

Abstract
Maintaining the stability of the replication forks is one of the main tasks of the DNA damage response. Specifically, checkpoint mechanisms detect stressed forks and prevent their collapse. In the published report reviewed here we have shown that defective chromatin assembly in cells lacking either H3K56 acetylation or the chromatin assembly factors CAF1 and Rtt106 affects the integrity of advancing replication forks, despite the presence of functional checkpoints. This loss of replication intermediates is exacerbated in the absence of Rad52, suggesting that collapsed forks are rescued by homologous recombination and providing an explanation for the accumulation of recombinogenic DNA damage displayed by these mutants. These phenotypes mimic those obtained by a partial reduction in the pool of available histones and are consistent with a model in which defective histone deposition uncouples DNA synthesis and nucleosome assembly, thus making the fork more susceptible to collapse. Here, we review these findings and discuss the possibility that defects in the lagging strand represent a major source of fork instability in chromatin assembly mutants.
Keywords: Asf1, H3K56 acetylation, Okazaki fragment, Rad27, genetic instability, homologous recombination, nucleosome assembly, replication fork
Introduction
The DNA damage response functions as a barrier that prevents genetic instability, a hallmark of cancer development.1 The integrity of the replication fork is a central component of this response,2 and mutations that affect the mechanisms involved in the detection, stabilization and repair of stressed forks cause genome instability.3 So far, however, the analyses of these mechanisms have not really taken into account the fact that the advancing fork is rapidly assembled into chromatin through a cell cycle-regulated process that involves genetic and physical interactions between components of the replisome and chromatin assembly factors.4 This tight connection is required to coordinate the correct supply of histones at the fork in response to cellular and environmental changes.5-7 However, it is still largely unknown how the process of nucleosome assembly, and its connection with DNA synthesis, influence replication fork stability and consequently genome integrity.
A first approach to this problem came some years ago with the finding that cells, from yeast to humans, accumulate genetic instability when they are defective in chromatin assembly, as determined by increased frequencies of recombination and gross chromosomal rearrangements (GCRs) in these cells.8-15 However, the fact that chromatin assembly is also associated with DNA replication–independent processes known to influence genome integrity, such as DNA repair and transcription, leaves open the question of how genetic instability is linked to problems in histone deposition. In fact, a general phenotype of cells with mutations in chromatin assembly factors is that they are highly sensitive to DNA damaging agents.10,11,13,16-21
Nucleosome Assembly Maintains Replication Fork Stability
To directly address the impact that defective chromatin assembly has on replication fork stability, we followed the fate of replication intermediates from the early replication origin ARS305 by 2D gel analysis in yeast cells partially depleted of histone H4 (t::HHF2 cells).22 In these cells, the demand for a huge amount of histones during replication impairs chromatin assembly. This is rapidly followed by defects in the replisome integrity and a loss of replication intermediates, despite the presence of functional replication initiation and checkpoint activation. Taken together, these results highlight the importance that a fine-tuned regulation of the available pool of histones has on maintaining genome integrity, and suggest that the process of histone deposition plays an essential role in assuring the integrity of replication forks.
To validate this hypothesis, we took advantage of some important findings about the mechanism of histone deposition mediated by the acetylation of newly synthesized histone H3 at lysine 56. This modification (H3K56ac) is catalyzed by the histone acetyltransferase Rtt109 upon presentation of the H3/H4 heterodimer by the histone chaperone Asf1.11,13,19 In a seminal work, Zhang, Verreault and colleagues showed that H3K56ac promotes nucleosome assembly at the fork by increasing the affinity of H3 for two chromatin assembly factors, CAF1 and Rtt106, with redundant functions in histone deposition.16 We have shown that cells lacking either H3K56 acetylation (asf1Δ, rtt109Δ and H3K56R) or CAF1 and Rtt106 (that is, cac1Δ rtt106Δ, but not cac1Δ or rtt106Δ single mutants) displayed a similar loss of replication intermediates. In these mutants the chromatin structure of ARS305 was not altered (our unpublished results), and the origin was properly activated.23 Besides, they were proficient in checkpoint activation.9,13,24-26 In fact, mutations that impaired the replicative checkpoint delayed progression through S phase in asf1Δ cells,27 suggesting that they affect DNA replication by different mechanisms. Therefore, the loss of replication intermediates in chromatin assembly mutants is not due to defective replication initiation or checkpoint activation. Of note, cac1Δ rtt106Δ cells are proficient in H3K56 acetylation,16 and the loss of fork integrity in the triple mutant asf1Δ cac1Δ rtt106Δ was similar to that displayed by asf1Δ and cac1Δ rtt106Δ.23 Thus, the loss of fork integrity was not due to the lack of H3K56 acetylation per se but rather to defective nucleosome assembly.
