

Abstract
Autosomal dominant cutis laxa (ADCL) is characterized by a typical facial appearance and generalized loose skin folds, occasionally associated with aortic root dilatation and emphysema. We sequenced exons 28–34 of the ELN gene in 5 probands with ADCL features and found 5 de novo heterozygous mutations: c.2296_2299dupGCAG (CL-1), c.2333delC (CL-2), c.2137delG (CL-3), c.2262delA (monozygotic twin CL-4 and CL-5) and c.2124del25 (CL-6). Four probands (CL-1, -2, -3, -6) presented with progressive aortic root dilatation. CL-2 and CL-3 also had bicuspid aortic valves. CL-2 presented with severe emphysema. Electron microscopy revealed elastic fiber fragmentation and diminished dermal elastin deposition. RT-PCR studies showed stable mutant mRNA in all patients. Exon 32 skipping explains a milder phenotype in patients with exon 32 mutations. Mutant protein expression in fibroblast cultures impaired deposition of tropoelastin onto microfibril-containing fibers, and enhanced tropoelastin coacervation and globule formation leading to lower amounts of mature, insoluble elastin. Mutation-specific effects also included endoplasmic reticulum stress and increased apoptosis. Increased pSMAD2 staining in ADCL fibroblasts indicated enhanced transforming growth factor beta (TGFβ) signaling. We conclude that ADCL is a systemic disease with cardiovascular and pulmonary complications, associated with increased TGFβ signaling and mutation-specific differences in endoplasmic reticulum stress and apoptosis.
Keywords: ELN, CL, connective tissue, skin, aneurysm, emphysema
Introduction
Cutis laxa (CL) comprises a heterogeneous group of acquired and inherited connective tissue disorders characterized by loose, redundant skin folds. The syndromic forms of CL include X-linked, autosomal dominant and recessive forms, which differ in severity and spectrum of associated clinical manifestations (de Schepper, et al., 2003; Milewicz, et al., 2000). Many related disorders are described and several forms of cutis laxa remain unclassified.
The X-linked form, also known as occipital horn syndrome (MIM# 304150), includes mild mental retardation, skeletal manifestations including the pathognomonic occipital horns, and bladder diverticulae (Tsukahara, et al., 1994) and is caused by mutations in the ATP7A gene (MIM# 300011) encoding a copper transporter (Kaler, et al., 1994). Autosomal recessive CL (ARCL) comprise at least three subtypes, with typically severe emphysema and life threatening vascular lesions in ARCL type I (MIM# 219100); developmental delay, microcephaly and skeletal abnormalities in ARCL type IIA (MIM# 219200) and the related entities geroderma osteodysplasticum (GO; MIM# 231070) and wrinky skin syndrome (WSS; MIM# 278250); ARCL type III, also known as de Barsy syndrome (MIM# 219150), associated with corneal abnormalities. In type I, mutations in the genes encoding fibulin-5 and fibulin-4 (FBLN5; MIM# 604580; EFEMP2; MIM# 604633), and most recently in the gene encoding latent transforming growth factor beta-binding protein 4 (LTBP4; MIM# 604710; Urban-Rifkin-Davis syndrome; MIM# 613177) have been identified (Hucthagowder, et al., 2006; Loeys, et al., 2002; Urban, et al., 2009). Mutations in ATP6V0A2 (MIM# 611716) have been shown to cause type II recessive cutis laxa and WSS (Kornak, et al., 2008).
Mutations in PYCR1 (MIM# 179035) encoding the mitochondrial pyrroline-5-carboxylate reductase 1 were identified in a similar patient population as well as in patients with GO and de Barsy syndrome (Guernsey, et al., 2009; Reversade, et al., 2009). Patients with GO may also have mutations in SCYL1BP1 (MIM# 607983), encoding a soluble Golgin protein that binds Rab6 (Hennies, et al., 2008). Other cutis laxa-related entities include a neurocutaneous syndrome with cataract, skeletal abnormalities and severe mental retardation (MIM# 612652) (Bicknell, et al., 2008) caused by defects in the ALDH18A1 (MIM# 138250) gene encoding a pyrroline-5-carboxylate synthase, and MACS syndrome (microcephaly, alopecia, cutis laxa and scoliosis, MIM# 613075) caused by mutations in RIN2 (MIM# 610222) (Basel-Vanagaite, et al., 2009), a ubiquitously expressed protein that interacts with Rab5.
Autosomal dominant cutis laxa (ADCL; MIM# 123700) presents with generalized lax skin folds resulting in a prematurely aged appearance. It is generally considered as a milder disease without the severe involvement of internal organs and normal neuromotor development (de Schepper, et al., 2003; Marchase, et al., 1980; Nahas, et al., 1999; Sarkar, et al., 2002). However, aortic and pulmonary abnormalities have been occasionally reported in patients with ADCL (Brown, et al., 1982; Szabo, et al., 2006; Urban, et al., 2005). Histopathology in ADCL is characterized by disorganized and often shortened elastic fibers that are predicted to result in a weakened and less elastic connective tissue (Szabo, et al., 2006). In most cases, patients carry frameshift mutations at the 3′-end of the elastin gene (ELN; MIM# 130160), predicted to result in a mutant tropoelastin (TE) protein with an extended carboxy-terminal missense peptide sequence (Rodriguez-Revenga, et al., 2004; Szabo, et al., 2006; Tassabehji, et al., 1998; Urban, et al., 2005; Zhang, et al., 1999), but mutations have also been identified in FBLN5 and exon 25 of the ELN gene (Graul-Neumann, et al., 2008; Markova, et al., 2003).
Little is known concerning the molecular mechanisms causing ADCL, in part because of the scarcity of patient tissue samples. Previously, both dominant negative effects and gain-of-function with enhanced proteolysis have been suggested for elastin mutations (Urban, et al., 2005). Molecular insights into this rare disease could be relevant for far more common diseases that are also characterized by elastic fiber fragmentation and are associated with aortic and pulmonary damage such as familial thoracic aortic aneurysms and emphysema (Robinson, et al., 2006; Shifren and Mecham, 2006). Conversely, more common monogenic diseases with degeneration of the elastic fibers, like Marfan syndrome (MFS; MIM# 154700), caused by mutations in fibrillin-1 (FBN1; MIM# 134797) (Dietz, et al., 1991), a main component of microfibrils and, as such, of elastic fibers, could provide helpful insights into the pathogenesis in cutis laxa. Enhanced transforming growth factor beta (TGFβ) signaling has been reported as a major disease-causing mechanism in MFS (Neptune, et al., 2003), a finding with direct therapeutic consequences (Habashi, et al., 2006). We report 5 new probands with de novo ELN mutations in the 3′ terminus of which four presented with internal organ involvement. We show enhanced TGFβ signaling, and demonstrate that mutation-specific differences in intracellular mutant protein processing cause different levels of endoplasmic reticulum (ER) stress and apoptosis.
Materials and Methods
Patients and samples
All patients were evaluated by a clinical geneticist at the referring center. Skin specimens were obtained from patients CL-1 (inguinal during inguinal surgery, 4 months), CL-2 (abdomen, 2 years), CL-3 (face during cosmetic surgery, 6 years) and CL-4 (inner upper arm, 3 years). Part of the skin specimens were prepared for electron microscopy (see below). For immunohistochemistry and electron microscopy, we used a skin biopsy from the outer upper arm of a 5 year old female control. We used age-, sex- and passage- matched controls (biopsies taken at the inner upper arm) for immunocytochemistry and the insoluble elastin assay. No fibroblast cultures were available for CL-2, CL-5 and CL-6. Skin fibroblasts were cultured in Dulbecco’s modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) in the presence of 100 I.U./ml penicillin, 100 μg/ml streptomycin and 0.25 μg/ml amphotericin. Experiments were performed between passages 2 to 5. Appropriate informed consent, including specific consent to publish clinical pictures (Fig. 1) was obtained from all patients involved in the study.
Figure 1.

