/a-z/integrity), GeneGo Metabase (version 6.2, http://www.genego.com, accessed January 2011), and GVKBio (at http://www.gvkbio.com/, accessed January 2011). In addition, we also compared predictions to the ChEMBL_1129 and Drugbank 3.048 databases. For comparison across data sources, compounds were represented using the non-stereo specific part of InChiKeys.49
For prospective evaluation of the remaining predictions we used binding and functional assay data from internal Novartis profiling efforts, carried out in parallel to the SEA study. For some targets, functional assays were also available. Full concentration-responses curves were plotted for any compound with at least 50% inhibition or activity at the maximal tested concentration (30 μM, Supplementary Figure S1). For detailed assay descriptions, see Supplementary Methods and Supplementary Table S2.
Comparison to a 1NN model
We evaluated two 1NN models, using either ECFP_4 or Daylight fingerprints. Each drug was compared to all reference ligands of a target. The highest Tc value resulting from that comparison was assigned to the drug-target pair. For each drug, we identified the lowest Tc value that yielded valid SEA predictions using the respective fingerprint and collected all drug-target pairs with Tc scores above that threshold, irrespective of the SEA E-value. We counted the predictions confirmed in the proprietary databases or by experiment at Novartis. We calculated an adjusted hit rate:
| Equation 1 |
The additional count for both numerator and denominator distinguishes cases where no predictions were confirmed, but one method or the other predicted fewer targets. For example, SEA predicted four targets for bezafibrate, none of which were confirmed (Supplementary Table S4). However, at the corresponding Tc threshold of 0.37, the ECFP_4 1NN model identified 12 potential targets, none of which were confirmed. The adjusted fraction for SEA is 0.2 (= (0+1)/(4+1)), while the adjusted fraction for the 1NN model is 0.077 (1/13). We monitored the average adjusted hit rate for ten similarity threshold bins ranging from 0 to 1.
BLAST Target comparison
To investigate how closely the predicted targets were related to already known primary or off-targets, we calculated a target similarity matrix for all known and predicted targets found in our study. Amino acid sequences of all targets were assembled from UniProt.50 Sequences were compared in a pair-wise manner using BLASTp as implemented in Pipeline Pilot (version 8, http://www.accelrys.com).51 Target sequence similarity was quantified using BLAST E-values. Target pairs with values smaller than 10-5 were considered related by sequence.
Target and drug promiscuity
Targets were classified using ChEMBL’s target taxonomy, which consists of eight levels. The first three levels were used here in order to distinguish between small molecule and peptide GPCRs, as well as voltage and ligand gated ion channels (Supplementary Table S5). In-house and literature drug-target annotations were combined and annotations with IC50 <30 μM were counted as hits. Lipophilicity of drugs was assessed by calculating AlogP values in Pipeline Pilot. Negative values correspond to hydrophilic, and positive values to lipophilic compounds.
Associations between targets and adverse drug reactions
Adverse drug reactions were extracted from the World Drug Index (WDI, http://thomsonreuters.com/products_services/science/science_products/a-z/world_drug_index/, accessed March 2011) and mapped to preferred terms from the Medicinal Dictionary for Regulatory Affairs (MedDRA).52 MedDRA organizes adverse reaction terms in a hierarchy reaching from low-level terms to system organ classes at the highest level. Original WDI terms were first mapped to low-level terms in the MedDRA hierarchy using text mining components in Pipeline Pilot (version 8). Low-level terms serve as synonyms for preferred terms in MedDRA. These preferred terms were used to uniquely identify each adverse event. For example, the low-level terms “dry mouth” and “xerostomia” both map to the preferred term “dry mouth”. This resulted in 1,685 unique ADR terms, 2,760 unique drug structures with ADR annotations, and a total number of 51,101 drug-ADR pairs. Using drug-target associations from databases used for testing predictions, we enumerated all target-ADR pairs (681,797 total). The assessment was done separately for binding, antagonist, and agonist annotations. Assuming that each ADR could potentially occur due to any of the targets hit by the drug, we enumerated all possible target-ADR pairs for each drug. Target-ADR pairs occurring more than ten times were retained. The number of observations for each unique pair was then compared to the expected number of observations given the overall distribution of activity and adverse effect annotations. An enrichment score was calculated for each target-ADR pair:
| Equation 2 |
Where p is the co-occurrence of target X and ADR Y, A is the number of times ADR Y was linked to any drug-target pair, T is the number of times target X was linked with any drug-ADR pair, and P is the total number of target-ADR pairs.
