

Abstract
Energy homeostasis involves a complex network of hypothalamic and extra-hypothalamic neurons that transduce hormonal, nutrient and neuronal signals into responses that ultimately match caloric intake to energy expenditure and thereby promote stability of body fat stores. Growing evidence suggests that rather than reflecting a failure to regulate caloric intake, common forms of obesity involve fundamental changes to this homeostatic system that favor the defense of an elevated level of body adiposity. This article reviews emerging evidence that during high-fat feeding, obesity pathogenesis involves fundamental alteration of hypothalamic systems that regulate food intake and energy expenditure.
Keywords: leptin, insulin, inflammation, nutrient, innate immunity, hypothalamus
Introduction
Obesity affects more than 300 million humans worldwide and projections for the next 20 years predict a global prevalence of up to 20%, amounting to more than one billion obese individuals by the year 20301. As a major risk factor for diabetes mellitus, hypertension, dyslipidemia, arteriosclerosis, as well as joint and skeletal deterioration, respiratory failure, and certain types of cancer,2, 3, 4, 5 the burden of disease posed by the looming obesity epidemic is staggering. Unfortunately, lifestyle or behavioral interventions designed to correct nutritional overload and increase physical activity have yet to halt or even slow the increase of obesity prevalence. Similarly, the few drugs currently approved for obesity treatment are limited by both side effects and lack of efficacy,6, 7, 8 and although bariatric surgery can achieve substantial reduction of body weight and improve metabolic control, it is an invasive and costly intervention that is not uniformly effective and realistically can be offered to only a tiny fraction of those affected.6, 9
The highly integrated and redundant nature of neurocircuits involved in energy homeostasis offers important insight into the question of why common forms of obesity have proven so difficult to treat. Energy homeostasis is the biological process that promotes stability in the amount of body energy stored as fat, and weight loss triggers robust activation of this homeostatic system in obese as well as in lean individuals. This response in turn limits the capacity to sustain voluntary weight loss and explains why non-surgical interventions typically cannot sustain weight loss of 5-7%—an amount that activates compensatory responses that limit further weight loss and favor recovery of lost weight. Stated differently, obesity arises not simply via passive accumulation of excess calories as fat but involves an upward re-setting of the biologically defended level of body fat.
In light of these considerations, it is evident that an improved understanding of how obesity occurs in the context of a homeostatic regulatory system is crucial to the development of more effective and safer therapeutic options. Here, we highlight recent findings from rodent models of obesity induced by high-fat (HF) feeding that offer insight into how the hypothalamus is affected by—and contributes to—common forms of obesity. Although our focus is directed to the hypothalamus, our intent is not to diminish the crucial role that brain regions other than the hypothalamus have in energy homeostasis. The integrative function of the hindbrain, for example, provides connectivity between gut, hormonal and nutrient signals, and the interested reader is referred elsewhere for reviews on this topic.10, 11 Yet obesity pathogenesis is tied more closely to neuronal systems in the hypothalamus than other brain areas, which offers a compelling rationale for the focus of this review.
Hypothalamic control of feeding and thermogenesis: early studies
Hypothalamic lesioning studies undertaken between the late 1930s and the early 1960s12, 13, 14 offered the first evidence of a role for the brain in body weight control and the pathogenesis of obesity and diabetes.15 On the basis of these and subsequent studies, the lateral and ventromedial hypothalamic nuclei were identified as potentially important brain areas controlling food intake and energy expenditure.16 One limitation of a lesion-based approach is that results can be misleading if the brain area targeted contains neurons with opposing effects on energy balance. Such is the case for the hypothalamic arcuate nucleus, which contains key neuronal subsets for both increasing and decreasing food intake and weight gain. Consequently, lesion of this brain area has a rather unremarkable impact on these parameters,17 despite the predominant role that neurons in this brain area were subsequently shown to have in energy homeostasis.
Based in part on evidence that manipulation of specific areas of the hypothalamus can reliably alter feeding behavior, energy metabolism and body mass, two major theories emerged in the 1950s to explain the physiology of this system. According to Kennedy,18 energy stored in adipose tissue is communicated to the hypothalamus through circulating signals that ultimately serve to adjust food intake in response to variation in energy depot size. On the other hand, Mayer19 proposed that variation in plasma or tissue glucose concentrations is a key signal informing the central nervous system (CNS) about energy availability. These two hypotheses were subsequently termed the ‘adipostatic’ and ‘glucostatic’ models of energy homeostasis.
Although there is little question that reduced glucose availability constitutes a potent stimulus to food intake, the glucostatic theory of Mayer cannot reliably explain adjustments of feeding behavior that occur in the absence of hypoglycemia and promote stability of body fat stores in normal animals. By comparison, the adipostat theory received compelling early support from parabiosis experiments, in which two animals are physically joined to one another such that they share a common circulation. The first of these was published by Hervey20 and showed that when rats previously made obese by hypothalamic lesion were parabiosed to lean control rats, the latter (but not the former) ate less and lost weight. This finding was interpreted as evidence that rats with hypothalamic obesity have high levels of a circulating factor capable of reducing intake and body weight in normal animals (presumably, the hypothalamic lesion interferes with the capacity to respond to the signal).
Coleman21 subsequently demonstrated that parabiosis of lean mice to ob/ob mice, a model of monogenic obesity, reduced food intake and body mass in the obese mice, but not the lean controls. Thus, Coleman21 hypothesized that ob/ob mice are obese because they lack a circulating anorexic factor that is present in normal animals. When db/db mice were parabiosed to lean mice, by comparison, the lean mice ate less and lost weight, whereas the mutant animals were unaffected, suggesting that db/db mice produce, but cannot respond to, the same circulating anorexic factor. Confirmation of this hypothesis came 21 years later, with the demonstration that the ob locus encodes the adipocyte hormone leptin, whereas the leptin receptor is encoded by the db locus.
Insulin comes on the scene
Meanwhile, the demonstration that plasma insulin enters the brain and acts centrally to reduce food intake and body weight, and that insulin receptors are concentrated in CNS areas involved in the control of food intake, suggested that circulating insulin acts in the brain as an adiposity negative-feedback signal.22, 23 Consistent with this hypothesis, insulin secretion from the endocrine pancreas is regulated by feeding and its blood levels vary in direct proportion to body adiposity. Insulin also crosses the blood-brain barrier to enter the brain, and insulin receptors are concentrated in hypothalamic areas implicated in energy balance regulation. Moreover, insulin inhibits food intake following direct administration into the hypothalamus or adjacent third ventricle,24 whereas neuron-specific deletion of insulin receptors induces a modest increase of body fat mass.25
During the 1980s and early 1990’s, a number of studies explored the role of insulin in the control of feeding and adiposity, including the demonstration of an inhibitory effect of this hormone on neuronal firing in a hypothalamic slice preparation,26 the existence of a transport system for systemic insulin to reach the brain27, 28 and that insulin in the cerebrospinal fluid gains access to the hypothalamus,29 and that kinetic analysis of insulin uptake from plasma to the cerebrospinal fluid established the existence of a saturable transport mechanism for brain insulin delivery in vivo.30
Brain insulin action also influences autonomic function in a variety of ways. For example, injection of insulin directly into the lateral hypothalamus stimulates parasympathetic outflow to the pancreas, while injection of insulin in the ventromedial hypothalamus has the opposite effect.31 More recent studies suggest that by increasing hepatic vagal outflow, insulin signaling in the arcuate nucleus enhances liver insulin sensitivity and in turn lowers hepatic glucose production.32, 33 A thermogenic action of insulin was also reported following hypothalamic injection, an effect accompanied by increased firing of sympathetic nerves innervating brown adipose tissue,34 and recent work has begun to clarify the hypothalamic neuronal subsets and signal-transduction mechanisms underlying this effect.35
A major and persisting source of confusion surrounding the hypothesis that insulin action in the brain reduces food intake and body weight while also lowering hepatic glucose production and increasing thermogenesis stems from evidence that following systemic insulin administration, the subsequent fall in glucose levels potently increases food intake36, 37 while also increasing liver glucose production and reducing sympathetically driven thermogenesis. Thus, insulin-induced hypoglycemia potently overrides virtually all of insulin’s central effects, an observation that for many years has confounded research in this field.