Clinical presentation of patients CL-1, CL-2, CL-3, CL-4 and 5, and CL-6. Note the variable severity of generalized cutis laxa, and the typical facial characteristics including a premature aged appearance and a long philtrum (CL-3, CL-4, CL-5) and large ears (CL-4, CL-5).
Molecular analysis
Genomic DNA was extracted from blood samples by standard procedures, followed by touchdown PCR amplification of exons 28 through 34 of the ELN gene using forward and reverse primers located in the flanking introns (Supp. Table S1). The PCR products were analyzed by gel electrophoresis and visualized by ethidium bromide staining on 2% agarose gels. All amplimers were directly sequenced with an ABI PRISM 3730 automated sequencer (Applied Biosystems, Foster City, CA) using the PCR primers and BigDye terminator cycle sequencing chemistry. These sequences were compared to the wild type sequence as submitted to Ensembl Accession number ENST0000358929. The nucleotides were numbered starting from the first base of the initiation codon (ATG) of the cDNA reference sequence, according to journal guidelines (www.hgvs.org/mutnomen). The initiation codon is codon 1. Amino acid residues are numbered from the first methionine residue (reference sequence ENSP00000351807), according to journal guidelines. All mutations were confirmed to be de novo and absent in a panel of 100 normal control individuals.
RNA studies
RNA was extracted from fibroblast cultures using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA), and cDNA was synthesized using Superscript II Reverse Transcriptase Kit with hexamer primers (Invitrogen, Carlsbad, CA) in a total volume of 20 μl. First strand cDNA (1:10 dilution) was amplified using a oligonucleotide sense and antisense primers complementary to exon 29 (AGCCAAAGCTGCTGCCAAAG) and the 3′-untranslated region (3′UTR) (CATGGGATGGGGTTACAAAG), respectively (Szabo, et al., 2006). The sense primer was labeled using a fluorescent M13 primer tail (CACGACGTTGTAAAACGAC) at its 5′-end. Fragments were run with a 500 basepair ROX ladder (Gelcompany, San Francisco, CA) on an Applied Biosystems Prism 3130 genetic analyzer (Applied Biosystems). The data were processed using Genescan software (Applied Biosystems).
To determine the identity of mutant mRNA species, cDNA was prepared from fibroblast cultures as described above. RT-PCR amplimers were generated with previously described primers complementary to exon 29 and the 3′UTR using a Kapa hotstart Taq polymerase. Cloning into a TOPO TA vector (Invitrogen), transformation of TOP10F’ competent E. coli cells (Invitrogen), plating, selection and growth of selected colonies were done according to the manufacturer’s instructions (Invitrogen). Plasmid DNA was isolated using the Qiaprep Spin Miniprep (Qiagen) and sequenced.
Elastin staining
Semi-thin sections (1 micrometer thick) of the resin embedded skin biopsies were incubated with 1% aqueous methylene blue for 2–3 min at 80 degree Celsius (on a hot plate), rinsed with water and dried. Using this method, elastic material is stained darker blue than collagen bundles.
Electron microscopy
All specimens were fixed for at least 2h at room temperature in 3% glutaraldehyde solution in 0.1M cacodylate buffer pH 7.4, cut into pieces of ca. 1mm3, washed in buffer, postfixed for 1 h at 4° in 1% osmium tetroxide or in 0.5% ruthenium tetroxide, rinsed in water, dehydrated through graded ethanol solutions, transferred into propylene oxide, and embedded in epoxy resin (glycidether 100). Semithin and ultrathin sections were cut with an ultramicrotome (Reichert Ultracut E). Ultrathin sections were treated with uranyl acetate and lead citrate, and examined with an electron microscope (Philips EM 400).
Generation of a -1 frameshift mutant tropoelastin (fmTE) specific antibody
To generate an antibody specific to -1 frameshift mutant tropoelastin (fmTE), we chose a peptide antigen encoded by exon 34 and the 3′-UTR of ELN as a result of an upstream – 1 frameshift. The peptide, VGPAWGKLVAGRENELPRTPDSRPHQRQC, was synthesized on an ABI431 peptide synthesizer using FMOC chemistry and the sequence was confirmed by mass spectrometry. The antibody was generated in rabbits immunized with the peptide coupled to rabbit serum albumin. The specificity of the antibody was verified by ELISA assays (not shown) and by staining tissue sections of transgenic and control mice with the fmTE antibody and a control TE antibody (Supp. Fig. S1).
Immunostaining
Cells were grown for 3 weeks postconfluency for staining with elastin/fibrillin-1, mutant elastin/fibrillin-1 and pSMAD2. Staining for elastin/BIP, peIF2α and Caspase-3 was performed at 1 week postconfluency. Details about the antibodies used in this study are represented in Supp. Table S2. In brief, confluent cultures were washed with PBS once and fixed in 4% paraformaldehyde in PBS at 4°C for 30 min. After several washes in PBS, cell cultures were incubated in 3% bovine serum albumin (BSA) in either Tris-buffered saline (TBS, 20 mM Tris/HCl, pH 7.4, and 0.15 M NaCl) for non-permeabilized cells or TBS/0.05% Tween for permeabilized cells for 1 h. Cell cultures were then incubated overnight at 4°C with primary antibody in TBS or TBS/0.05% Tween. After several washes, cell cultures were incubated with secondary antibody and nuclear stain (Hoechst 33258) in PBS or PBS/Tween 0.05% for 1 hour at room temperature in the dark. Cell cultures were washed several times, mounted with Gel/Mount (Biomeda, Foster City, CA, USA) and visualized with an Axioskop BX60 microscope (Olympus, Center Valley, PA, USA).
Insoluble elastin assay
The insoluble elastin protocol was carried out as described previously (Urban, et al., 2002) with slight modifications. In brief, for an early and late time point (4 and 8 days), fibroblasts were plated in 60 mm culture dishes (500,000 cells/dish) and grown to confluency. Along with fresh media, 20 μCi of [3H]-leucine was added to each dish and the cell cultures were either incubated for the next 4 days or maintained in a time course as long as 8 days in the presence of [3H]-leucine (fresh media was added at day 4). At each time point of the course, triplicate cultures belonging to each experimental group were terminated, and insoluble elastin was assessed. After the removal of the media, cell layers were washed in 0.1 M acetic acid, scraped in 0.1 N NaOH and sedimented by centrifugation (10 min at 16000 x g). After removal of the supernatant, the pellets were boiled (at 97°C) for 45 min in 0.5 ml of 0.1 N NaOH to dissolve all ECM components except the insoluble elastin. After centrifugation (10 min at 16000 x g) the supernatant were mixed with 2 mL of scintillation fluid and measured in a Beckman Coulter LS6500 scintillation counter. Next, the pellets were boiled in 200 ml of 5.7 N HCl for 1 h to dissolve the insoluble elastin. The samples were mixed with scintillation fluid and measured in a scintillation counter (Hinek and Rabinovitch, 1994; Hinek, et al., 1993). Final results were expressed as follows:
with IE: insoluble elastin, cpm: counts per minute, ECM: extracellular matrix proteins except insoluble elastin. In this way, insoluble elastin was normalized for total extracellular matrix production.
Purification of recombinant human TE
A recombinant full-length human TE and a mutant elastin cDNA with a -1 frameshift in exon 32 (c.2262delA, fmTE), which was extended with a 72-base pair sequence at the C-terminus of elastin, were prepared as previously described (Wachi, et al., 2005). Each construct was inserted into a bacterial expression pTrcHis-TOPO vector (Invitrogen). The bacteria were grown to the mid-log phase at 37oC, and IPTG (isopropyl-(-D-thiogalactoside) was added to the culture to 1 mM in order to induce expression. Purified TE or fmTE was resuspended in SDS-PAGE sample buffer (62.5 mM, Tris, pH 6.8, 0.4% (w/v) SDS, 10% (v/v) glycerol, and 0.003% (w/v) bromophenol blue) including 100 mM DTT. Samples were run on SDS-PAGE gels and subjected to Western blot analysis to verify the purity of the recombinant proteins.