To assess the statistical significance of found associations, we applied the Chi-square test for association based on contingency tables calculated for each unique target-ADR pair with an ef score greater than one. The false discovery rate was controlled using Benjamini-Hochberg correction in R (version 2.12, http://www.r-project.org).53 P-values and q-values (i.e. p-values corrected for multiple hypothesis testing), as well as the X2 statistic were calculated using the R statistical package. 3,257 associations with a q-Value of < 0.05 were retained (Supplementary Table S5).
Adverse reactions associated with predicted targets
Enrichment factors of predicted target-ADR pairs were compared to association of ADRs with any known targets of each drug. We prioritized adverse reactions that were stronger associated with the predicted than with any known target (i.e., had a higher ef score). In order to further prioritize adverse reactions likely due to the newly predicted target we extracted pharmacokinetic data from Thompson Reuters Integrity. Maximal plasma concentration (Cmax) and cumulative concentration (AUC) values measured in humans were assembled. Activity data were assembled from quantitative sources (ChEMBL_11 and GVKBio) for drugs that were not part of the predictions, but shared ADRs with the prediction drugs. Drugs were identified for each prediction and associated ADR that satisfied following three criteria: 1. They shared the ADR with the prediction drug. 2. They were not more than ten times more active at the predicted target. 3. Their Cmax value and/or AUC value was not more than ten times higher than for the prediction drug.
Platelet aggregation inhibition
Human blood samples from 6 healthy volunteer male donors were used to perform platelet aggregometry with a Multiplate® impedance aggregometer (Dynabyte Medical, Munich, Germany) as follows: Chlorotrianisene or indomethacin were added to whole blood at final concentrations of 0.5, 5 and 50 μM, and incubated at room temperature for 10 min; platelet aggregation was induced with collagen (1μg/mL) and measured at 37°C for 15 min; control aggregations were measured with vehicle only, and with acetylsalicylic acid (250 μM). Statistical analysis was performed using two-tailed t-tests and p ≤0.05 was considered significant. A detailed description can be found in the Supplementary Methods.
Supplementary Material
Acknowledgments
EL is a presidential postdoctoral fellow supported by the Education Office of the Novartis Institutes for Biomedical Research (co-mentors LU and BKS). Supported by US National Institute of Health grants GM71896 (to BKS and J. Irwin), AG002132 (to S. Prusiner and BKS), and GM93456 (to MJK), and by QB3 Rogers Family Foundation “Bridging-the-Gap” Award (to MJK).
Footnotes
Competing Interests: The authors declare competing financial interests.
Author Contributions: SEA calculations undertaken by MJK, target-ADR associations, networks, and promiscuity analysis by EL. In vitro assays were directed by SW, JH and LU. PK/PD experiments conducted by EW and PL. Platelet aggregation study designed and carried out by LU and SC; chlorotrianisene solubility and aggregation conducted by AKD. Project conceived and planned by BKS, JJ and LU. Overall analysis and writing largely by EL, MJK, BKS and LU; all authors contributed to the manuscript.
References
- 1.Giacomini KM, et al. When good drugs go bad. Nature. 2007;446:975–977. doi: 10.1038/446975a. [DOI] [PubMed] [Google Scholar]
- 2.Arrowsmith J. Trial watch: phase III and submission failures: 2007–2010. Nat Rev Drug Discov. 2011;10:87. doi: 10.1038/nrd3375. [DOI] [PubMed] [Google Scholar]
- 3.Arrowsmith J. Trial watch: Phase II failures: 2008–2010. Nat Rev Drug Discov. 2011;10:328–329. doi: 10.1038/nrd3439. [DOI] [PubMed] [Google Scholar]
- 4.Boyer S. The use of computer models in pharmaceutical safety evaluation. Altern Lab Anim. 2009;37:467–475. doi: 10.1177/026119290903700505. [DOI] [PubMed] [Google Scholar]
- 5.Wang D, Wong D, Wang M, Cheng Y, Fitzgerald GA. Cardiovascular hazard and non-steroidal anti-inflammatory drugs. Curr Opin Pharmacol. 2005;5:204–210. doi: 10.1016/j.coph.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 6.Antunes AMM, et al. Protein adducts as prospective biomarkers of nevirapine toxicity. Chem Res Toxicol. 2010;23:1714–1725. doi: 10.1021/tx100186t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rothman RB, et al. Evidence for possible involvement of 5-HT(2B) receptors in the cardiac valvulopathy associated with fenfluramine and other serotonergic medications. Circulation. 2000;102:2836–2841. doi: 10.1161/01.cir.102.23.2836. [DOI] [PubMed] [Google Scholar]