The physiological importance of insulin as an adiposity negative-feedback signal remains uncertain, and available data suggest that of the two, leptin has the predominant role. Noteworthy in this context is that the anorexigenic action of insulin is not observed uniformly across laboratories.38 Interestingly, this action appears to be boosted by the simultaneous action of leptin,39 suggesting neuronal cross-talk involving these two hormonal systems (discussed below).
The identification of leptin
The hypothesis forwarded by Coleman in the early 1970s was confirmed in 1994 by the identification of leptin through positional cloning of the ob gene locus,40 followed shortly thereafter by cloning of the leptin receptor.41 Leptin is produced primarily in adipose tissue and circulates at levels directly proportional to whole-body adiposity.40 Upon entering the hypothalamus, leptin binds to and activates its cognate receptor, LepR, mediating leptin action on multiple parameters of brain function ranging from neuronal firing rate and neurotransmitter/neuropeptide production and release to effects on feeding, thermogenesis, reproductive and other aspects of neuroendocrine function, as well as metabolic regulation.41, 42 The importance of this hormone in physiological control of energy balance was firmly established by the findings that (1) ob/ob mice are leptin-deficient owing to mutation of the leptin gene,40 whereas db/db mice are autosomal recessive for mutation of LepR, and (2) the obese phenotype of ob/ob, but not db/db, mice is reversed by leptin replacement. Importantly, genetic leptin deficiency also causes severe obesity in humans that is remedied by leptin replacement therapy.43
The LepR is a type 1 cytokine receptor that, upon ligand binding, induces the tyrosine kinase catalytic activity of an associated enzyme, Janus kinase 2 (JAK2), resulting in the activation of a number of downstream proteins involved in the transduction of the leptin signal. The first step of LepR signaling involves JAK2-mediated tyrosine phosphorylation of residues 985, 1077 and 1138 in the intracellular portion of the receptor.44 Phosphorylated tyrosine 985 recruits and activates the SHP2/ERK pathway;45 phosphorylated tyrosine 1077 is involved in the activation of signal transducer and activator of transcription 5 (STAT5)46; and phosphorylated tyrosine 1138 leads to the activation of STAT345, 47 Each of these pathways influences expression of distinct genes, the nature of which can differ across different cell types. Binding of leptin to LepR can also activate/inactivate several additional targets such as Fyn, phosphatidylinositol 3-kinase, mammalian target of rapamycin and AMP-activated protein kinase by mechanisms not completely elucidated.39, 48, 49, 50, 51, 52, 53 Through these signal-transduction cascades, leptin can effectively cross-talk with other hormones, cytokines and nutrient-sensing systems.54
The relevance of leptin and insulin as adiposity factors
Although peptides of the gut, such as cholecystokinin, somatostatin, ghrelin, glucose-dependent insulinotropic peptide and glucose-dependent insulinotropic peptide 155, 56 nutrients, such as glucose, fatty acids and amino acids;50, 57 and hormones, such as glucagon, adiponectin, glucocorticoids and estradiol,54, 58 can each influence feeding behavior, leptin and, perhaps to a lesser extent, insulin, are the only humoral signals known to fulfill criteria set forth for an adiposity negative-feedback signal. Some studies have shown that the activity of leptin in the hypothalamus is enhanced by insulin, and vice versa implying the potential for both hormones to act upon a shared subset of neurons or signaling pathways.39, 53, 59 More specifically, both the biological response to leptin, as determined by food intake suppression, as well as the transduction of its signal, measured as activation of phosphatidylinositol 3-kinase or STAT3, are significantly increased if leptin and insulin act together.39 To the extent that the CNS effects of both hormones involve overlapping mechanisms, dysfunction of such shared pathways may cause resistance to both hormones and thereby predispose to obesity.60
Although examples of this type of convergence exist, it is now clear that insulin and leptin have decidedly different effects on at least some neuronal subsets. For example, insulin appears to hyperpolarize a subset of hypothalamic pro-opiomelanocortin neurons in the arcuate nucleus, whereas leptin depolarizes them.61 Indeed, Elmquist and colleagues have suggested that insulin and leptin act on entirely distinct subsets of pro-opiomelanocortin neurons.62 Although the notion of convergent neuronal insulin and leptin signaling in energy homeostasis therefore remains somewhat speculative and controversial, a compelling case can be made that obesity-associated metabolic impairment causing insulin resistance also causes leptin resistance, and vice versa. Indeed, obesity induced by HF feeding is associated with neuronal resistance to the actions of both hormones.
Hypothalamic resistance to leptin and insulin and obesity pathogenesis
Shortly after leptin’s discovery, its promise as an obesity therapeutic was undermined by evidence that the great majority of obese humans are hyperleptinemic, and that leptin administration fails to produce sustained body mass reduction in most cases.63, 64 Indeed, exogenous leptin is currently employed therapeutically only for patients with rare loss-of-function mutations of the gene encoding leptin or with equally rare lipodystrophic disorders.65
On the basis of the clinical parallel to hyperinsulinemia in type 2 diabetes mellitus, most obese humans are believed to be resistant to leptin.66 However, reliable assays of leptin action in humans have yet to emerge, and definitive cellular evidence of leptin resistance in humans is still awaited. In experimental obesity, however, substantial progress has been made in both the characterization of and mechanisms underlying leptin and insulin resistance in the hypothalamus.39, 67, 68
The leptin-induced reduction of spontaneous food intake in lean animals occurs during a time window of 2–12 h, causing a reduction of caloric intake of up to 50% during that time.69 In obese animals, this anorexic effect is diminished varying degrees, depending on the background strain and mechanism underlying the obesity,39, 70, 71 among other factors. A more direct measure of leptin action involves the evaluation of STAT3 tyrosine phosphorylation in response to intracerebroventricular (ICV) or systemic leptin injection,39 and biochemical insulin action can similarly be assessed by measurement of IRS1/IRS2 tyrosine phosphorylation following ICV injection of this hormone.67 In each case, determination is performed by immunoblotting of extracts of hypothalamus or other tissues.
Simply showing hypothalamic resistance to insulin or leptin in obese animals, however, does little to address the issue of cause and effect,69 and the question of whether such resistance is essential to obesity pathogenesis (or is merely a consequence of the obese state) remains a matter of intense investigation. Inhibition or reversal of resistance by genetic,72, 73 pharmacological74, 75 or behavioral approaches, including food restriction76 or increased physical activity,77, 78 is typically associated with reduced adiposity, implicating a role for hypothalamic resistance to these hormones in obesity pathogenesis. Yet the question as to whether this leptin/insulin resistance is a cause or a result of obesity is complex and is only beginning to be answered.