Coacervation assay
Coacervation was assayed by monitoring turbidity using light scattering at 400 nm with a UV spectrometer and JASCO V-500 software (Nihonbunkou, Tokyo, Japan) as previously described (Wachi, et al., 2008). In brief, the cuvette holder was connected to a re-circulating water bath in order to control the temperature of samples. Light scattering by each solution was monitored every 0.5 min while the temperature was increased from 15 °C to 45 °C at a rate of 1 °C/min. Both TE and the fmTE were used at concentrations of 6.25 μM and 12.5 μM in PBS, and as a mixture at a concentration of 6.25 μM each in PBS. Reference samples of pure recombinant TE and fmTE proteins were studied at concentrations of 12.5 μM in PBS.
Statistics
Immunostaining data were quantified for each coverslip by counting all positive cells in 10 high power fields containing at least a total of 300 nuclei. This number was divided by the total number of nuclei counted. Patients were compared to age, sex and passage matched controls using the Fisher’s exact test. For the insoluble elastin assay, results were analyzed using the paired, one-tailed student t-test.
Results
Clinical reports
Patient CL-1
Patient 1 is a 4 year old Caucasian boy. He was born to unrelated, healthy parents after an uneventful pregnancy at 42 weeks. Birth weight, length and head circumference were 4150g (75th percentile), 56 cm (97th percentile) and 36 cm (75th percentile), respectively. Cutis laxa was noted immediately at birth. At the age of three months, he presented with generalized cutis laxa with marked skin folds on face, neck, trunk, abdomen and limbs, but not on palms and soles. He had an aged appearance with loose redundant skin folds of the cheeks and chin, a high forehead, a long philtrum and large ear lobes. External genitals were normal. Growth and neuromotor development were appropriate. There was a large right-sided direct inguinal hernia, which needed surgical correction. Wound healing was normal. Pulmonary evaluation and echocardiography were within normal limits.
At age 3 years 5 months (Fig. 1, CL-1), he measured 100 cm (75–90th percentile) and weighed 14.8 kg (90th percentile). Clinical examination was unchanged, but a hoarse voice was noted. During exercise he experienced mild dyspnoea. Echocardiography revealed borderline enlarged sinuses of Valsalva and minimal aortic and mitral valve insufficiency. At age 6, the pear-shaped dilation of the aortic root progressed to a Z score of 3.7 (25 mm, weight 20 kg, height 123 cm). During the following year, the diameter at the sinuses of valsalva did not change (Z-score 2.5, height 127 cm, weight 21 kg) despite the fact that the patient was not under β-blocker therapy.
Patient CL-2
Patient CL-2 was born to unrelated, healthy parents after an uneventful pregnancy at 40 weeks. Birth weight was 3500 g (50th percentile), length 50 cm (25–50th percentile). Marked cutis laxa was immediately observed. Loose redundant skin folds were generalized but more prominent in the face, neck and abdomen (Fig. 1, CL-2). She had an aged appearance with a coarse face, a long philtrum and large dysplastic ear lobes. She had bilateral inguinal hernias. Growth and neuromotor development were normal. A slight strabismus was noted. At the age of 12, cosmetic surgery (face-lift) was planned but cancelled because preoperative assessment revealed severe chronic obstructive pulmonary disease (total lung capacity 118%, residual volume 209%, one second forced expiratory volume/forced vital capacity 42.9 % predicted) and a dilated aortic root. MRI-angiography showed a bicuspid aortic valve, a dilated aortic root of 41 mm (Z score = 7, weight 66.9 kg, height 164.5 cm) at the sinuses of Valsalva compressing the left atrium and the right ventricular outflow tract without significant hemodynamic changes. The diameter of the sinotubular junction was normal (25 mm) but the ascending aorta was enlarged up to 32–33 mm at the pulmonary bifurcation. The aortic arch was elongated and somewhat tortuous, but not enlarged. The diameter of the descending aorta was normal. Despite treatment with β-blocking agents (2.5 mg bisoprolol once daily) the aortic root further dilated to 45 mm at age 17 years, but remained stable when normalized to body surface area (1.95 m2). Aortic root replacement was recommended, but so far declined by the patient and her parents because of the high anesthetic risk due to the severity of the chronic obstructive pulmonary disease.
Patient CL-3
Patient 3 is a 17-year old female patient born to unrelated, healthy parents. She was reported previously at the age of 6 years (Jung, et al., 1996). She was noted to have generalized, loose, redundant skin folds at birth. During infancy, she suffered from several lung infections. At the age of six years, she presented with marked joint hypermobility and prominent facial characteristics with an aged appearance, large ears, beaked nose, and long philtrum. She had a deep, husky voice attributed to slack vocal chords observed by laryngoscopy. Blood and urine analysis including serum copper, ceruloplasmin and α1-antitrypsin were in normal ranges (Jung, et al., 1996). The karyotype was normal. She had a bicuspid aortic valve and during further follow-up, she developed a progressive dilation of the aortic root. At age 13, the diameter of the sinuses of valsalva measured 31 mm (Z-score 2.7, height 156 cm, weight 40 kg). The aortic dimensions normalized at the sinotubular junction, and more distally a second enlargement of the ascending aorta occurred (29 mm). At age 17, the aortic measurements were 39 mm at the sinuses of Valsalva (Z-score 4.2 for a height of 172 cm and weight 57 cm). At that time a mitral valve prolapse was noted. She was normotensive (113/77 mmHg right axillary artery). She had several cosmetic surgeries to correct skin folds and ears. Wound healing was normal but the skin folds rapidly recurred (Fig. 1, CL-3).
Patient CL-4 and CL-5
Patients CL-4 and CL-5 are 3-year old identical twins born at term to unrelated healthy parents. They presented at birth with generalized skin folds and typical facial characteristics including large ears, a coarse face and a long philtrum. Both had a deep and husky voice (Fig. 1, CL-4, CL-5). To date (at the age of 6), growth and neuromotor development was normal, and neither of them has developed any cardiovascular or pulmonary complications. Echocardiography showed normal mitral valve morphology and normal aortic root measurements in both (CL-4: aortic root 2.14 cm (Z-score 1.95), CL-5: aortic root 1.98 cm (Z-score 1.45).
Patient CL-6
This 7 year old Caucasian boy was the single child of unrelated parents. He was born following an uneventful pregnancy with Caesarean section at 39 weeks of gestation. Birth weight was 3570 g (25th percentile), length 50 cm (10th percentile), head circumference 35 cm (25th percentile). Growth and psychomotor development was adequate. Skin laxity was first noticed at the age of seven months. Subsequent follow-up showed recurrent but uncomplicated upper airway infections and recurrent balanitis. At age 2, abdominal, renal and pelvic ultrasound were normal. At age 5 (Fig. 1, CL-6), a cystourethrogram and chest X-ray were normal, but an echocardiography showed a pear-shaped dilatation of the aortic root of 27 mm (Z score = 4.7, weight 22 kg, height 118 cm).
Diminished and disorganized elastin deposition onto the microfibril bundles
Light microscopic examination of skin biopsies of patients CL-1 CL-2 and CL-3 showed severe rarefaction of elastic tissue compared to control (Fig. 2). Electron microscopic findings in the same patients (Fig. 2) revealed a reduced number of elastic fibers with diminished amounts of amorphous elastic material due to reduced and disorganized elastin deposition onto microfibrillar bundles, compared to a control. The amorphous component of the elastic fibers showed extensive branching and fragmentation and was not properly associated with the microfibrils. A particular finding was the increasing electron density from inner to outer regions of the elastic material and the separate elastic globules. Patent CL-4 showed similar, but milder abnormalities both by light and by electron microscopy (Fig. 2).
Figure 2.