Previous ChIP analyses have shown that cells defective in H3K56 acetylation lose their replisome integrity in the presence of hydroxyurea (HU).28,29 Since this drug causes fork stalling by depleting dNTPs, this result suggested a role for H3K56 acetylation in keeping the stability of stalled forks. We confirmed this loss of fork integrity by 2D gel analysis and extended it to cells lacking CAF1 and Rtt106. Remarkably, the loss of replication intermediates was higher in the presence than in the absence of HU except for nucleosome assembly mutants lacking the recombinational repair protein Rad52, which is involved in the rescue of the collapsed replication forks. This suggest that the HU does not increase fork instability but rather prevents the restart of collapsed forks.23 In agreement with this, asf1Δ and cac1Δ rtt106Δ cells were not affected in resuming DNA replication upon HU arrest but progressed to the following cell cycle, with a normal checkpoint recovery.23,30 In summary, the H3K56ac-dependent CAF1 and Rtt106 histone deposition pathways provide redundant mechanisms for stabilizing advancing, but not stalled, replication forks.
Genome Integrity in Chromatin Assembly Mutants
Defective histone deposition causes the collapse and likely the breakage of the fork (Fig. 1). Indeed, H3K56 acetylation mutants accumulated GCRs, as well as breaks and contractions at CAG/CTG tracts that are mediated by the double-strand break (DSB) repair mechanism of non-homologous end joining.15,31 Importantly, the frequency of GCRs is exacerbated in the absence of Rad52,15 suggesting that most DSBs are properly processed by homologous recombination with the sister chromatid. Correspondingly, chromatin assembly mutants that lack Rad52 displayed a higher loss of replication intermediates. Along this line, the loss of replication intermediates in t::HHF2 and asf1Δ mutants was associated with an accumulation of Holliday junctions in the presence of HU, which inhibits DNA synthesis and thereby the restart of the rescued fork22,23(Fig. 1). Likewise, H3K56 acetylation mutants can repair DSBs by both ectopic and sister-chromatid recombination9,26,32,33and spontaneously accumulate sister-chromatid recombination products.9 Altogether, these results suggest that the accumulation of recombinogenic DNA damage in histone deposition mutants is associated with the repair of broken replication forks.
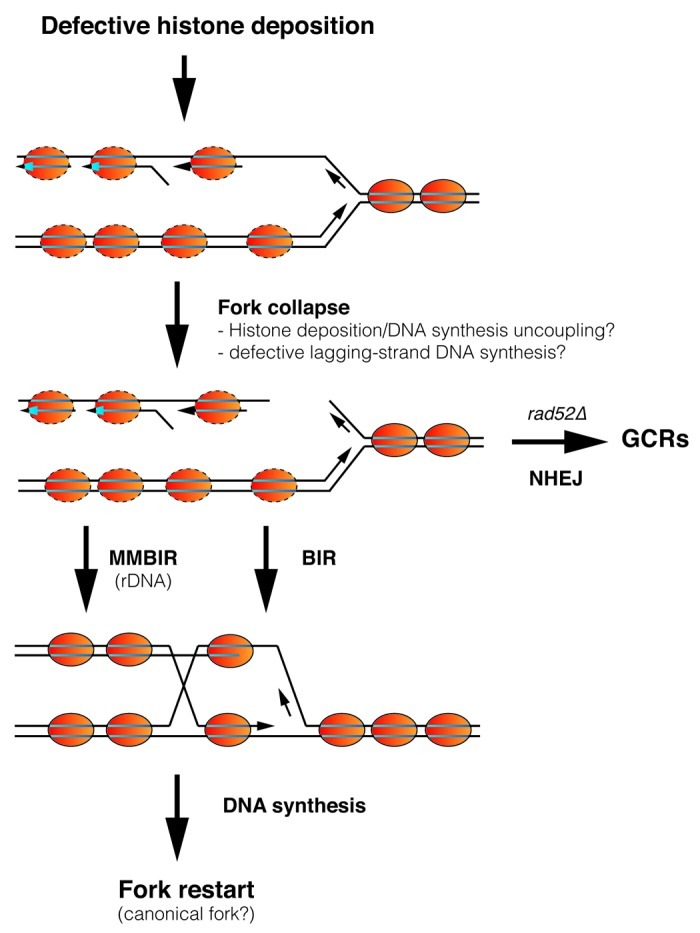