There is little question that genetic interventions that impair CNS insulin/leptin signaling predispose to obesity.79, 80 Conversely, neuron-specific disruption of genes encoding proteins that terminate signaling by these hormones—and are implicated as molecular mediators of leptin/insulin resistance (for example, SOCS3)—protects against diet-induced obesity (DIO) in mouse models,73 strengthening the hypothesis that impaired signaling by these hormones is required for obesity to occur. Combined with the observation that the hypothalamus becomes resistant to adipostatic hormone action relatively early in the course of HF feeding,81 available data are suggestive of a causal role for hypothalamic leptin/insulin resistance in the genesis of at least some forms of obesity. What has been less clear is (1) how a change of diet impairs neuronal signaling by these hormones, (2) the identity of the affected neurons, and-perhaps most perplexing-(3) how consumption of an HF diet leads to an apparently permanent increase in the defended level of body weight. That is, why does not a period of caloric restriction or switch to a low-fat diet return the defended level of body weight to its original, reduced value? Although this recovery of normal weight can occur in rodent models, it is clearly the exception in human obesity.
Mechanisms underlying hypothalamic resistance to leptin and insulin
Several molecular mechanisms have been identified with the potential to explain hypothalamic leptin and/or insulin resistance. Although these mechanisms are in some ways redundant, and more than one may be active in a given individual, each has the potential to independently impair leptin and/or insulin signaling (Figure 1), and several are directly linked to weight gain during HF feeding.
Figure 1.
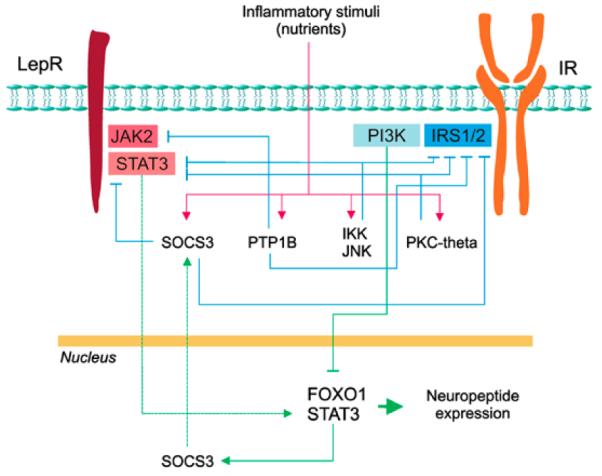
Mechanisms capable of causing leptin and insulin resistance in the hypothalamus. Molecular mediators of resistance to leptin and insulin in the hypothalamus include SOCS3, which can be induced by inflammatory or hormone signaling, as well as PTP1B, IKK, JNK and PKC-theta, all induced by inflammatory signaling. Mechanisms regulating these proteins are indicated by the red lines, their connection with gene transcription/protein expression is indicated by green lines, and inhibition of leptin/insulin signaling is indicated by blue lines.
Induction of SOCS3 expression following leptin receptor activation has a physiological role to limit both the magnitude and duration of the leptin signal,82 and several observations implicate upregulation of hypothalamic SOCS3 expression in the pathogenesis of obesity-associated leptin resistance. SOCS3 belongs to the Suppressor of Cytokine Signaling family of proteins, and its expression can be induced both by leptin-mediated STAT3 transcriptional activity and by other mechanisms73, 83 (see below). In most forms of both genetic and DIO, increased levels of SOCS3 can be detected in hypothalamic neurons, a response that in turn can blunt leptin’s neuronal actions. Although the extent to which this response contributes to the development of common forms of obesity remains unresolved,73 transient overexpression of SOCS3 clearly impairs leptin signal transduction and favors weight gain, while hypothalamic knockdown of this protein protects against DIO.83, 84 Similarly, genetic ‘knock-in’ of a leptin receptor mutant that cannot bind SOCS3 results in mice that are protected against DIO.85
PTP1B is a phosphatase that, similar to SOCS3, is hypothesized to function as a physiological inhibitor of both leptin and insulin signal transduction, and similar to SOCS3, its expression is increased in the hypothalamus of obese rodents.86, 87 By dephosphorylating both JAK2 and key insulin signal-transduction molecules, this enzyme terminates signaling by both leptin and insulin.86, 88 As both hypothalamic knockout of the PTP1B gene and its inhibition by an antisense oligonuleotides protect against DIO,86, 89, 90 obesity induced by HF feeding appears to require intact PTP1B function. Conversely, pro-opiomelanocortin neuron-specific deletion of PTP1B leads to reduced adiposity and increased energy expenditure through a leptin-dependent mechanism that depends, at least in part, on reduced AMP-activated protein kinase activity in the hypothalamus.91, 92, 93
These observations raise the fundamental question of how HF feeding increases hypothalamic SOCS3 and PTP1B activity in rodent models of DIO. In this context, much attention has recently focused on pathways of cellular inflammation. Just as it occurs in the liver, muscle or adipose tissue, overnutrition induced by HF feeding also activates inflammatory signaling pathways in mediobasal hypothalamus. One such mechanism involves c-Jun-N-terminal kinase (JNK), a serine/threonine kinase activated in response to numerous inflammatory and environmental factors.94 In DIO, hypothalamic JNK activation is increased, whereas pharmacological inhibition of JNK activity restores intact hypothalamic leptin and insulin signal transduction and reduces weight gain.74 In addition, JNK knockout increases insulin signal transduction in mouse hypothalamus.95
Another key intracellular proinflammatory signal transducer is the serine kinase IKK,96 which activates the transcription factor nuclear factor-κB in response to a wide array of microbial, chemical and immunological stimuli and produces a global increase of inflammatory transcriptional activity.96 Recent data show that IKK is activated in the hypothalamus of obese rodents and that hypothalamic disruption of IKK expression protects mice against DIO. Conversely, induced expression and activation of IKK in the hypothalamus leads to both excessive weight gain and a state of hypothalamic leptin/insulin resistance.97 Similarly, pharmacological inhibition of hypothalamic IKK improves hypothalamic insulin action and reduces food intake in rats.98
Increased expression of the serine/threonine kinase protein kinase C-theta is another mechanism implicated in the impairment of adipostatic signaling in the hypothalamus of animals consuming an HF diet.99 Activation of this enzyme can be induced by the long-chain saturated fatty acid palmitate, an effect that has been documented specifically in AgRP neurons in the hypothalamic arcuate nucleus. This response in turn impairs hypothalamic insulin action and predisposes to increased adiposity, whereas knockdown of protein kinase C-theta in discrete hypothalamic areas protects against DIO.99 Taken together, emerging evidence suggests that multiple inflammatory responses are mounted in the hypothalamus during HF feeding, and that these responses are sufficient to explain, at least in part, both excess weight gain and associated hypothalamic resistance to adiposity signals.
Role of hypothalamic inflammation in leptin and insulin resistance
Clear evidence linking inflammation in peripheral tissues to insulin resistance in the setting of obesity and type 2 diabetes100 led to speculation that hypothalamic resistance to adipostatic hormones might reflect an extension to the brain of responses to nutrient excess observed in peripheral tissues. In adipose tissue, for example, obesity induced by HF feeding induces proinflammatory cytokine gene expression that in turn is associated with macrophage recruitment and activation. Soluble inflammatory factors produced in adipose and other tissues during HF feeding are hypothesized in turn to cause systemic inflammation and whole-body insulin resistance. According to this model, hypothalamic inflammation could arise as a consequence of systemic inflammation.
Such a scenario, however, is inconsistent with evidence that hypothalamic induction of proinflammatory cytokines and associated hypothalamic insulin and leptin resistance precedes by many weeks the appearance of such responses in adipose tissue.81 Hypothalamic responses to HF feeding, therefore, seem unlikely to be driven by systemic inflammation, raising the possibility that a direct mechanism mediates the hypothalamic response to nutrient excess during HF feeding. Should these effects occur early following the switch to an obesigenic diet, excess weight gain might arise, at least in part, as a consequence of impaired hypothalamic function.