- 8.Roy M, Dumaine R, Brown AM. HERG, a primary human ventricular target of the nonsedating antihistamine terfenadine. Circulation. 1996;94:817–823. doi: 10.1161/01.cir.94.4.817. [DOI] [PubMed] [Google Scholar]
- 9.Curran ME, et al. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- 10.Ji ZL, et al. Drug Adverse Reaction Target Database (DART): proteins related to adverse drug reactions. Drug Saf. 2003;26:685–690. doi: 10.2165/00002018-200326100-00002. [DOI] [PubMed] [Google Scholar]
- 11.Kuhn M, Campillos M, Letunic I, Jensen LJ, Bork P. A side effect resource to capture phenotypic effects of drugs. Mol Syst Biol. 2010;6:343. doi: 10.1038/msb.2009.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matthews EJ, Frid AA. Prediction of drug-related cardiac adverse effects in humans--A: Creation of a database of effects and identification of factors affecting their occurrence. Reg Tox Pharmacol. 2010;56:247–275. doi: 10.1016/j.yrtph.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 13.Yang X, et al. Kinase inhibition-related adverse events predicted from in vitro kinome and clinical trial data. J Biomed Inform. 2010;43:376–384. doi: 10.1016/j.jbi.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol. 2008;4:682–690. doi: 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- 15.Zhang JX, et al. DITOP: drug-induced toxicity related protein database. Bioinformatics. 2007;23:1710–1712. doi: 10.1093/bioinformatics/btm139. [DOI] [PubMed] [Google Scholar]
- 16.Yang L, Luo H, Chen J, Xing Q, He L. SePreSA: a server for the prediction of populations susceptible to serious adverse drug reactions implementing the methodology of a chemical-protein interactome. Nucleic Acids Res. 2009;37:W406–412. doi: 10.1093/nar/gkp312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee S, Lee KH, Song M, Lee D. Building the process-drug-side effect network to discover the relationship between biological processes and side effects. BMC Bioinformatics. 2011;12 (Suppl 2):S2. doi: 10.1186/1471-2105-12-S2-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schreiber G, Keating AE. Protein binding specificity versus promiscuity. Curr Opin Struct Biol. 2011;21:50–61. doi: 10.1016/j.sbi.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oprea TI, et al. Associating Drugs, Targets and Clinical Outcomes into an Integrated Network Affords a New Platform for Computer-Aided Drug Repurposing. Mol Inf. 2011;30:100–111. doi: 10.1002/minf.201100023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paolini GV, Shapland RHB, van Hoorn WP, Mason JS, Hopkins AL. Global mapping of pharmacological space. Nat Biotechnol. 2006;24:805–815. doi: 10.1038/nbt1228. [DOI] [PubMed] [Google Scholar]
- 21.Scheiber J, et al. Mapping adverse drug reactions in chemical space. J Med Chem. 2009;52:3103–3107. doi: 10.1021/jm801546k. [DOI] [PubMed] [Google Scholar]
- 22.Bender A, et al. Analysis of pharmacology data and the prediction of adverse drug reactions and off-target effects from chemical structure. ChemMedChem. 2007;2:861–873. doi: 10.1002/cmdc.200700026. [DOI] [PubMed] [Google Scholar]
- 23.Campillos M, Kuhn M, Gavin AC, Jensen LJ, Bork P. Drug target identification using side-effect similarity. Science. 2008;321:263–266. doi: 10.1126/science.1158140. [DOI] [PubMed] [Google Scholar]
- 24.Tatonetti NP, et al. Detecting Drug Interactions From Adverse-Event Reports: Interaction Between Paroxetine and Pravastatin Increases Blood Glucose Levels. Clin Pharmacol Ther. 2011 doi: 10.1038/clpt.2011.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keiser MJ, et al. Predicting new molecular targets for known drugs. Nature. 2009;462:175–181. doi: 10.1038/nature08506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keiser MJ, et al. Relating protein pharmacology by ligand chemistry. Nat Biotechnol. 2007;25:197–206. doi: 10.1038/nbt1284. [DOI] [PubMed] [Google Scholar]
- 27.Hert J, Keiser MJ, Irwin JJ, Oprea TI, Shoichet BK. Quantifying the relationships among drug classes. J Chem Inf Model. 2008;48:755–765. doi: 10.1021/ci8000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Azzaoui K, et al. Modeling promiscuity based on in vitro safety pharmacology profiling data. ChemMedChem. 2007;2:874–880. doi: 10.1002/cmdc.200700036. [DOI] [PubMed] [Google Scholar]