Figure 1. Replication fork collapse and rescue by defective nucleosome assembly. Replication fork advance is rapidly followed by nucleosome assembly of the newly synthesized DNA through a process that requires physical interactions between components of the replisome and chromatin assembly factors. A defect in the supply of histones causes some of the forks to collapse and break, likely by uncoupling of the processes of DNA synthesis and histone deposition. The temporal association between Okazaki fragment maturation and nucleosome assembly supports the idea that the lagging strand is more susceptible to breakage under conditions of defective chromatin assembly. Homologous recombination can efficiently repair the broken fork by BIR, although additional, non-recombinational microhomology-mediated BIR (MMBIR) mechanisms can also operate on rDNA. In some cases, the broken fork is repaired by nonhomologous end joining, leading to GCRs and genetic instability, an event that is triggered in the absence of homologous recombination. See text for more details.
Chromatin assembly mutants progress properly through S phase, despite the strong drop in replication intermediates (for ARS305, this is approximately 50% of the wild type), indicating that repair by recombination is highly efficient. Indeed, t::HHF2 and asf1Δ mutants are very sick in the absence of Rad52.22,23 The repair of the fork might occur by a two-ended DSB repair process upon arrival of the oncoming fork, although the kinetic analysis of replication intermediates suggests that the fork is rescued by break-induced replication (BIR), using the one-ended DSB generated by the breakage to invade the sister chromatid and to prime new DNA synthesis (Fig. 1). Remarkably, Rad52 is not essential for viability, suggesting that additional mechanisms may participate in the rescue of the broken forks. Indeed, cells lacking Asf1 undergo repeat expansions at the rDNA that require the BIR-specific replication factor Pol32 but that, unexpectedly, are independent of the recombination proteins Rad51 and Rad52. These results can be explained by fork breakage at the lagging-strand template with the subsequent annealing to the lagging-strand template of another replication fork through a microhomology-mediated BIR mechanism.34
In contrast to asf1Δ and rtt109Δ, cac1Δ rtt106Δ displays low sensitivity to drugs that interfere with the advance of the replication fork, such as the alkylating agent methyl methanesulfonate (MMS) or the topoisomerase I inhibitor camptothecin (CPT).23 This suggests that H3K56 acetylation plays a role in tolerating replicative DNA damage that is independent of its role in histone deposition. Accordingly, a H3 mutant containing glutamic acid rather than lysine at position 56 was able to bind to CAF1 and Rtt106 and still displayed sensitivity to MMS and CPT.35 Thus, H3K56 acetylation has a role in DNA repair that is subsequent to its deposition in chromatin. In this regard, it has been shown that H3K56 acetylation is required for the recombinational repair of DNA damage generated by MMS and CPT through its recruitment of the Rtt101 ubiquitin ligase complex.32,33,36
Fork Instability in Nucleosome Assembly Mutants: A Matter of Lagging-Strand Synthesis?