Consistent with this hypothesis is evidence that in rodent models, DIO is attenuated by local inhibition of any of several mechanisms underlying hypothalamic leptin/insulin resistance, as described above,72, 73, 74, 75, 89, 97, 99 whereas food intake and adiposity increase in a manner that mimics HF feeding following hypothalamic overexpression of inducers of leptin/insulin resistance.83, 95, 97 Similarly, low-grade hypothalamic inflammation induced by central administration of a very low dose of tumor necrosis factor-α can induce local resistance to leptin/insulin and modulate neurotransmitter expression toward an obesity-like pattern,101 whereas hypothalamic administration of an antitumor necrosis factor--α monoclonal antibody reduces body mass gain and increases thermogenesis during HF feeding.72
These findings add to growing evidence implicating hypothalamic inflammation in the genesis of local leptin/insulin resistance, which in turn predisposes to obesity in animal models. Available evidence suggests that in most instances, obesity-associated hypothalamic inflammation develops before or in parallel with (and is not secondary to) systemic inflammation, although the mechanisms and cell types involved in this response remain poorly understood.
Mechanisms leading to hypothalamic inflammation in obesity
Of the various potential mechanisms underlying hypothalamic inflammation in the context of HF feeding, we highlight two here (Figure 2). One of these involves the pattern recognition receptor, Toll-like receptor 4 (TLR4), a component of the innate immune system that in the CNS is expressed predominantly by microglia, the resident macrophage of the brain.102 In rodent models of DIO, hypothalamic TLR4 expression and activity are increased leading to signal transduction through MyD88,75, 103 an intracellular protein that couples TLR4 (or interleukin-1R) to intracellular inflammatory signaling cascades. Local activation of TLR4 during HF feeding can induce expression of inflammatory mediators, causing neuronal resistance to leptin and insulin.75 The finding that TLR4 activation in brain can be induced by ICV infusion of long-chain saturated fatty acids75 raises the possibility that dietary fat content might underlie hypothalamic TLR4 induction during HF feeding, although further study is required to assess the utility of centrally administered free fatty acids as a model of the CNS effects of HF feeding. Conversely, disruption of TLR4 or Myd88 signaling by genetic or immunological approaches reduces leptin/insulin resistance and can protect against DIO,75, 103 suggesting that the above sequence of events may be required for obesity to occur during HF feeding. These observations also highlight the need for an improved understanding of interactions between neurons and microglia (as well as other glial cells) in the hypothalamus and elsewhere during HF feeding.
Figure 2.
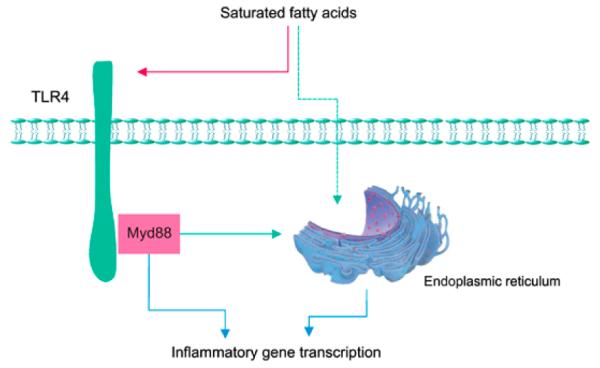
Depiction of cellular inflammation induced by either TLR4 activation or endoplasmic reticulum stress (blue lines). TLR4 activity is hypothesized to increase in response to long-chain fatty acids (red line). Induction of hypothalamic endolasmic reticulum stress involves mechanisms that remain poorly understood by may include direct activation by nutrients and/or activation through TLR4 (green lines).
Endoplasmic reticulum stress (ER stress) is a second cellular process that can cause inflammation during HF feeding. Correct folding of nascent proteins is essential for the maintenance of cell viability,100 and when this process is compromised by any of a variety of infectious, chemical or metabolic factors, ER stress can result.100 Recent studies implicate this phenomenon as a mechanism driving inflammatory gene transcription and hepatic insulin resistance in rodent models of obesity,100 and a similar mechanism was recently reported in the hypothalamus, further highlighting similarities in the response of peripheral tissues and hypothalamus to nutrient excess during HF feeding.75, 97 Similar to TLR4 activation, hypothalamic ER stress can also be induced by direct exposure to long-chain saturated fatty acids during ICV infusion,75, 97 whereas inhibiting hypothalamic ER stress with a chemical chaperone protects against DIO.97
Whether ER stress is a primary driver or secondary responder in the genesis of hypothalamic inflammation during HF feeding is uncertain. The finding that inhibition of TLR4 is sufficient to ameliorate ER stress in rodent models treated with either an HF diet or with ICV infusion of saturated fatty acids suggests that hypothalamic ER stress may lie downstream of TLR4 activation.75 This possibility is further supported by evidence that inhibition of IKK (an enzyme that couples TLR4-MyD88 signaling to activation of nuclear factor-κB activation) in mice with DIO also reduces hypothalamic ER stress, although the reduction of food intake and body weight in this model confounds interpretation of causal relationships between these various mechanisms.97 Thus, additional studies are needed to assess the relative importance of ER stress, activation of TLR4 and other as yet unidentified mechanisms involved in the genesis of hypothalamic inflammation during HF feeding, as well as to identify the cell types underlying these responses.
Hypothalamic parameters affected by DIO
Beyond the extent of weight gain during HF feeding, the impact on energy homeostasis can be measured by determining if the body weight ‘set point’ was increased. Of the two, the latter measure is a more important gauge of whether a ‘fixed change’ of underlying regulatory mechanisms has occurred. In some rodent studies, elevated body fat mass during HF is completely reversed by switching back to regular chow, whereas in others, the reversal is incomplete, and numerous factors most likely contribute to this variability. Yet the defended level of body adiposity appears to be permanently elevated in most obese humans, and a greater focus on this issue in preclinical studies is needed if its underlying pathophysiology is to be better understood.
Although changes of hypothalamic neuropeptide gene expression have provided essential insight into the functional organization of the energy homeostasis system, a clear pattern linking such responses to obesity pathogenesis is still awaited.54, 104 Changes in neuronal firing rate offer perhaps a more direct measure of altered hypothalamic function in obesity. Both leptin and insulin modulate synaptic release of neurotransmitters and neuropeptides by altering the firing of specific neuronal sub-populations of the hypothalamus.62, 105, 106 Included among these are neurons whose membrane potential is regulated by adenosine triphosphate-sensitive-K+ channels, which are sensitive to both nutrient- and hormone-related inputs. The observation that expression of these channels is reduced in the hypothalamus of rats with DIO, and that this effect is associated with altered firing of hypothalamic neurons,107, 108 offers an example of how obesity can fundamentally change neuronal function. Whether such changes contribute to obesity pathogenesis or are simply a consequence of pathological weight gain awaits further study.
A growing number of studies have used genetic strategies to delete genes in distinct neuronal subsets and then assess the consequences for obesity susceptibility. Examples include mice lacking LepR or related signal-transduction molecules selectively in neurons of the hypothalamic arcuate nucleus or VMN, which have clear effects on body weight gain and susceptibility to DIO.105, 109 Additionally, HF feeding increases apoptosis of hypothalamic neurons,76 an outcome that appears more dependent on diet composition (for example, saturated fat content) than on caloric intake or weight gain per se. Another factor affecting neuronal apoptosis during HF feeding is genetic background, with increased neuronal apoptosis being observed in obesity-prone strains.76 How this phenomenon might be related to hypothalamic inflammation and obesity pathogenesis awaits further study, as does its relationship to hypothalamic neurogenesis, which has been reported following the death of specific neuronal subsets.110, 111
An additional area of recent interest regarding the hypothalamic response to HF feeding involves changes in neuronal wiring and/or synaptic plasticity. As in other regions of the brain, hypothalamic neurons undergo continuous rewiring,112, 113 and factors such as leptin, nutrients and age, among others, can influence this process.113, 114, 115, 116 Not surprisingly, therefore, synaptic plasticity in key hypothalamic neuronal systems is reportedly altered during HF feeding.113, 114 Along with neuronal apoptosis, this type of ‘fixed’ change in the organization of hypothalamic neurocircuits has the potential to explain not only how HF feeding raises the defended level of body fat mass in susceptible rodent strains, but also how this defense of elevated body fat mass persists even after the obese animal (or human) returns to a low-fat diet.