- 29.Gaulton A, et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2011 doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laggner C, et al. Chemical informatics and target identification in a zebrafish phenotypic screen. Nat Chem Biol. 2012;8:144–146. doi: 10.1038/nchembio.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shelley JC, et al. Epik: a software program for pK(a) prediction and protonation state generation for drug-like molecules. J Comput Aided Mol Des. 2007;21:681–691. doi: 10.1007/s10822-007-9133-z. [DOI] [PubMed] [Google Scholar]
- 32.Muchmore SW, et al. Application of belief theory to similarity data fusion for use in analog searching and lead hopping. J Chem Inf Model. 2008;48:941–948. doi: 10.1021/ci7004498. [DOI] [PubMed] [Google Scholar]
- 33.Yildirim MA, Goh KI, Cusick ME, Barabási AL, Vidal M. Drug-target network. Nat Biotechnol. 2007;25:1119–1126. doi: 10.1038/nbt1338. [DOI] [PubMed] [Google Scholar]
- 34.Marshall V, Grosset DG. Role of dopamine transporter imaging in the diagnosis of atypical tremor disorders. Mov Disord. 2003;18 (Suppl 7):S22–27. doi: 10.1002/mds.10574. [DOI] [PubMed] [Google Scholar]
- 35.Kuo CC, Huang RC, Lou BS. Inhibition of Na(+) current by diphenhydramine and other diphenyl compounds: molecular determinants of selective binding to the inactivated channels. Mol Pharmacol. 2000;57:135–143. [PubMed] [Google Scholar]
- 36.Schoen RT, Vender RJ. Mechanisms of nonsteroidal anti-inflammatory drug-induced gastric damage. Am J Med. 1989;86:449–458. doi: 10.1016/0002-9343(89)90344-6. [DOI] [PubMed] [Google Scholar]
- 37.Kong SX, Hatoum HT, Zhao SZ, Agrawal NM, Geis SG. Prevalence and cost of hospitalization for gastrointestinal complications related to peptic ulcers with bleeding or perforation: comparison of two national databases. Am J Manag Care. 1998;4:399–409. [PubMed] [Google Scholar]
- 38.Perrone MG, Scilimati A, Simone L, Vitale P. Selective COX-1 inhibition: A therapeutic target to be reconsidered. Curr Med Chem. 2010;17:3769–3805. doi: 10.2174/092986710793205408. [DOI] [PubMed] [Google Scholar]
- 39.Akarasereenont P, Tripatara P, Chotewuttakorn S, Palo T, Thaworn A. The effects of estrone, estradiol and estriol on platelet aggregation induced by adrenaline and adenosine diphosphate. Platelets. 2006;17:441–447. doi: 10.1080/09537100600745302. [DOI] [PubMed] [Google Scholar]
- 40.Norris LA, Bonnar J. Effect of oestrogen dose on whole blood platelet activation in women taking new low dose oral contraceptives. Thromb Haemost. 1994;72:926–930. [PubMed] [Google Scholar]
- 41.Leeson PD, Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat Rev Drug Discov. 2007;6:881–890. doi: 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- 42.Peters JU, Schnider P, Mattei P, Kansy M. Pharmacological promiscuity: dependence on compound properties and target specificity in a set of recent Roche compounds. ChemMedChem. 2009;4:680–686. doi: 10.1002/cmdc.200800411. [DOI] [PubMed] [Google Scholar]
- 43.Cosgrove BD, et al. Cytokine-associated drug toxicity in human hepatocytes is associated with signaling network dysregulation. Mol Biosyst. 2010;6:1195–1206. doi: 10.1039/b926287c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mestres J, Gregori-Puigjané E, Valverde S, Solé RV. Data completeness--the Achilles heel of drug-target networks. Nat Biotechnol. 2008;26:983–984. doi: 10.1038/nbt0908-983. [DOI] [PubMed] [Google Scholar]
- 45.Mestres J, Gregori-Puigjané E, Valverde S, Solé RV. The topology of drug-target interaction networks: implicit dependence on drug properties and target families. Mol Biosyst. 2009;5:1051–1057. doi: 10.1039/b905821b. [DOI] [PubMed] [Google Scholar]
- 46.Rogers D, Hahn M. Extended-connectivity fingerprints. J Chem Inf Model. 2010;50:742–754. doi: 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- 47.James C, Weininger D, Delany J. Daylight theory manual - Daylight 4.91. 2005. [Google Scholar]
- 48.Wishart DS, et al. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008;36:D901–906. doi: 10.1093/nar/gkm958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stein S, Heller S, Tchekhovski D. An Open Standard for Chemical Structure Representation - The IUPAC Chemical Identifier. Nimes International Chemical Information Conference Proceedings; 2003. pp. 131–143. [Google Scholar]
- 50.The UniProt Consortium. Ongoing and future developments at the Universal Protein Resource. Nucleic Acids Res. 2010;39:D214–D219. doi: 10.1093/nar/gkq1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brown EG, Wood L, Wood S. The medical dictionary for regulatory activities (MedDRA) Drug Saf. 1999;20:109–117. doi: 10.2165/00002018-199920020-00002. [DOI] [PubMed] [Google Scholar]
- 53.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J R Stat Soc Series B (Methodological) 1995;57:289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.