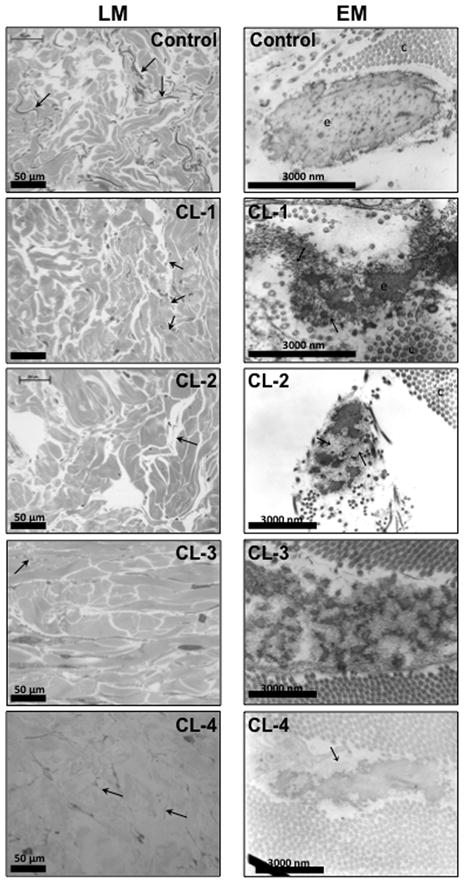
Light microscopic (LM) images of semi-thin methylene blue-stained sections of skin biopsies from a control (female, 15 years old), CL-1, CL-2, CL-3, and CL-4. Using this method, elastic fibers are stained darker than collagen bundles. Arrows indicate elastic fibers (dark grey). Elastic fibers are scarce in each ADCL sample. Electron microscopic (EM) images of a control sample, CL-1, CL-2, CL-3 and CL-4; c, collagen fibers; e, elastin; arrow, microfibrils not embedded in elastin. Patients show elastic fibers with diminished amounts and abnormal morphology of amorphous elastin, including extensive branching and fragmentation and a lack of proper association with the microfibrils. The electron density increases from inner to outer regions of the elastic material and there are separate elastic globules. In patient CL-4, similar, but milder abnormalities are present.
COOH-terminal ELN mutations produce stable mutant transcripts in ADCL
Direct sequencing of the probands’ DNA revealed 5 different de novo frameshift mutations in exon 30 of patients CL-6 (c.2124del25, p.G708fsX746, p.G708fsX792) and CL-3 (c.2137delG, p.A713fsX754, p.A713fsX800), in exon 32 of CL-4 and CL-5 (c.2262delA, p.L754fsX818) and in exon 33 of CL-1 (c.2296_2299dupGCAG, p.A767fsX816) and CL-2 (c.2333delC, p.P778fsX818) (Fig. 3A). One mutation, c.2262delA, was recurrent (Tassabehji, et al., 1998). Patients CL-4 and CL-5 were identical twins and had the same mutation. Four of the five ELN mutations) generated -1 frameshifts, whereas one produced a +1 frameshift. Irrespective of the location or type, each mutation was predicted to result in the replacement of the normal C-terminus of TE with a missense peptide sequence (Fig. 3B). In patients CL-2 and CL-3, α1-antitrypsin deficiency was excluded to be a predisposing factor for the development of severe emphysema by the finding of an M/M genotype and phenotype through direct sequencing of the SERPINA1 gene, and normal serum α1-antitrypsin measurement, respectively (Jung, et al., 1996).
Figure 3.

A: Schematic representation of the 3′-end of ELN with the location of the mutations identified in each patient. The coding exons are drawn to scale but the 3′-untranslated region (3′-UTR, hatched) and the introns are not. B: Schematic representation of the structure of wildtype (WT) and mutant (MT) mRNA products with semi-quantitative analysis. ELN transcripts in ADCL and control fibroblasts were studied using a oligonucleotide sense and antisense primer complementary to exon 29 and the 3′-UTR, respectively (Szabo, et al., 2006). M13 fluorescently labeled PCR-based amplicons were run on a genetic analyzer and data were processed using Genescan software (Applied Biosystems). Electropherograms are shown next to schematic representations of mRNA species identified in the control and in each patient. Cross-hatched bars indicate sequences coding for missense or read-through peptides generated by each frame shift mutation. Normal ELN sequence is shown by open bars. For patients only the products of the mutant allele are drawn, but electropherograms show both WT and MT products. Identities, sizes and % abundance relative to total ELN mRNA in each sample are shown next to each peak. In control fibroblasts exon 32 is spliced out (-e32) in about 75% of the transcripts (peak at 342 nucleotides). In CL-3 there is markedly higher levels of the mutant mRNA in transcripts lacking exon 32. Full-length transcripts of the wild type and mutant allele are present at low quantities. In patient CL-4, there is higher abundance of mutant mRNA in the full-length transcript (transcripts without exon 32 are all wild type). In patient CL-1, increased levels of the 4-nucleotide larger mutant mRNA is observed.
A complex interplay of the location of the mutation, naturally occurring alternative splicing of exon 32, mutation-induced exon skipping and nonsense-mediated decay (NMD) can influence the outcome of frameshift mutations at the 3′-end of ELN (Szabo, et al., 2006). Therefore, we studied the mRNA products of wild type and mutant alleles by semi-quantitative fragment analysis with and without treatment with cycloheximide, an inhibitor of NMD (Fig. 3B, Supp. Table S3). We verified the composition of the expected splicing products (with versus without exon 32) by cloning and subsequent sequencing of the different cDNA fragments produced by control and CL-1, CL-3 and CL-4 fibroblasts. We identified all fragments as predicted by fragment analysis, while no other sequences were detected (data not shown). Removal of exon 32 by alternative splicing was observed in control fibroblasts in about 70% of the transcripts (peak at 342 base pairs).
In patient CL-3 (c.2137delG mutation in exon 30), inclusion of exon 32 yielded a premature termination codon in exon 32 (p.A713fsX754) and thus a truncated open reading frame in 7.1% of ELN mRNA (Fig 3B, CL-3 MT). When exon 32 was spliced out, the transcript contained an extended C-terminal missense sequence of 61 amino acids (AA) with a read-through into the 3′-UTR (p.A713fsX800) adding 25 AA (63.3% of ELN mRNA) (Fig. 3B, CL-3 MT-e32). CL-3 fibroblasts showed markedly elevated levels of mutant mRNA 70.4%, compared to 29.6% of wildtype (WT) mRNA. Remarkably, the abundance of exon 32 containing (full-length) transcripts was lower in CL-3 fibroblasts than in control, CL-1 or CL-4 fibroblasts. The c.2137delG mutation introduced a premature termination codon into exon 32, which may have activated NMD eliminating exon 32 containing transcripts. Although cycloheximide raised the full-length MT message level by 55% (Supp. Table S3), it also elevated the level of the full-length WT mRNA. Thus, reduced levels of full length mRNA in CL-3 may be more related to inter-individual variation in alternative splicing than to NMD. This finding is consistent with the know reduction of the effectiveness of NMD close to the 3′-end of transcripts.
In patient CL-4 (mutation c.2262delA in exon 32), the -1 frameshift mutation resulted in C-terminal altered sequence of 28 AA (p.L754fsX818) and extension with 25 AA in the full-length transcript accounting for 54.3% of ELN mRNA (Fig. 3B). When exon 32 was spliced out, a normal elastin mRNA was produced, indistinguishable from the –e32 splice product of the wildtype allele. CL-4 fibroblasts also showed significantly higher abundance mutant mRNA (Supp. Table S3). Moreover, we noted that in CL-4 fibroblasts the abundance of the full-length transcript was higher than the -e32 transcript, unlike any other CL or control fibroblasts we studied to date.
Patient CL-1 had a duplication of 4 nucleotides in exon 33 (c.2296_2299dupGCAG). This frameshift mutation resulted in a C-terminal missense sequence of 26 AA and an extension of 23 AA irrespective of exon 32 splicing (p.P778fsX818). Analysis of mRNA also revealed higher levels of mutant mRNA (Fig. 3B). In conclusion, all patients showed increased abundance of stable mutant mRNA, under both cycloheximide treated and untreated conditions.