Concluding remarks
Although many brain systems participate in energy homeostasis, changes of hypothalamic function have important potential to participate in the genesis of experimental obesity. With continued progress in our understanding of obesity-associated changes in hypothalamic neurocircuits in animal models, translation of these findings to human hypothalamus should become possible. This work may ultimately inform new approaches to the treatment of one of the most prevalent and threatening diseases of modern societies.
References
- 1.Kelly T, Yang W, Chen CS, Reynolds K, He J. Global burden of obesity in 2005 and projections to 2030. Int J Obes (Lond) 2008;32:1431–1437. doi: 10.1038/ijo.2008.102. | Article | PubMed | ChemPort |. [DOI] [PubMed] [Google Scholar]
- 2.Daniels SR. Complications of obesity in children and adolescents. Int J Obes (Lond) 2009;33(Suppl 1):S60–S65. doi: 10.1038/ijo.2009.20. [DOI] [PubMed] [Google Scholar]
- 3.Tsiros MD, Olds T, Buckley JD, Grimshaw P, Brennan L, Walkley J, et al. Health-related quality of life in obese children and adolescents. Int J Obes (Lond) 2009;33:387–400. doi: 10.1038/ijo.2009.42. [DOI] [PubMed] [Google Scholar]
- 4.Anandacoomarasamy A, Caterson I, Sambrook P, Fransen M, March L. The impact of obesity on the musculoskeletal system. Int J Obes (Lond) 2008;32:211–222. doi: 10.1038/sj.ijo.0803715. [DOI] [PubMed] [Google Scholar]
- 5.Haslam DW, James WP. Obesity. Lancet. 2005;366:1197–1209. doi: 10.1016/S0140-6736(05)67483-1. [DOI] [PubMed] [Google Scholar]
- 6.Ross R, Bradshaw AJ. The future of obesity reduction: beyond weight loss. Nat Rev Endocrinol. 2009;5:319–325. doi: 10.1038/nrendo.2009.78. [DOI] [PubMed] [Google Scholar]
- 7.Neovius M, Narbro K. Cost-effectiveness of pharmacological anti-obesity treatments: a systematic review. Int J Obes (Lond) 2008;32:1752–1763. doi: 10.1038/ijo.2008.189. [DOI] [PubMed] [Google Scholar]
- 8.Hainer V, Toplak H, Mitrakou A. Treatment modalities of obesity: what fits whom? Diabetes Care. 2008;31(Suppl 2):S269–S277. doi: 10.2337/dc08-s265. [DOI] [PubMed] [Google Scholar]
- 9.Tice JA, Karliner L, Walsh J, Petersen AJ, Feldman MD. Gastric banding or bypass? A systematic review comparing the two most popular bariatric procedures. Am J Med. 2008;121:885–893. doi: 10.1016/j.amjmed.2008.05.036. [DOI] [PubMed] [Google Scholar]
- 10.Grill HJ. Distributed neural control of energy balance: contributions from hindbrain and hypothalamus. Obesity (Silver Spring) 2006;14(Suppl 5):216S–221S. doi: 10.1038/oby.2006.312. [DOI] [PubMed] [Google Scholar]
- 11.Grill HJ, Hayes MR. The nucleus tractus solitarius: a portal for visceral afferent signal processing, energy status assessment and integration of their combined effects on food intake. Int J Obes (Lond) 2009;33(Suppl 1):S11–S15. doi: 10.1038/ijo.2009.10. [DOI] [PubMed] [Google Scholar]
- 12.Brooks C. The hypothalamus and obesity. Med J Aust. 1948;1:327–331. doi: 10.5694/j.1326-5377.1948.tb97167.x. [DOI] [PubMed] [Google Scholar]
- 13.Kennedy GC. Experimental hypothalamic obesity. Proc R Soc Med. 1951;44:899–902. doi: 10.1177/003591575104401017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sunderman FW, Haymaker W. Hypothermia and elevated serum magnesium in a patient with facial hemangioma extending into the hypothalamus. Am J Med Sci. 1947;213:562–571. doi: 10.1097/00000441-194705000-00006. [DOI] [PubMed] [Google Scholar]
- 15.Katsuki S, Hirata Y, Horino M, Ito M, Ishimoto M, Makino N, et al. Obesity and hyperglycemia induced in mice by goldthioglucose. Diabetes. 1962;11:209–215. [PubMed] [Google Scholar]
- 16.Anand BK, Dua S, Shoenberg K. Hypothalamic control of food intake in cats and monkeys. J Physiol. 1955;127:143–152. doi: 10.1113/jphysiol.1955.sp005244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanaka K, Shimada M, Nakao K, Kusunoki T. Hypothalamic lesion induced by injection of monosodium glutamate in suckling period and subsequent development of obesity. Exp Neurol. 1978;62:191–199. doi: 10.1016/0014-4886(78)90050-x. [DOI] [PubMed] [Google Scholar]
- 18.Kennedy GC. The role of depot fat in the hypothalamic control of food intake in the rat. Proc R Soc Lond B Biol Sci. 1953;140:578–596. doi: 10.1098/rspb.1953.0009. [DOI] [PubMed] [Google Scholar]
- 19.Mayer J. The glucostatic theory of regulation of food intake and the problem of obesity. Bull New Engl Med Cent. 1952;14:43–49. [PubMed] [Google Scholar]
- 20.Hervey GR. The effects of lesions in the hypothalamus in parabiotic rats. J Physiol. 1959;145:336–352. doi: 10.1113/jphysiol.1959.sp006145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coleman DL. Effects of parabiosis of obese with diabetes and normal mice. Diabetologia. 1973;9:294–298. doi: 10.1007/BF01221857. [DOI] [PubMed] [Google Scholar]
- 22.Margolis RU, Altszuler N. Insulin in the cerebrospinal fluid. Nature. 1967;215:1375–1376. doi: 10.1038/2151375a0. [DOI] [PubMed] [Google Scholar]
- 23.Havrankova J, Roth J, Brownstein M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature. 1978;272:827–829. doi: 10.1038/272827a0. [DOI] [PubMed] [Google Scholar]
- 24.Hatfield JS, Millard WJ, Smith CJ. Short-term influence of intra-ventromedial hypothalamic administration of insulin on feeding in normal and diabetic rats. Pharmacol Biochem Behav. 1974;2:223–226. doi: 10.1016/0091-3057(74)90056-2. [DOI] [PubMed] [Google Scholar]
- 25.Bruning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, et al. Role of brain insulin receptor in control of body weight and reproduction. Science. 2000;289:2122–2125. doi: 10.1126/science.289.5487.2122. [DOI] [PubMed] [Google Scholar]
- 26.Shibata S, Liou SY, Ueki S, Oomura Y. Inhibitory action of insulin on suprachiasmatic nucleus neurons in rat hypothalamic slice preparations. Physiol Behav. 1986;36:79–81. doi: 10.1016/0031-9384(86)90077-6. [DOI] [PubMed] [Google Scholar]; Baskin DG, Brewitt B, Davidson DA, Corp E, Paquette T, Figlewicz DP, et al. Quantitative autoradiographic evidence for insulin receptors in the choroid plexus of the rat brain. Diabetes. 1986;35:246–249. doi: 10.2337/diab.35.2.246. [DOI] [PubMed] [Google Scholar]
- 27.Stein LJ, Dorsa DM, Baskin DG, Figlewicz DP, Ikeda H, Frankmann SP, et al. Immunoreactive insulin levels are elevated in the cerebrospinal fluid of genetically obese Zucker rats. Endocrinology. 1983;113:2299–2301. doi: 10.1210/endo-113-6-2299. [DOI] [PubMed] [Google Scholar]
- 28.Baskin DG, Woods SC, West DB, van Houten M, Posner BI, Dorsa DM, et al. Immunocytochemical detection of insulin in rat hypothalamus and its possible uptake from cerebrospinal fluid. Endocrinology. 1983;113:1818–1825. doi: 10.1210/endo-113-5-1818. [DOI] [PubMed] [Google Scholar]