How defective H3K56ac/CAF1/Rtt106–mediated histone deposition causes fork instability is still unknown, but the fact that H3K56 acetylation is a mark of newly synthesized histones that is removed at the end of S phase17 suggests that the problem lies in the deposition of histones synthesized de novo rather than in recycling the parental histones. Additionally, problems associated specifically with the H3K56ac deposition pathways can be rule out, since a partial reduction in the pool of available histones displays similar phenotypes of fork integrity and recombination. One possibility to explore is that a defect in the supply of histones uncouples DNA synthesis and histone deposition, thus exposing unassembled and unstable DNA fragments behind the fork that might directly break or form nuclease-processed DNA structures. Although fork breakage and its repair by recombination can occur at both the leading and lagging strands, the later appears to be more susceptible to problems during DNA replication.37 Along this line, several reports suggest that the lagging strand is unstable under conditions of defective nucleosome assembly. The first is related to the accumulation of breaks and CAG/CTG contractions in asf1Δ and rtt109Δ, but not in rtt101Δ, cells.31 These contractions are associated with hairpin-like DNA structures, which are more likely to be formed at the lagging-strand template, as they require single-stranded DNA regions. Another piece of evidence comes from the aforementioned analysis of repeat instability at the rDNA that supports a mechanism initiated by breakage at the lagging strand template.34 Finally, asf1Δ cells accumulate Polα at stalled forks and are highly sensitive to mutations in this polymerase.26,28
While the leading strand is synthesized as a continuous DNA fragment, the lagging strand is synthesized in short (150–200 nt) DNA sequences called Okazaki fragments. Each one is synthesized by the DNA polymerase Polδ from a primer formed by 7–14 ribonucleotides followed by 10–20 deoxyribonucleotides. Consequently, the synthesis of the complete lagging strand requires that the RNA primers are removed and the DNA fragments are subsequently joined. This maturation step is performed primarily by the nuclease FEN-1 (Rad27 in Saccharomyces cerevisiae) that cleaves the 5′ single-stranded flap structure generated upon displacement of the RNA primer by the upstream lagging DNA synthesis. This cleavage generates a nick that is sealed by the DNA ligase I.38 Therefore, defective Okazaki fragments maturation is expected to generate an accumulation of nicks that might lead to template strand breakage and fork collapse. Indeed, rad27Δ cells accumulate high rates of homologous recombination and GCRs and display a unique duplication mutator phenotype thought to occur by DNA repair upon incorrect Okazaki fragments maturation.39,40 Consistent with this idea, rad27Δ displays replication defects and, importantly, these defects are epistatic over those displayed by asf1Δ (our unpublished results), suggesting that Rad27 and Asf1 prevent the loss of replication intermediates through the same mechanism.
In this frame, nucleosome assembly might facilitate the maturation of the Okazaki fragments. This is a conceivable possibility, since nucleosome assembly occurs as soon as there is sufficient DNA behind the fork,41,42 and since unprocessed Okazaki fragments are associated with pre-nucleosomal particles.43 Indeed, human FEN-1 and ligase I can efficiently process DNA flap structures assembled into nucleosomes in vitro.44 Notably, the cleavage activity of hFEN-1 on nucleosome substrates requires nucleosomal DNA mobility;45 this mobility could be provided by H3K56 acetylation, as this modification breaks a histone-DNA interaction that modulates chromatin compaction.17 Along these lines, replication fork collapse in nucleosome assembly mutants might be due to defective Okazaki fragment maturation and the accumulation of unstable single-stranded DNA regions.
Future Perspectives
Although promising, a relationship between nucleosome assembly and Okazaki fragment maturation remains speculative and requires an in-depth study of lagging-strand DNA replication as well as a detailed analysis of nucleosome positioning at the fork under conditions of defective nucleosome assembly. Besides, we cannot rule out other scenarios like a loss of physical tension in the replisome architecture as a source of fork instability. The observation that chromatin assembly mutants complete DNA replication despite the loss of replication intermediates could be due to the formation of non-canonical replication forks. Alternative but not exclusive possibilities include altered programs of replication initiation and elongation, and this should be analyzed on a genome-wide scale. These studies may also provide information about the existence of chromosome regions that are more susceptible to replication problems due to defective nucleosome assembly. Finally, several observations have linked nucleosome assembly with cancer progression, such as the accumulation in breast cancer of the Asf1b isoform required for proliferation,46 and the association of chromatin assembly genes with chromosome regions deleted in some tumors.14 This link is further strengthened by the observation that defective H3K56 acetylation accelerates aging in yeast cells.47
Acknowledgments
We thank J.C. Reyes and R.E. Wellinger for critical reading of the manuscript. The research was funded by the Spanish Ministry of Science (BFU2009-09036).