Mutant TE is deposited in the ECM, results in clumping and reduced deposition of insoluble elastin
Elastic fiber formation is believed to depend on the deposition of TE on fibrillin microfibrils and subsequent crosslinking to form insoluble elastin. Colocalization immunofluorescent staining for fibrillin-1 and elastin on cultured patient fibroblasts shows less dense elastin deposition into the elastic fibers, but more globular deposits consisting of elastin only (patients CI95%: 2.28 ± 0.64 gobules/nucleus versus control CI95%: 1.35 ± 0.21 globules/nucleus) (Fig. 4). Co-localization experiments of fibrillin-1 and mutant elastin showed more prominent intracellular and globular extracellular staining and less robust extracellular fibrillar staining in both patient CL-3 and CL- 4 (Supp. Fig. S2).
Figure 4.
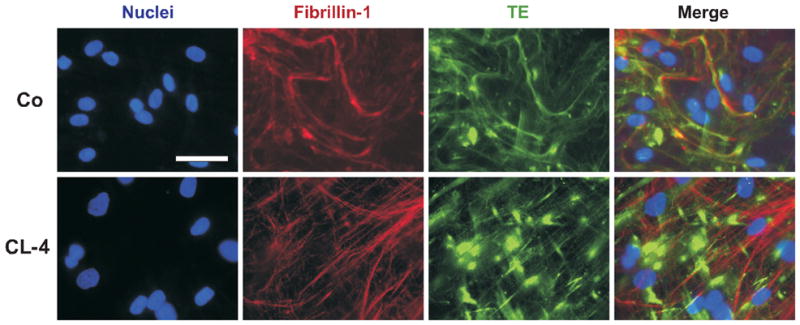
Representative colocalization immunofluorescent staining for fibrillin-1 (red) and TE (green) of non-permeabilized cell cultures of patient CL-4 and a matching control (Co). In the patient, elastin was less densely deposited in fibers, and showed significantly enhanced aggregation in clumps. Nuclei were stained blue. Scale bar: 25 μm.
In order to elucidate the reason of enhanced elastin globule formation, we assessed temperature-dependent coacervation of normal TE and fmTE in vitro. Despite lower overall hydropathy (which generally inhibits coacervation) of fmTE, its coacervation temperature was lower than that of TE (Fig. 5A), consistent with increased self-association of mutant TE yielding globular aggregates. Moreover, the coacervation temperature of an equimolar mixture of TE and fmTE were close to fmTE alone, showing a dominant effect of fmTE on TE coacervation (Fig. 5B).
Figure 5.
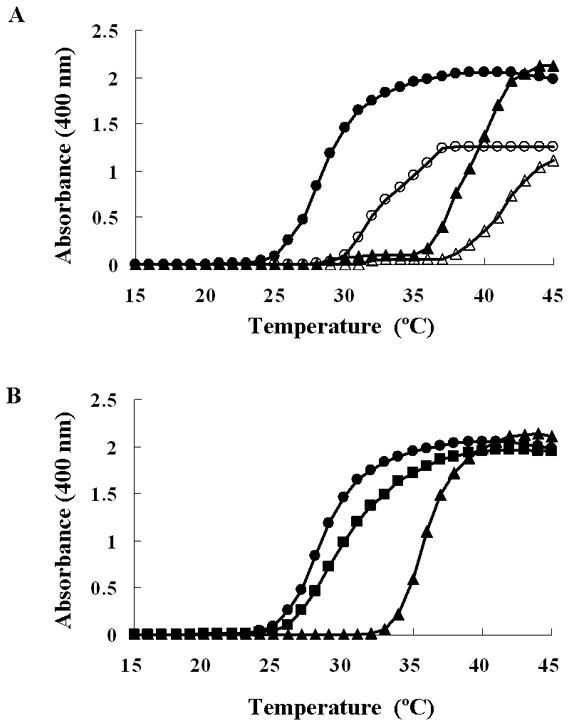
A: Coacervation of TE and fmTE. TE (triangles) and fmTE (circles) were diluted to a concentration of 6.25 μM (open symbols) and 12.5 μM (closed symbols) in PBS. Light scattering was monitored every 0.5 min while raising the temperature form 15 °C to 45 °C. B: Coacervation of a mixture of TE and fmTE was studied at a concentration of 6.25 μM each in PBS. As reference, pure TE and fmTE protein solutions were evaluated at concentration of 12.5 μM in PBS.
To test whether altered coacervation affects the amount of mature, crosslinked elastin in the ECM, we analyzed isolated insoluble elastin from metabolically labeled cell cultures relative to the amount of other intra- and extracellular proteins (as a measure for cell metabolism). We found significantly lower amounts of insoluble elastin in ADCL cells compared to controls on day 4 and 8 (Fig. 6).
Figure 6.
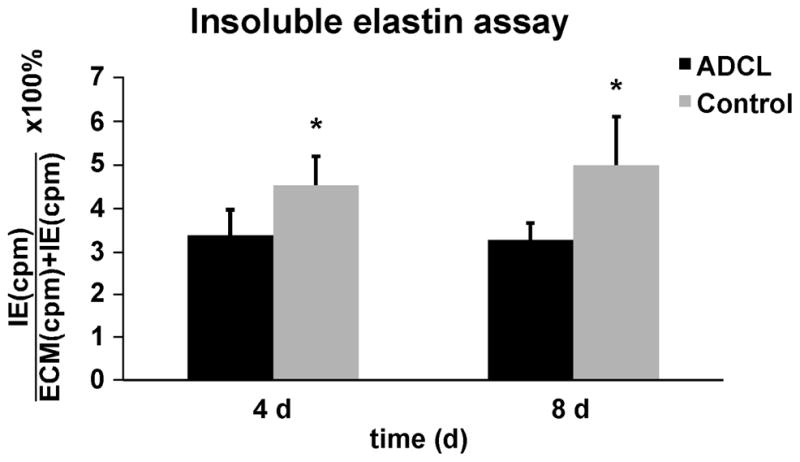
Biochemically isolated insoluble elastin was measured form metabolically labeled cell cultures and normalized to all intra- and extracellular proteins in the cell layer (as a measure for cell metabolism) and cell count. Bars represent the averaged values of insoluble elastin in all ADCL and matched control fibroblasts. There was a significant reduction of mature, crosslinked, insoluble elastin after 4 and 8 days in ADCL patients. *p value < 0.01 using the paired one-tailed student T-test. Error bars show the standard deviation of the mean.
Elastic fiber disorganization in ADCL results in upregulation of TGFβ signaling
Elastic fibers constitute of an elastic core embedded in a microfibrillar structure. These microfibrils also serve as important extracellular storage sites for TGFβ. We therefore tested whether the disruption of the elastic fibers influences TGFβ signaling. Staining for pSMAD2 in fibroblast cultures of all patients showed significant upregulation of the TGFβ signaling pathway (Fig. 7A, B).
Figure 7.

A: p-SMAD2 staining in fibroblast cultures. All patients (CL-1, CL-3, and CL-4) showed increased proportion of p-SMAD2 positive cells (p<0.001) indicating enhanced TGFβ signaling compared to matching controls (Co-1, Co-3 and Co-4). Scale bar: 50 μm. B: Quantitative morphometry of pSMAD2 staining in fibroblasts of ADCL patients and their matching controls. Quantitative morphometry of C: BiP; D: peIF2α; E: caspase-3 staining in fibroblasts of ADCL patients and matching controls. Patients CL-3 and CL-4, but not patient CL- 1, showed upregulation of the chaperone BiP co-localizing with TE in the ER. peIF2α and caspase-3 are significantly upregulated in patient CL-3, but not in CL-1 or CL-4. **: p<0.001; *: p<0.01; NS: not significant.
Intracellular retention of fmTE and unfolded protein response
Recently, a transgenic mouse model for ADCL expressing a human elastin minigene with a -1 frameshift in exon 30 showed intracellular retention of fmTE, induction of unfolded protein response (UPR) and initiation of the apoptotic cascade in lung tissue (Hu, et al., 2010). Therefore, we investigated the UPR pathway in our ADCL fibroblast lines. The chaperone binding protein (BiP) was upregulated in varying degrees in fibroblast cell cultures of patients CL-1 (mutation in exon 33) (p=0.14), CL-3 (mutation in exon 30) (p<0.01) and CL-4 (mutation in exon 32) (p<0.001), (Fig. 7C, Supp. Fig. S2). BiP co-localized with TE in the ER, indicating misfolding (Supp. Fig. S3). We then assessed phosphorylated eukaryotic translation initiation factor (peIF2α), a marker for ER stress, which was only significantly upregulated in patient CL-3 (Fig. 7D). This latter observation paralleled the findings for Caspase-3, a marker for apoptosis (Fig. 7E).