- 29.Schwartz MW, Sipols A, Kahn SE, Lattemann DF, Taborsky GJ, Jr, Bergman RN, et al. Kinetics and specificity of insulin uptake from plasma into cerebrospinal fluid. Am J Physiol. 1990;259(3 part 1):E378–E383. doi: 10.1152/ajpendo.1990.259.3.E378. [DOI] [PubMed] [Google Scholar]
- 30.Oomura Y, Kita H. Insulin acting as a modulator of feeding through the hypothalamus. Diabetologia. 1981;20(Suppl):290–298. [PubMed] [Google Scholar]
- 31.Obici S, Feng Z, Arduini A, Conti R, Rossetti L. Inhibition of hypothalamic carnitine palmitoyltransferase-1 decreases food intake and glucose production. Nat Med. 2003;9:756–761. doi: 10.1038/nm873. [DOI] [PubMed] [Google Scholar]
- 32.Lam TK, Gutierrez-Juarez R, Pocai A, Rossetti L. Regulation of blood glucose by hypothalamic pyruvate metabolism. Science. 2005;309:943–947. doi: 10.1126/science.1112085. [DOI] [PubMed] [Google Scholar]
- 33.Sakaguchi T, Takahashi M, Bray GA. Diurnal changes in sympathetic activity. Relation to food intake and to insulin injected into the ventromedial or suprachiasmatic nucleus. J Clin Invest. 1988;82:282–286. doi: 10.1172/JCI113584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanchez-Alavez M, Tabarean IV, Osborn O, Mitsukawa K, Schaefer J, Dubins J, et al. Insulin causes hyperthermia by direct inhibition of warm-sensitive neurons. Diabetes. 2010;59:43–50. doi: 10.2337/db09-1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lotter EC, Woods SC. Injections of insulin and changes of body weight. Physiol Behav. 1977;18:293–297. doi: 10.1016/0031-9384(77)90136-6. [DOI] [PubMed] [Google Scholar]
- 36.Brandes JS. Insulin induced overeating in the rat. Physiol Behav. 1977;18:1095–1102. doi: 10.1016/0031-9384(77)90017-8. [DOI] [PubMed] [Google Scholar]
- 37.Jessen L, Clegg DJ, Bouman SD. Evaluation of the lack of anorectic effect of intracerebroventricular insulin in rats. Am J Physiol Regul Integr Comp Physiol. 2010;298:R43–R50. doi: 10.1152/ajpregu.90736.2008. [DOI] [PubMed] [Google Scholar]
- 38.Carvalheira JB, Siloto RM, Ignacchitti I, Brenelli SL, Carvalho CR, Leite A, et al. Insulin modulates leptin-induced STAT3 activation in rat hypothalamus. FEBS Lett. 2001;500:119–124. doi: 10.1016/s0014-5793(01)02591-1. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 40.Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, et al. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 41.Chua SC, Jr, White DW, Wu-Peng XS, Liu SM, Okada N, Kershaw EE, et al. Phenotype of fatty due to Gln269Pro mutation in the leptin receptor (Lepr) Diabetes. 1996;45:1141–1143. doi: 10.2337/diab.45.8.1141. [DOI] [PubMed] [Google Scholar]
- 42.Farooqi IS, Matarese G, Lord GM, Keogh JM, Lawrence E, Agwu C, et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. 2002;110:1093–1103. doi: 10.1172/JCI15693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Villanueva EC, Myers MG., Jr Leptin receptor signaling and the regulation of mammalian physiology. Int J Obes (Lond) 2008;32(Suppl 7):S8–12. doi: 10.1038/ijo.2008.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li C, Friedman JM. Leptin receptor activation of SH2 domain containing protein tyrosine phosphatase 2 modulates Ob receptor signal transduction. Proc Natl Acad Sci USA. 1999;96:9677–9682. doi: 10.1073/pnas.96.17.9677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gong Y, Ishida-Takahashi R, Villanueva EC, Fingar DC, Munzberg H, Myers MG., Jr The long form of the leptin receptor regulates STAT5 and ribosomal protein S6 via alternate mechanisms. J Biol Chem. 2007;282:31019–31027. doi: 10.1074/jbc.M702838200. [DOI] [PubMed] [Google Scholar]
- 46.Banks AS, Davis SM, Bates SH, Myers MG., Jr Activation of downstream signals by the long form of the leptin receptor. J Biol Chem. 2000;275:14563–14572. doi: 10.1074/jbc.275.19.14563. [DOI] [PubMed] [Google Scholar]
- 47.Gao S, Kinzig KP, Aja S, Scott KA, Keung W, Kelly S, et al. Leptin activates hypothalamic acetyl-CoA carboxylase to inhibit food intake. Proc Natl Acad Sci USA. 2007;104:17358–17363. doi: 10.1073/pnas.0708385104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Girasol A, Albuquerque GG, Mansour E, Araujo EP, Degasperi G, Denis RG, et al. Fyn mediates leptin actions in the thymus of rodents. PLoS One. 2009;4:e7707. doi: 10.1371/journal.pone.0007707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ropelle ER, Pauli JR, Fernandes MF, Rocco SA, Marin RM, Morari J, et al. A central role for neuronal AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) in high-protein diet-induced weight loss. Diabetes. 2008;57:594–605. doi: 10.2337/db07-0573. [DOI] [PubMed] [Google Scholar]
- 50.Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC, et al. Hypothalamic mTOR signaling regulates food intake. Science. 2006;312:927–930. doi: 10.1126/science.1124147. [DOI] [PubMed] [Google Scholar]
- 51.Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. 2004;428:569–574. doi: 10.1038/nature02440. [DOI] [PubMed] [Google Scholar]
- 52.Niswender KD, Morrison CD, Clegg DJ, Olson R, Baskin DG, Myers MG, Jr, et al. Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: a key mediator of insulin-induced anorexia. Diabetes. 2003;52:227–231. doi: 10.2337/diabetes.52.2.227. [DOI] [PubMed] [Google Scholar]
- 53.Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443:289–295. doi: 10.1038/nature05026. [DOI] [PubMed] [Google Scholar]
- 54.Moran TH. Gut peptides in the control of food intake. Int J Obes (Lond) 2009;33(Suppl 1):S7–10. doi: 10.1038/ijo.2009.9. [DOI] [PubMed] [Google Scholar]
- 55.Badman MK, Flier JS. The gut and energy balance: visceral allies in the obesity wars. Science. 2005;307:1909–1914. doi: 10.1126/science.1109951. [DOI] [PubMed] [Google Scholar]
- 56.Woods SC, Seeley RJ, Cota D. Regulation of food intake through hypothalamic signaling networks involving mTOR. Annu Rev Nutr. 2008;28:295–311. doi: 10.1146/annurev.nutr.28.061807.155505. [DOI] [PubMed] [Google Scholar]
- 57.Coope A, Milanski M, Araujo EP, Tambascia M, Saad MJ, Geloneze B, et al. AdipoR1 mediates the anorexigenic and insulin/leptin-like actions of adiponectin in the hypothalamus. FEBS Lett. 2008;582:1471–1476. doi: 10.1016/j.febslet.2008.03.037. [DOI] [PubMed] [Google Scholar]
- 58.Niswender KD, Baskin DG, Schwartz MW. Insulin and its evolving partnership with leptin in the hypothalamic control of energy homeostasis. Trends Endocrinol Metab. 2004;15:362–369. doi: 10.1016/j.tem.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 59.Niswender KD, Schwartz MW. Insulin and leptin revisited: adiposity signals with overlapping physiological and intracellular signaling capabilities. Front Neuroendocrinol. 2003;24:1–10. doi: 10.1016/s0091-3022(02)00105-x. [DOI] [PubMed] [Google Scholar]