Glossary
Abbreviations:
- GCRs
gross chromosomal rearrangements
- H3K56ac
acetylated lysine 56 of histone H3
- HU
hydroxyurea
- DSB
double-strand break
- MMS
methyl methanesulfonate
- CPT
camptothecin
- BIR
break-induced replication
- FEN-1
flap endonuclease 1
Note
A work that demonstrates a interconnection between lagging-strand synthesis and chromatin assembly was published while this manuscript was in press.48
Footnotes
Previously published online: www.landesbioscience.com/journals/BioArchitecture/article/19737
References
- 1.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–5. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 2.Tercero JA, Longhese MP, Diffley JFX. A central role for DNA replication forks in checkpoint activation and response. Mol Cell. 2003;11:1323–36. doi: 10.1016/S1097-2765(03)00169-2. [DOI] [PubMed] [Google Scholar]
- 3.Kolodner RD, Putnam CD, Myung K. Maintenance of genome stability in Saccharomyces cerevisiae. Science. 2002;297:552–7. doi: 10.1126/science.1075277. [DOI] [PubMed] [Google Scholar]
- 4.Ransom M, Dennehey BK, Tyler JK. Chaperoning histones during DNA replication and repair. Cell. 2010;140:183–95. doi: 10.1016/j.cell.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gunjan A, Verreault AA. A Rad53 kinase-dependent surveillance mechanism that regulates histone protein levels in S. cerevisiae. Cell. 2003;115:537–49. doi: 10.1016/S0092-8674(03)00896-1. [DOI] [PubMed] [Google Scholar]
- 6.Groth A, Ray-Gallet D, Quivy J-P, Lukas J, Bartek J, Almouzni G. Human Asf1 regulates the flow of S phase histones during replicational stress. Mol Cell. 2005;17:301–11. doi: 10.1016/j.molcel.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 7.Groth A, Corpet A, Cook AJL, Roche D, Bartek J, Lukas J, et al. Regulation of replication fork progression through histone supply and demand. Science. 2007;318:1928–31. doi: 10.1126/science.1148992. [DOI] [PubMed] [Google Scholar]
- 8.Prado F, Aguilera A. Partial depletion of histone H4 increases homologous recombination-mediated genetic instability. Mol Cell Biol. 2005;25:1526–36. doi: 10.1128/MCB.25.4.1526-1536.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prado F, Cortés-Ledesma F, Aguilera A. The absence of the yeast chromatin assembly factor Asf1 increases genomic instability and sister chromatid exchange. EMBO Rep. 2004;5:497–502. doi: 10.1038/sj.embor.7400128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burgess RJ, Zhou H, Han J, Zhang Z. A role for Gcn5 in replication-coupled nucleosome assembly. Mol Cell. 2010;37:469–80. doi: 10.1016/j.molcel.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han J, Zhou H, Horazdovsky B, Zhang K, Xu R-M, Zhang Z. Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science. 2007;315:653–5. doi: 10.1126/science.1133234. [DOI] [PubMed] [Google Scholar]
- 12.Endo M, Ishikawa Y, Osakabe K, Nakayama S, Kaya H, Araki T, et al. Increased frequency of homologous recombination and T-DNA integration in Arabidopsis CAF-1 mutants. EMBO J. 2006;25:5579–90. doi: 10.1038/sj.emboj.7601434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Driscoll R, Hudson A, Jackson SP. Yeast Rtt109 promotes genome stability by acetylating histone H3 on lysine 56. Science. 2007;315:649–52. doi: 10.1126/science.1135862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ye X, Franco AA, Santos H, Nelson DM, Kaufman PD, Adams PD. Defective S phase chromatin assembly causes DNA damage, activation of the S phase checkpoint, and S phase arrest. Mol Cell. 2003;11:341–51. doi: 10.1016/S1097-2765(03)00037-6. [DOI] [PubMed] [Google Scholar]