Discussion
To date, cutis laxa syndromes have been classified mainly on both clinical grounds and mode of inheritance (de Schepper, et al., 2003). Clinical overlap and related, unclassified disorders illustrate many shortcomings of this classification, as illustrated by the fact that 4 out of 5 probands were initially diagnosed with recessive cutis laxa. The recent progress in the molecular elucidation of cutis laxa subtypes will improve the clinical definition of these disorders and will facilitate genotype-phenotype correlations. In this study, we demonstrate that in ADCL, formerly considered as a more benign disease, patients are also at risk for severe aortic and pulmonary complications (including aortic root dilatation and emphysema), blurring the clinical distinction from type I recessive cutis laxa. Facial characteristics were the most distinguishing and ubiquitously present feature in all patients, with a long philtrum, large ears, a husky voice and often a beaked nose. Generally, patients had severe, generalized and congenital cutis laxa, but we also documented progression of an initially milder phenotype in one proband.
We also demonstrate significant association of bicuspid aortic valve (BAV) with ADCL. The population prevalence of BAV is 5/1075 (Tutar, et al., 2005) and the prevalence in our group of ADCL patients is 2/6. Applying Fisher’s exact test to these numbers yields p=0.0005, indicating a highly significant association. In addition, the presence of BAV in ADCL patients was associated with dilatation of both the aortic root and of the ascending aorta past the sinotubular junction. In contrast, ADCL patients without BAV only had dilatation of the aortic root. This has important implications of the cardiovascular follow up and surgical treatment of aortic aneurysms in ADCL.
Molecular heterogeneity has been suggested in ADCL (Graul-Neumann, et al., 2008; Markova, et al., 2003). Our data now suggest that, at least in the majority of cases, ADCL can be attributed to a mutant TE in which the C-terminus is replaced by an extended missense peptide sequence as a result of a frameshift (fmTE). The observation of a first +1 frameshift (CL-1) in a patient with a similar ADCL phenotype suggests that the molecular mechanisms act largely independent of the amino acid composition at the 3′ terminus.
The mechanisms by which an extended elastin protein disrupts elastic fiber assembly and leads to the observed phenotype have been a subject of discussion. Matrix deposition of mutant TE was shown for a partial tandem duplication in the elastin gene (Urban, et al., 2005), and confirmed in this report for the more common frameshift mutations. This carboxy-terminus of TE has been implicated in microfibril associated glycoprotein binding (Brown-Augsburger, et al., 1994) and in interaction with cell-surface glycosaminoglycans (Broekelmann, et al., 2005), both important in elastic fiber formation.
Time-lapse imaging studies showed that the assembly of elastic fibers was a complex process with initial micro-assembly of TE monomers into globular aggregates, followed by a cell-directed, dynamic macro-assembly step that fashioned large-scale fibrillar structures (Czirok, et al., 2006; Kozel, et al., 2006). We observe an accumulation of globular elastin aggregates in fibroblast cultures from patients with ADCL, suggesting that fmTE disrupts later stages of the elastin assembly process. Decreased deposition of insoluble elastin by ADCL cells provides further evidence for this notion. Finally, our electron microscopic results demonstrate the abnormal elastin assembly, characterized by poor integration of elastin with microfibrils.
Our studies also clarify the exact mechanism by which fmTE disrupts elastin assembly. We demonstrate that fmTE has increased self-association properties manifested by a lower coacervation temperature resulting in enhanced globule formation. Moreover, fmTE had a dominant effect on lowering the coacervation temperature in mixtures of fmTE and nTE providing a molecular basis for the autosomal dominant inheritance of ADCL. Other studies focusing on fmTE showed reduced binding to fibrillin-1 and fibulin-5 and, as a result, impaired elastin accumulation on microfibrils in cell based assays (Sato, et al., 2006). Taken together, these results suggest that a combination of increased aggregation of fmTE and its decreased binding to microfibrils leads to a poor integration of the elastin and microfibrils in the elastic fibers of patients with ADCL. Importantly, impaired association of TE with microfibrils and abnormal aggregation of TE have been shown to be part of the molecular mechanisms leading to multiple forms of cutis laxa, including ARCL1 caused by mutations in FBLN5 (Hu, et al., 2006), and ARCL2 caused by mutations in ATP6V0A2 (Hucthagowder, et al., 2009). These studies, together with the present report demonstrate shared disease mechanisms in multiple CL syndromes.
A recently established transgenic mouse model, generated by pronuclear injection of a minigene encoding a human elastin gene with a -1 frameshift in exon 30, showed intracellular retention and initiation of the apoptotic cascade in lung tissue (Hu, et al., 2010). In this report, we observed mutation-specific induction of the unfolded protein response (UPR). The mutation in exon 33 (CL-1) resulted in no measurable increase in any of the UPR markers studied. This mutation is a +1 frameshift leading to a 49 AA-long missense peptide sequence. The mutation in exon 32, a −1 frameshift predicted to generate a 53 AA-long missense peptide sequence led to elevated BiP recruitment to fmTE, a sensitive, early measure of UPR, but no significant elevation of peIF2a or of Caspase-3, late markers of severe UPR. Finally, the mutation in exon 30, a −1 frameshift mutation predicted to generate 86 missense AA, caused elevation in both early (BiP) and late (peIF2a, Caspase-3) markers of UPR. Thus we conclude that both the length and the primary structure of the missense amino acid sequence (depending on the type of frame shift) can influence the extent of the UPR response to ELN mutations. Developmental defects through increased apoptosis was also shown in ARCL type II, caused both by ATP6V0A2 and PYCR1 mutations (Hucthagowder, et al., 2009; Reversade, et al., 2009) and may therefore represent another common mechanism in cutis laxa. However, the observed allele-specific differences in the extent of ER stress alone do not explain the observed clinical variability in pulmonary and aortic involvement among patients.
In MFS, another disease of the elastic fibers with aortic dilatation and emphysema as important clinical manifestations, disruption of fibrillin-1 results in enhanced release of active TGFβ from the extracellular matrix, inducing disorganization of the arterial wall (Habashi, et al., 2006) and failure of differentiation of the terminal alveolar septae (Neptune, et al., 2003). Both the histological aberrations and the clinical manifestations are prevented by antagonizing TGFβ signaling. We also found highly increased levels of pSMAD2 staining in fibroblast cultures of ADCL patients, consistent with increased TGFβ activity. TGFβ is released by stretching the ECM and upon traction from myofibroblasts (Wipff, et al., 2007). We speculate that due to failure of proper elastic fiber organization in ADCL, a more compliant ECM results in higher amounts of released TGFβ. In further support of these data, a transgene mouse model of ADCL that develops emphysema displays decreased lung stiffness, increased stretch at physiological pressures and increased TGFβ signaling (Hu, et al., 2010). Therefore, increased TGFβ activity likely accounts for aortic root dilatation and emphysema and opens perspectives for treatment with losartan (Habashi, et al., 2006). It should be noted that patients with exon 32 deletions have less severe internal organ involvement (p < 0.005, Supp. Table S4). Indeed, exon 32 skipping results in a partial rescue and better elastic fiber formation (Fig. 2, Fig. 4). Hence, this extracellular matrix might preserve its resilient characteristics better in lung and aortic tissues reducing the amount of activated TGFβ upon stretching.
In conclusion, these data provide new insights in the clinical and molecular findings in ADCL. Partly retained mutant elastin may predispose to ER stress and apoptosis, while partly secreted mutant elastin results in impaired elastic fiber formation and reduced elastin quality. Enhanced TGFβ release from this extracellular matrix clinically manifests in aortic dilatation and pulmonary emphysema. Therefore these data may also be relevant for more common diseases of the elastic fiber including isolated aortic aneurysms and emphysema.