- 60.Hill JW, Elias CF, Fukuda M, Williams KW, Berglund ED, Holland WL, et al. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab. 2010;11:286–297. doi: 10.1016/j.cmet.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Williams KW, Margatho LO, Lee CE, Choi M, Lee S, Scott MM, et al. Segregation of acute leptin and insulin effects in distinct populations of arcuate proopiomelanocortin neurons. J Neurosci. 2010;30:2472–2479. doi: 10.1523/JNEUROSCI.3118-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pijl H, Toornvliet AC, Meinders AE. Serum leptin in normal-weight and obese humans. N Engl J Med. 1996;334:1544. doi: 10.1056/NEJM199606063342314. [DOI] [PubMed] [Google Scholar]
- 63.Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med. 1995;1:1155–1161. doi: 10.1038/nm1195-1155. [DOI] [PubMed] [Google Scholar]
- 64.Bluher S, Mantzoros CS. Leptin in humans: lessons from translational research. Am J Clin Nutr. 2009;89:991S–997S. doi: 10.3945/ajcn.2008.26788E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schwartz MW, Peskind E, Raskind M, Boyko EJ, Porte D., Jr Cerebrospinal fluid leptin levels: relationship to plasma levels and to adiposity in humans. Nat Med. 1996;2:589–593. doi: 10.1038/nm0596-589. [DOI] [PubMed] [Google Scholar]
- 66.Torsoni MA, Carvalheira JB, Pereira-Da-Silva M, de Carvalho-Filho MA, Saad MJ, Velloso LA. Molecular and functional resistance to insulin in hypothalamus of rats exposed to cold. Am J Physiol Endocrinol Metab. 2003;285:E216–E223. doi: 10.1152/ajpendo.00031.2003. [DOI] [PubMed] [Google Scholar]
- 67.Schwartz MW, Baskin DG, Bukowski TR, Kuijper JL, Foster D, Lasser G, et al. Specificity of leptin action on elevated blood glucose levels and hypothalamic neuropeptide Y gene expression in ob/ob mice. Diabetes. 1996;45:531–535. doi: 10.2337/diab.45.4.531. [DOI] [PubMed] [Google Scholar]
- 68.Myers MG, Jr, Leibel RL, Seeley RJ, Schwartz MW. Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol Metab. 2010;21:643–651. doi: 10.1016/j.tem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Frederich RC, Hamann A, Anderson S, Lollmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med. 1995;1:1311–1314. doi: 10.1038/nm1295-1311. [DOI] [PubMed] [Google Scholar]
- 70.Halaas JL, Boozer C, Blair-West J, Fidahusein N, Denton DA, Friedman JM. Physiological response to long-term peripheral and central leptin infusion in lean and obese mice. Proc Natl Acad Sci USA. 1997;94:8878–8883. doi: 10.1073/pnas.94.16.8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Romanatto T, Roman EA, Arruda AP, Denis RG, Solon C, Milanski M, et al. Deletion of tumor necrosis factor-alpha receptor 1 (TNFR1) protects against diet-induced obesity by means of increased thermogenesis. J Biol Chem. 2009;284:36213–36222. doi: 10.1074/jbc.M109.030874. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 72.Mori H, Hanada R, Hanada T, Aki D, Mashima R, Nishinakamura H, et al. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat Med. 2004;10:739–743. doi: 10.1038/nm1071. [DOI] [PubMed] [Google Scholar]
- 73.De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, et al. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology. 2005;146:4192–4199. doi: 10.1210/en.2004-1520. [DOI] [PubMed] [Google Scholar]
- 74.Milanski M, Degasperi G, Coope A, Morari J, Denis R, Cintra DE, et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J Neurosci. 2009;29:359–370. doi: 10.1523/JNEUROSCI.2760-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Moraes JC, Coope A, Morari J, Cintra DE, Roman EA, Pauli JR, et al. High-fat diet induces apoptosis of hypothalamic neurons. PLoS One. 2009;4:e5045. doi: 10.1371/journal.pone.0005045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Flores MB, Fernandes MF, Ropelle ER, Faria MC, Ueno M, Velloso LA, et al. Exercise improves insulin and leptin sensitivity in hypothalamus of Wistar rats. Diabetes. 2006;55:2554–2561. doi: 10.2337/db05-1622. [DOI] [PubMed] [Google Scholar]
- 77.Ropelle ER, Flores MB, Cintra DE, Rocha GZ, Pauli JR, Morari J, et al. IL-6 and IL-10 anti-inflammatory activity links exercise to hypothalamic insulin and leptin sensitivity through IKKbeta and ER stress inhibition. PLoS Biol. 2010;8:e1000465. doi: 10.1371/journal.pbio.1000465. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 78.Cohen P, Zhao C, Cai X, Montez JM, Rohani SC, Feinstein P, et al. Selective deletion of leptin receptor in neurons leads to obesity. J Clin Invest. 2001;108:1113–1121. doi: 10.1172/JCI13914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gao Q, Wolfgang MJ, Neschen S, Morino K, Horvath TL, Shulman GI, et al. Disruption of neural signal transducer and activator of transcription 3 causes obesity, diabetes, infertility, and thermal dysregulation. Proc Natl Acad Sci USA. 2004;101:4661–4666. doi: 10.1073/pnas.0303992101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Prada PO, Zecchin HG, Gasparetti AL, Torsoni MA, Ueno M, Hirata AE, et al. Western diet modulates insulin signaling, c-Jun N-terminal kinase activity, and insulin receptor substrate-1ser307 phosphorylation in a tissue-specific fashion. Endocrinology. 2005;146:1576–1587. doi: 10.1210/en.2004-0767. [DOI] [PubMed] [Google Scholar]
- 81.Bjorbaek C, El-Haschimi K, Frantz JD, Flier JS. The role of SOCS-3 in leptin signaling and leptin resistance. J Biol Chem. 1999;274:30059–30065. doi: 10.1074/jbc.274.42.30059. [DOI] [PubMed] [Google Scholar]
- 82.Howard JK, Flier JS. Attenuation of leptin and insulin signaling by SOCS proteins. Trends Endocrinol Metab. 2006;17:365–371. doi: 10.1016/j.tem.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 83.Howard JK, Cave BJ, Oksanen LJ, Tzameli I, Bjorbaek C, Flier JS. Enhanced leptin sensitivity and attenuation of diet-induced obesity in mice with haploinsufficiency of Socs3. Nat Med. 2004;10:734–738. doi: 10.1038/nm1072. [DOI] [PubMed] [Google Scholar]
- 84.Bjornholm M, Munzberg H, Leshan RL, Villanueva EC, Bates SH, Louis GW, et al. Mice lacking inhibitory leptin receptor signals are lean with normal endocrine function. J Clin Invest. 2007;117:1354–1360. doi: 10.1172/JCI30688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, Haj F, Wang Y, Minokoshi Y, et al. PTP1B regulates leptin signal transduction in vivo. Dev Cell. 2002;2:489–495. doi: 10.1016/s1534-5807(02)00148-x. [DOI] [PubMed] [Google Scholar]