- 15.Myung K, Pennaneach V, Kats ES, Kolodner RD. Saccharomyces cerevisiae chromatin-assembly factors that act during DNA replication function in the maintenance of genome stability. Proc Natl Acad Sci U S A. 2003;100:6640–5. doi: 10.1073/pnas.1232239100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Q, Zhou H, Wurtele H, Davies B, Horazdovsky B, Verreault A, et al. Acetylation of histone H3 lysine 56 regulates replication-coupled nucleosome assembly. Cell. 2008;134:244–55. doi: 10.1016/j.cell.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Masumoto H, Hawke D, Kobayashi R, Verreault A. A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature. 2005;436:294–8. doi: 10.1038/nature03714. [DOI] [PubMed] [Google Scholar]
- 18.Tsubota T, Berndsen CE, Erkmann JA, Smith CL, Yang L, Freitas MA, et al. Histone H3-K56 acetylation is catalyzed by histone chaperone-dependent complexes. Mol Cell. 2007;25:703–12. doi: 10.1016/j.molcel.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tyler JK, Adams CR, Chen S-R, Kobayashi R, Kamakaka RT, Kadonaga JT. The RCAF complex mediates chromatin assembly during DNA replication and repair. Nature. 1999;402:555–60. doi: 10.1038/990147. [DOI] [PubMed] [Google Scholar]
- 20.Choy JS, Kron SJ. NuA4 subunit Yng2 function in intra-S-phase DNA damage response. Mol Cell Biol. 2002;22:8215–25. doi: 10.1128/MCB.22.23.8215-8225.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qin S, Parthun MR. Histone H3 and the histone acetyltransferase Hat1p contribute to DNA double-strand break repair. Mol Cell Biol. 2002;22:8353–65. doi: 10.1128/MCB.22.23.8353-8365.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clemente-Ruiz M, Prado F. Chromatin assembly controls replication fork stability. EMBO Rep. 2009;10:790–6. doi: 10.1038/embor.2009.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clemente-Ruiz M, González-Prieto R, Prado F. Histone H3K56 acetylation, CAF1, and Rtt106 coordinate nucleosome assembly and stability of advancing replication forks. PLoS Genet. 2011;7:e1002376. doi: 10.1371/journal.pgen.1002376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Emili A, Schieltz DM, Yates JR, 3rd, Hartwell LH. Dynamic interaction of DNA damage checkpoint protein Rad53 with chromatin assembly factor Asf1. Mol Cell. 2001;7:13–20. doi: 10.1016/S1097-2765(01)00150-2. [DOI] [PubMed] [Google Scholar]
- 25.Hu F, Alcasabas AA, Elledge SJ. Asf1 links Rad53 to control of chromatin assembly. Genes Dev. 2001;15:1061–6. doi: 10.1101/gad.873201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramey CJ, Howar S, Adkins M, Linger J, Spicer J, Tyler JK. Activation of the DNA damage checkpoint in yeast lacking the histone chaperone anti-silencing function 1. Mol Cell Biol. 2004;24:10313–27. doi: 10.1128/MCB.24.23.10313-10327.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kats ES, Albuquerque CP, Zhou H, Kolodner RD. Checkpoint functions are required for normal S-phase progression in Saccharomyces cerevisiae RCAF- and CAF-I-defective mutants. Proc Natl Acad Sci U S A. 2006;103:3710–5. doi: 10.1073/pnas.0511102103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Franco AA, Lam WM, Burgers PM, Kaufman PD. Histone deposition protein Asf1 maintains DNA replisome integrity and interacts with replication factor C. Genes Dev. 2005;19:1365–75. doi: 10.1101/gad.1305005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han J, Zhou H, Li Z, Xu R-M, Zhang Z. Acetylation of lysine 56 of histone H3 catalyzed by RTT109 and regulated by ASF1 is required for replisome integrity. J Biol Chem. 2007;282:28587–96. doi: 10.1074/jbc.M702496200. [DOI] [PubMed] [Google Scholar]
- 30.Collins SR, Miller KM, Maas NL, Roguev A, Fillingham J, Chu CS, et al. Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature. 2007;446:806–10. doi: 10.1038/nature05649. [DOI] [PubMed] [Google Scholar]