Supplementary Material
Acknowledgments
We thank the families and patients for their interest and cooperation in this research. BL is a senior researcher of the Fund of Scientific Research – Flanders. This work was supported by the National Institutes of Health (HL090648 to Z.U and HL084944 to Z.U. and R.P.M), a Methusalem grant from the Flemmish Government and the Ghent University (01M01108).
References
- Basel-Vanagaite L, Sarig O, Hershkovitz D, Fuchs-Telem D, Rapaport D, Gat A, Isman G, Shirazi I, Shohat M, Enk CD, Birk E, Kohlhase J, Matysiak-Scholze U, Maya I, Knopf C, Peffekoven A, Hennies HC, Bergman R, Horowitz M, Ishida-Yamamoto A, Sprecher E. RIN2 deficiency results in macrocephaly, alopecia, cutis laxa, and scoliosis: MACS syndrome. Am J Hum Genet. 2009;85:254–263. doi: 10.1016/j.ajhg.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bicknell LS, Pitt J, Aftimos S, Ramadas R, Maw MA, Robertson SP. A missense mutation in ALDH18A1, encoding Delta1-pyrroline-5-carboxylate synthase (P5CS), causes an autosomal recessive neurocutaneous syndrome. Eur J Hum Genet. 2008;16:1176–1186. doi: 10.1038/ejhg.2008.91. [DOI] [PubMed] [Google Scholar]
- Broekelmann TJ, Kozel BA, Ishibashi H, Werneck CC, Keeley FW, Zhang L, Mecham RP. Tropoelastin interacts with cell-surface glycosaminoglycans via its COOH-terminal domain. J Biol Chem. 2005;280(49):40939–40947. doi: 10.1074/jbc.M507309200. [DOI] [PubMed] [Google Scholar]
- Brown FR, 3rd, Holbrook KA, Byers PH, Stewart D, Dean J, Pyeritz RE. Cutis laxa. Johns Hopkins Med J. 1982;150:148–153. [PubMed] [Google Scholar]
- Brown-Augsburger P, Broekelmann T, Mecham L, Mercer R, Gibson MA, Cleary EG, Abrams WR, Rosenbloom J, Mecham RP. Microfibril-associated glycoprotein binds to the carboxyl-terminal domain of tropoelastin and is a substrate for transglutaminase. J Biol Chem. 1994;269:28443–28449. [PubMed] [Google Scholar]
- Czirok A, Zach J, Kozel BA, Mecham RP, Davis EC, Rongish BJ. Elastic fiber macro-assembly is a hierarchical, cell motion-mediated process. J Cell Physiol. 2006;207:97–106. doi: 10.1002/jcp.20573. [DOI] [PubMed] [Google Scholar]
- de Schepper S, Loeys B, de Paepe A, Lambert J, Naeyaert JM. Cutis laxa of the autosomal recessive type in a consanguineous family. Eur J Dermatol. 2003;13:529–533. [PubMed] [Google Scholar]
- Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, Puffenberger EG, Hamosh A, Nanthakumar EJ, Curristin SM, Stetten G, Meyers DA, Francomano CA. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- Graul-Neumann LM, Hausser I, Essayie M, Rauch A, Kraus C. Highly variable cutis laxa resulting from a dominant splicing mutation of the elastin gene. Am J Med Genet A. 2008;146A:977–983. doi: 10.1002/ajmg.a.32242. [DOI] [PubMed] [Google Scholar]
- Guernsey DL, Jiang H, Evans SC, Ferguson M, Matsuoka M, Nightingale M, Rideout AL, Provost S, Bedard K, Orr A, Dubé MP, Ludman M, Samuels ME. Mutation in pyrroline-5-carboxylate reductase 1 gene in families with cutis laxa type 2. Am J Hum Genet. 2009;85:120–129. doi: 10.1016/j.ajhg.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, Podowski M, Neptune ER, Halushka MK, Bedja D, Gabrielson K, Rifkin DB, Carta L, Ramirez F, Huso DL, Dietz HC. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennies HC, Kornak U, Zhang H, Egerer J, Zhang X, Seifert W, Kuhnisch J, Budde B, Natebus M, Brancati F, Wilcox WR, Müller D, Kaplan PB, Rajab A, Zampino G, Fodale V, Dallapiccola B, Newman W, Metcalfe K, Clayton-Smith J, Tassabehji M, Steinman B, Barr FA, Nürnberg P, Wieacker P, Mundlos S. Gerodermia osteodysplastica is caused by mutations in SCYL1BP1, a Rab-6 interacting golgin. Nat Genet. 2008;40:1410–1412. doi: 10.1038/ng.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinek A, Rabinovitch M. 67-kD elastin-binding protein is a protective “companion” of extracellular insoluble elastin and intracellular tropoelastin. J Cell Biol. 1994;126:563–574. doi: 10.1083/jcb.126.2.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinek A, Rabinovitch M, Keeley F, Okamura-Oho Y, Callahan J. The 67-kD elastin/laminin-binding protein is related to an enzymatically inactive, alternatively spliced form of beta-galactosidase. J Clin Invest. 1993;91:1198–1205. doi: 10.1172/JCI116280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, Loeys BL, Coucke PJ, De Paepe A, Mecham RP, Choi J, Davis EC, Urban Z. Fibulin-5 mutations: mechanisms of impaired elastic fiber formation in recessive cutis laxa. Hum Mol Genet. 2006;15:3379–3386. doi: 10.1093/hmg/ddl414. [DOI] [PubMed] [Google Scholar]
- Hu Q, Shifren A, Sens C, Choi J, Szabo Z, Starcher BC, Knutsen RH, Shipley JM, Davis EC, Mecham RP, Urban Z. Mechanisms of emphysema in autosomal dominant cutis laxa. Matrix Biol. 2010;29:621–628. doi: 10.1016/j.matbio.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hucthagowder V, Morava E, Kornak U, Lefeber DJ, Fischer B, Dimopoulou A, Aldinger A, Choi J, Davis EC, Abuelo DN, Adamowicz M, Al-Aama J, Basel-Vanagaite L, Fernandez B, Greally MT, Gillessen-Kaesbach G, Kayserili H, Lemyre E, Tekin M, Türkmen S, Tüysuz B, Yuksel-Konuk B, Mundlos S, Van Maldergem L, Wevers RA, Urban Z. Loss-of-function mutations in ATP6V0A2 impair vesicular trafficking, tropoelastin secretion and cell survival. Hum Mol Genet. 2009;18:2149–2165. doi: 10.1093/hmg/ddp148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hucthagowder V, Sausgruber N, Kim KH, Angle B, Marmorstein LY, Urban Z. Fibulin-4: a novel gene for an autosomal recessive cutis laxa syndrome. Am J Hum Genet. 2006;78:1075–1080. doi: 10.1086/504304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung K, Ueberham U, Hausser I, Bosler K, John B, Linse R. Autosomal recessive cutis laxa syndrome. A case report. Acta Derm Venereol. 1996;76:298–301. doi: 10.2340/0001555576298301. [DOI] [PubMed] [Google Scholar]
- Kaler SG, Gallo LK, Proud VK, Percy AK, Mark Y, Segal NA, Goldstein DS, Holmes CS, Gahl WA. Occipital horn syndrome and a mild Menkes phenotype associated with splice site mutations at the MNK locus. Nat Genet. 1994;8:195–202. doi: 10.1038/ng1094-195. [DOI] [PubMed] [Google Scholar]
- Kornak U, Reynders E, Dimopoulou A, van Reeuwijk J, Fischer B, Rajab A, Budde B, Nurnberg P, Foulquier F, Lefeber D, Urban Z, Gruenewald S, Annaert W, Brunner HG, van Bokhoven H, Wevers R, Morava E, Matthijs G, Van Maldergem L, Mundlos S ARCL Debré Study Group. Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+-ATPase subunit ATP6V0A2. Nat Genet. 2008;40:32–34. doi: 10.1038/ng.2007.45. [DOI] [PubMed] [Google Scholar]