- 86.Morrison CD, White CL, Wang Z, Lee SY, Lawrence DS, Cefalu WT, et al. Increased hypothalamic protein tyrosine phosphatase 1B contributes to leptin resistance with age. Endocrinology. 2007;148:433–440. doi: 10.1210/en.2006-0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cheng A, Uetani N, Simoncic PD, Chaubey VP, Lee-Loy A, McGlade CJ, et al. Attenuation of leptin action and regulation of obesity by protein tyrosine phosphatase 1B. Dev Cell. 2002;2:497–503. doi: 10.1016/s1534-5807(02)00149-1. [DOI] [PubMed] [Google Scholar]
- 88.Picardi PK, Calegari VC, Prada Pde O, Moraes JC, Araujo E, Marcondes MC, et al. Reduction of hypothalamic protein tyrosine phosphatase improves insulin and leptin resistance in diet-induced obese rats. Endocrinology. 2008;149:3870–3880. doi: 10.1210/en.2007-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bence KK, Delibegovic M, Xue B, Gorgun CZ, Hotamisligil GS, Neel BG, et al. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med. 2006;12:917–924. doi: 10.1038/nm1435. [DOI] [PubMed] [Google Scholar]
- 90.Banno R, Zimmer D, De Jonghe BC, Atienza M, Rak K, Yang W, et al. PTP1B and SHP2 in POMC neurons reciprocally regulate energy balance in mice. J Clin Invest. 2010;120:720–734. doi: 10.1172/JCI39620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Martin TL, Alquier T, Asakura K, Furukawa N, Preitner F, Kahn BB. Diet-induced obesity alters AMP kinase activity in hypothalamus and skeletal muscle. J Biol Chem. 2006;281:18933–18941. doi: 10.1074/jbc.M512831200. [DOI] [PubMed] [Google Scholar]
- 92.Xue B, Pulinilkunnil T, Murano I, Bence KK, He H, Minokoshi Y, et al. Neuronal protein tyrosine phosphatase 1B deficiency results in inhibition of hypothalamic AMPK and isoform-specific activation of AMPK in peripheral tissues. Mol Cell Biol. 2009;29:4563–4573. doi: 10.1128/MCB.01914-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 94.Unger EK, Piper ML, Olofsson LE, Xu AW. Functional role of c-Jun-N-terminal kinase in feeding regulation. Endocrinology. 2010;151:671–682. doi: 10.1210/en.2009-0711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Israel A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol. 2010;2:a000158. doi: 10.1101/cshperspect.a000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135:61–73. doi: 10.1016/j.cell.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Posey KA, Clegg DJ, Printz RL, Byun J, Morton GJ, Vivekanandan-Giri A, et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol Endocrinol Metab. 2009;296:E1003–E1012. doi: 10.1152/ajpendo.90377.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Benoit SC, Kemp CJ, Elias CF, Abplanalp W, Herman JP, Migrenne S, et al. Palmitic acid mediates hypothalamic insulin resistance by altering PKC-theta subcellular localization in rodents. J Clin Invest. 2009;119:2577–2589. doi: 10.1172/JCI36714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Romanatto T, Cesquini M, Amaral ME, Roman EA, Moraes JC, Torsoni MA, et al. TNF-alpha acts in the hypothalamus inhibiting food intake and increasing the respiratory quotient--effects on leptin and insulin signaling pathways. Peptides. 2007;28:1050–1058. doi: 10.1016/j.peptides.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 101.Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, et al. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci USA. 2003;100:8514–8519. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kleinridders A, Schenten D, Konner AC, Belgardt BF, Mauer J, Okamura T, et al. MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab. 2009;10:249–259. doi: 10.1016/j.cmet.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.van den Pol AN. Weighing the role of hypothalamic feeding neurotransmitters. Neuron. 2003;40:1059–1061. doi: 10.1016/s0896-6273(03)00809-2. [DOI] [PubMed] [Google Scholar]
- 104.Dhillon H, Zigman JM, Ye C, Lee CE, McGovern RA, Tang V, et al. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron. 2006;49:191–203. doi: 10.1016/j.neuron.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 105.Sakaguchi T, Bray GA. Intrahypothalamic injection of insulin decreases firing rate of sympathetic nerves. Proc Natl Acad Sci USA. 1987;84:2012–2014. doi: 10.1073/pnas.84.7.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Levin BE, Brown KL, Dunn-Meynell AA. Differential effects of diet and obesity on high and low affinity sulfonylurea binding sites in the rat brain. Brain Res. 1996;739:293–300. doi: 10.1016/s0006-8993(96)00835-9. [DOI] [PubMed] [Google Scholar]
- 107.Spanswick D, Smith MA, Mirshamsi S, Routh VH, Ashford ML. Insulin activates ATP-sensitive K+ channels in hypothalamic neurons of lean, but not obese rats. Nat Neurosci. 2000;3:757–758. doi: 10.1038/77660. [DOI] [PubMed] [Google Scholar]
- 108.Plum L, Ma X, Hampel B, Balthasar N, Coppari R, Munzberg H, et al. Enhanced PIP3 signaling in POMC neurons causes KATP channel activation and leads to diet-sensitive obesity. J Clin Invest. 2006;116:1886–1901. doi: 10.1172/JCI27123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pierce AA, Xu AW. De novo neurogenesis in adult hypothalamus as a compensatory mechanism to regulate energy balance. J Neurosci. 2010;30:723–730. doi: 10.1523/JNEUROSCI.2479-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kokoeva MV, Yin H, Flier JS. Neurogenesis in the hypothalamus of adult mice: potential role in energy balance. Science. 2005;310:679–683. doi: 10.1126/science.1115360. [DOI] [PubMed] [Google Scholar]
- 111.Coleman CG, Wang G, Park L, Anrather J, Delagrammatikas GJ, Chan J, et al. Chronic intermittent hypoxia induces NMDA receptor-dependent plasticity and suppresses nitric oxide signaling in the mouse hypothalamic paraventricular nucleus. J Neurosci. 2010;30:12103–12112. doi: 10.1523/JNEUROSCI.3367-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chun SK, Jo YH. Loss of leptin receptors on hypothalamic POMC neurons alters synaptic inhibition. J Neurophysiol. 2010;104:2321–2328. doi: 10.1152/jn.00371.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Horvath TL. Synaptic plasticity in energy balance regulation. Obesity (Silver Spring) 2006;14(Suppl 5):228S–233S. doi: 10.1038/oby.2006.314. [DOI] [PubMed] [Google Scholar]
- 114.Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, et al. Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science. 2004;304:110–115. doi: 10.1126/science.1089459. [DOI] [PubMed] [Google Scholar]
- 115.Fenoglio KA, Chen Y, Baram TZ. Neuroplasticity of the hypothalamic-pituitary-adrenal axis early in life requires recurrent recruitment of stress-regulating brain regions. J Neurosci. 2006;26:2434–2442. doi: 10.1523/JNEUROSCI.4080-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]