- 31.Yang JH, Freudenreich CH. The Rtt109 histone acetyltransferase facilitates error-free replication to prevent CAG/CTG repeat contractions. DNA Repair (Amst) 2010;9:414–20. doi: 10.1016/j.dnarep.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duro E, Vaisica JA, Brown GW, Rouse J. Budding yeast Mms22 and Mms1 regulate homologous recombination induced by replisome blockage. DNA Repair (Amst) 2008;7:811–8. doi: 10.1016/j.dnarep.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 33.Wurtele H, Kaiser GS, Bacal J, St-Hilaire E, Lee E-H, Tsao S, et al. Histone H3 lysine 56 acetylation and the response to DNA replication fork damage. Mol Cell Biol. 2012;32:154–72. doi: 10.1128/MCB.05415-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Houseley J, Tollervey D. Repeat expansion in the budding yeast ribosomal DNA can occur independently of the canonical homologous recombination machinery. Nucleic Acids Res. 2011;39:8778–91. doi: 10.1093/nar/gkr589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Erkmann JA, Kaufman PD. A negatively charged residue in place of histone H3K56 supports chromatin assembly factor association but not genotoxic stress resistance. DNA Repair (Amst) 2009;8:1371–9. doi: 10.1016/j.dnarep.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roberts TM, Zaidi IW, Vaisica JA, Peter M, Brown GW. Regulation of rtt107 recruitment to stalled DNA replication forks by the cullin rtt101 and the rtt109 acetyltransferase. Mol Biol Cell. 2008;19:171–80. doi: 10.1091/mbc.E07-09-0961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zou H, Rothstein R. Holliday junctions accumulate in replication mutants via a RecA homolog-independent mechanism. Cell. 1997;90:87–96. doi: 10.1016/S0092-8674(00)80316-5. [DOI] [PubMed] [Google Scholar]
- 38.Zheng L, Shen B. Okazaki fragment maturation: nucleases take centre stage. J Mol Cell Biol. 2011;3:23–30. doi: 10.1093/jmcb/mjq048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tishkoff DX, Filosi N, Gaida GM, Kolodner RD. A novel mutation avoidance mechanism dependent on S. cerevisiae RAD27 is distinct from DNA mismatch repair. Cell. 1997;88:253–63. doi: 10.1016/S0092-8674(00)81846-2. [DOI] [PubMed] [Google Scholar]
- 40.Chen C, Kolodner RD. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat Genet. 1999;23:81–5. doi: 10.1038/12687. [DOI] [PubMed] [Google Scholar]
- 41.Sogo JM, Stahl H, Koller T, Knippers R. Structure of replicating simian virus 40 minichromosomes. The replication fork, core histone segregation and terminal structures. J Mol Biol. 1986;189:189–204. doi: 10.1016/0022-2836(86)90390-6. [DOI] [PubMed] [Google Scholar]
- 42.Lucchini R, Wellinger RE, Sogo JM. Nucleosome positioning at the replication fork. EMBO J. 2001;20:7294–302. doi: 10.1093/emboj/20.24.7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cusick ME, Lee KS, DePamphilis ML, Wassarman PM. Structure of chromatin at deoxyribonucleic acid replication forks: nuclease hypersensitivity results from both prenucleosomal deoxyribonucleic acid and an immature chromatin structure. Biochemistry. 1983;22:3873–84. doi: 10.1021/bi00285a024. [DOI] [PubMed] [Google Scholar]
- 44.Huggins CF, Chafin DR, Aoyagi S, Henricksen LA, Bambara RA, Hayes JJ. Flap endonuclease 1 efficiently cleaves base excision repair and DNA replication intermediates assembled into nucleosomes. Mol Cell. 2002;10:1201–11. doi: 10.1016/S1097-2765(02)00736-0. [DOI] [PubMed] [Google Scholar]
- 45.Jagannathan I, Pepenella S, Hayes JJ. Activity of FEN1 endonuclease on nucleosome substrates is dependent upon DNA sequence but not flap orientation. J Biol Chem. 2011;286:17521–9. doi: 10.1074/jbc.M111.229658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Corpet A, De Koning L, Toedling J, Savignoni A, Berger F, Lemaìtre C, et al. Asf1b, the necessary Asf1 isoform for proliferation, is predictive of outcome in breast cancer. EMBO J. 2011;30:480–93. doi: 10.1038/emboj.2010.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feser J, Truong D, Das C, Carson JJ, Kieft J, Harkness T, et al. Elevated histone expression promotes life span extension. Mol Cell. 2010;39:724–35. doi: 10.1016/j.molcel.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith DJ, Whitehouse I. Intrinsic coupling of lagging-strand synthesis to chromatin assembly. Nature. 2012;483:434–8. doi: 10.1038/nature10895. [DOI] [PMC free article] [PubMed] [Google Scholar]