- Kozel BA, Rongish BJ, Czirok A, Zach J, Little CD, Davis EC, Knutsen RH, Wagenseil JE, Levy MA, Mecham RP. Elastic fiber formation: a dynamic view of extracellular matrix assembly using timer reporters. J Cell Physiol. 2006;207:87–96. doi: 10.1002/jcp.20546. [DOI] [PubMed] [Google Scholar]
- Loeys B, Van Maldergem L, Mortier G, Coucke P, Gerniers S, Naeyaert JM, De Paepe A. Homozygosity for a missense mutation in fibulin-5 (FBLN5) results in a severe form of cutis laxa. Hum Mol Genet. 2002;11:2113–2118. doi: 10.1093/hmg/11.18.2113. [DOI] [PubMed] [Google Scholar]
- Marchase P, Holbrook K, Pinnell SR. A familial cutis laxa syndrome with ultrastructural abnormalities of collagen and elastin. J Invest Dermatol. 1980;75:399–403. doi: 10.1111/1523-1747.ep12523655. [DOI] [PubMed] [Google Scholar]
- Markova D, Zou Y, Ringpfeil F, Sasaki T, Kostka G, Timpl R, Uitto J, Chu ML. Genetic heterogeneity of cutis laxa: a heterozygous tandem duplication within the fibulin-5 (FBLN5) gene. Am J Hum Genet. 2003;72:998–1004. doi: 10.1086/373940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milewicz DM, Urban Z, Boyd C. Genetic disorders of the elastic fiber system. Matrix Biol. 2000;19:471–480. doi: 10.1016/s0945-053x(00)00099-8. [DOI] [PubMed] [Google Scholar]
- Nahas FX, Sterman S, Gemperli R, Ferreira MC. The role of plastic surgery in congenital cutis laxa: a 10-year follow-up. Plast Reconstr Surg. 1999;104:1174–1178. [PubMed] [Google Scholar]
- Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, Ramirez F, Sakai LY, Dietz HC. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet Nat Genet. 2003;33:407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- Reversade B, Escande-Beillard N, Dimopoulou A, Fischer B, Chng SC, Li Y, Shboul M, Tham PY, Kayserili H, Al-Gazali L, Sahwan M, Brancati F, Lee H, O’Connor BD, Schmidt-von Kegler M, Merriman B, Nelson SF, Masri A, Alkazaleh F, Guerra D, Ferrari P, Nanda A, Rajab A, Markie D, Gray M, Nelson J, Grix A, Somer A, Savarirayan R, Hanecke AR, Steichen E, Sillence D, Hausser I, Budde B, Nürnberg G, Nürnberg P, Seemann P, Kunkel D, Zambruno G, Dallapiccola B, Schuelke M, Robertson S, Hamamy H, Wollnik B, Van Maldergem L, Mundlos S, Kornak U. Mutations in PYCR1 cause cutis laxa with progeroid features. Nat Genet. 2009;41:1016–1021. doi: 10.1038/ng.413. [DOI] [PubMed] [Google Scholar]
- Robinson PN, Arteaga-Solis E, Baldock C, Collod-Beroud G, Booms P, De Paepe A, Dietz HC, Guo G, Handford PA, Judge DP, Kielty CM, Loeys B, Milewicz DM, Ney A, Ramirez F, Reinhardt DP, Tiedemann K, Whiteman P, Godfrey M. The molecular genetics of Marfan syndrome and related disorders. J Med Genet. 2006;43:769–787. doi: 10.1136/jmg.2005.039669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Revenga L, Iranzo P, Badenas C, Puig S, Carrio A, Mila M. A novel elastin gene mutation resulting in an autosomal dominant form of cutis laxa. Arch Dermatol. 2004;140:1135–1139. doi: 10.1001/archderm.140.9.1135. [DOI] [PubMed] [Google Scholar]
- Sarkar R, Kaur C, Kanwar AJ, Basu S. Cutis laxa in seven members of a north-Indian family. Pediatr Dermatol. 2002;19:229–231. doi: 10.1046/j.1525-1470.2002.00074.x. [DOI] [PubMed] [Google Scholar]
- Sato F, Wachi H, Starcher BC, Seyama Y. Biochemical analysis of elastic fiber formation with a frameshift-mutated tropoelastin (fmTE) at the C-terminus of tropoelastin. Journal of Health Science. 2006;52:259–267. [Google Scholar]
- Shifren A, Mecham RP. The stumbling block in lung repair of emphysema: elastic fiber assembly. Proc Am Thorac Soc. 2006;3:428–433. doi: 10.1513/pats.200601-009AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo Z, Crepeau MW, Mitchell AL, Stephan MJ, Puntel RA, Yin Loke K, Kirk RC, Urban Z. Aortic aneurysmal disease and cutis laxa caused by defects in the elastin gene. J Med Genet. 2006;43:255–258. doi: 10.1136/jmg.2005.034157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassabehji M, Metcalfe K, Hurst J, Ashcroft GS, Kielty C, Wilmot C, Donnai D, Read AP, Jones CJ. An elastin gene mutation producing abnormal tropoelastin and abnormal elastic fibres in a patient with autosomal dominant cutis laxa. Hum Mol Genet. 1998;7:1021–1028. doi: 10.1093/hmg/7.6.1021. [DOI] [PubMed] [Google Scholar]
- Tsukahara M, Imaizumi K, Kawai S, Kajii T. Occipital horn syndrome: report of a patient and review of the literature. Clin Genet. 1994;45:32–35. doi: 10.1111/j.1399-0004.1994.tb03986.x. [DOI] [PubMed] [Google Scholar]
- Tutar E, Ekici F, Atalay S, Nacar N. The prevalence of bicuspid aortic valve in newborns by echocardiographic screening. Am Heart J. 2005;150:513–515. doi: 10.1016/j.ahj.2004.10.036. [DOI] [PubMed] [Google Scholar]
- Urban Z, Gao J, Pope FM, Davis EC. Autosomal dominant cutis laxa with severe lung disease: synthesis and matrix deposition of mutant tropoelastin. J Invest Dermatol. 2005;124:1193–1199. doi: 10.1111/j.0022-202X.2005.23758.x. [DOI] [PubMed] [Google Scholar]
- Urban Z, Hucthagowder V, Schurmann N, Todorovic V, Zilberberg L, Choi J, Sens C, Brown CW, Clark RD, Holland KE, Marble M, Sakai LY, Dabovic B, Rifkin DB, Davis EC. Mutations in LTBP4 cause a syndrome of impaired pulmonary, gastrointestinal, genitourinary, musculoskeletal, and dermal development. Am J Hum Genet. 2009;85:593–605. doi: 10.1016/j.ajhg.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban Z, Riazi S, Seidl TL, Katahira J, Smoot LB, Chitayat D, Boyd CD, Hinek A. Connection between elastin haploinsufficiency and increased cell proliferation in patients with supravalvular aortic stenosis and Williams-Beuren syndrome. Am J Hum Genet. 2002;71:30–44. doi: 10.1086/341035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachi H, Nonaka R, Sato F, Shibata-Sato K, Ishida M, Iketani S, Maeda I, Okamoto K, Urban Z, Onoue S, Seyama Y. Characterization of the molecular interaction between tropoelastin and DANCE/fibulin-5. J Biochem. 2008;143:633–639. doi: 10.1093/jb/mvn014. [DOI] [PubMed] [Google Scholar]
- Wachi H, Sato F, Murata H, Nakazawa J, Starcher BC, Seyama Y. Development of a new in vitro model of elastic fiber assembly in human pigmented epithelial cells. Clin Biochem. 2005;38:643–653. doi: 10.1016/j.clinbiochem.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF- beta1 from the extracellular matrix. J Cell Biol. 2007;179:1311–1323. doi: 10.1083/jcb.200704042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang MC, He L, Giro M, Yong SL, Tiller GE, Davidson JM. Cutis laxa arising from frameshift mutations in exon 30 of the elastin gene (ELN) J Biol Chem. 1999;274:981–986. doi: 10.1074/jbc.274.2.981. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.