

Abstract
Immunotherapeutic ablation of lymphoma is a conceptually attractive treatment strategy that is the subject of intense translational research. Cytotoxic T lymphocytes (CTL) that are genetically modified to express CD19- or CD20-specific, single-chain-antibody-derived chimeric antigen receptors (CARs) display HLA-independent antigen-specific recognition/killing of lymphoma targets. Here, we describe our initial experience in applying CAR-redirected autologous CTL adoptive therapy to patients with recurrent lymphoma. Using plasmid vector electrotransfer/drug selection systems, cloned and polyclonal CAR+ CTL were generated from autologous peripheral blood mononuclear cells and expanded in vitro to cell numbers sufficient for clinical use. In two FDA-authorized trials, patients with recurrent diffuse large cell lymphoma (DLCL) were treated with cloned CD8+ CTL expressing a CD20-specific CAR (along with NeoR) following autologous HSCT, while patients with refractory follicular lymphoma (FL) were treated with polyclonal T cell preparations expressing a CD19-specific CAR (along with HyTK, a fusion of hygromycin resistance and HSV-1 thymidine kinase suicide genes) and low-dose s.c. rhuIL-2. A total of fifteen infusions were administered (five at 108cells/m2, seven at 109cells/m2, three at 2×109cells/m2) to four patients. Overt toxicities attributable to CTL administration were not observed. However, detection of transferred CTL in the circulation, measured by Q-PCR, was short (24hrs-7d), and cellular anti-transgene immune rejection responses were detected in two patients. These studies reveal the primary barrier to therapeutic efficacy is limited persistence and provide the rationale to prospectively define T cell populations intrinsically programmed for survival following adoptive transfer and to modulate the immune status of recipients to prevent/delay anti-transgene rejection responses.
Keywords: Cellular immunotherapy, Adoptive therapy, T cells, Chimeric antigen receptor, Clinical trial
INTRODUCTION
While conventional chemotherapy, radiation and antibody therapy can be efficacious in treating lymphoma, relapse and progressive disease are the major sources of patient morbidity and mortality (1, 2). Experimental evidence that the cellular immune system can eradicate lymphoma provides a basis for the development of therapies aimed at amplifying anti-tumor immune responses (3, 4). The adoptive transfer of lymphoma-specific T cells is one strategy to augment anti-lymphoma immunity. A significant challenge to executing this strategy is the isolation of T cells specifically reactive to lymphoma. Alternately, the ex vivo derivation of tumor-specific T lymphocytes by genetic modification to express tumor-targeting chimeric antigen receptors or CARs is now a rapidly evolving focus of translational cancer immunotherapy (5, 6). Antibody-based CARs are HLA-unrestricted and thus can be used in patient populations with target-antigen positive tumors.
We have constructed two CARs specific for the B cell lineage antigens CD20 and CD19 for the purpose of targeting lymphomas and leukemias (7, 8). When expressed in CTL, these CAR redirect effector cells to lyse B-lineage lymphoma targets (7, 8). Here we report our initial clinical experience in manufacturing and infusing autologous T cells expressing CD20R or CD19R in patients with relapsed B cell lymphoma under City of Hope-held FDA authorized trials BB-IND-8513/IRB 98142 and BB-IND-11411/IRB 01160, respectively.
MATERIALS AND METHODS
Patients
City of Hope Internal Review Board (IRB) protocols 98142 and 01160 were activated for patient accrual following IRB and Institutional Biological Safety Committee approval, Food and Drug Administration (FDA) authorization (BB-IND-8513 and BB-IND-11411 respectively) and NIH-Office of Biotechnology Activities registration (9907-330 and 0207-543 respectively). In brief, for IRB 98142, patients were eligible if they had immunohistopathologically documented CD20+ diffuse large cell lymphoma (DLCL) with a history of recurrent or refractory disease, but did not have central nervous system metastases. Following leukapheresis, patients began salvage/mobilization chemotherapy then underwent hematopoietic stem cell transplantation (HSCT), 28 days following which, the first of three escalating dose T cell infusions began. For IRB protocol 01160, patients were eligible if they had pathologically documented follicular lymphoma (FL) with evidence of progression after prior Rituximab therapy, and did not have central nervous system metastases or a history of allogeneic HSCT. These patients were enrolled no sooner than 3 weeks following their most recent cytotoxic chemotherapy.
Plasmid Vectors
The plasmid expression vectors encoding a) the CD20R chimeric immunoreceptor and the neomycin phosphotransferase cDNAs; and b) the CD19R chimeric immunoreceptor and the selection-suicide HyTK cDNAs have been previously described ((7, 8), see Fig. S1A). Briefly, the chimeric construct consists of VH and VL gene segments of the CD20-specific Leu-16 or CD19-specific FMC63 mAbs, an IgG hinge-CH2-CH3 region, a CD4 transmembrane region, and the cytoplasmic domain of the CD3ζ chain (Fig. S1B).
Isolation, Transfection, Selection, Cloning and Expansion of T Cells
The methods for OKT3-stimulation of peripheral blood mononuclear cells (PBMCs), their electroporation, selection, cloning (IRB 98142 only), and subsequent growth using the rapid expansion method (REM) consisting of recursive 14-day cycles of activation with OKT3, rhuIL-2 and PBMC/lymphoblastoid cell line (LCL) irradiated feeders has been previously described (9). The overall T cell product manufacturing schemas for each trial are depicted in Figure S1C.
Cell Product Quality Control Tests (QCTs)
A summary of the QCTs performed and the requisite test results for product release are summarized in Table S1.
Confirmation of plasmid vector integration (IRB 98142 only)
A single site of plasmid vector chromosomal integration was confirmed by Southern blot analysis of XbaI/HindIII digested T cell genomic DNA using a 420-basepair NeoR-specific probe generated using the pcDNA3.1(−) plasmid as a template (9). The pass criterion of this test was defined as detection of a single band.
Confirmation of CAR expression
Western blot analysis for CAR expression has been previously described (10). In brief, reduced whole cell lysates are subjected to Western blotting with an anti-human CD3-ζ (cytoplasmic tail) specific monoclonal antibody 8D3 (BD Pharmingen, San Diego, CA). This probe detects both the 16-kD endogenous ζ and the 66-kD CAR ζ. Pass criteria were defined as visualization of both the 16-kD and the 66-kD bands. Flow cytometric analysis for surface CAR expression was determined using a FITC-conjugated Fc-specific antibody (Jackson ImmunoResearch Laboratories, Inc., Westgrove, PA). Pass criteria were defined as unimodal positive staining for Fc compared to the FITC-conjugated isotype control (BD Biosciences, San Jose, CA).
Surface phenotype determination
T cell products were evaluated for cell-surface phenotype using standard staining and flow cytometric procedures with FITC-conjugated monoclonal Abs (BD Biosciences) followed by analysis on a FACS caliber (BD Biosciences). Pass criteria were defined as ≥ 90% positive staining for TCR-αβ and CD8 (IRB 98142) or CD3 (IRB 01160) compared to the isotype control. Independent of the QCT guidelines, other correlative surface markers included CD4 for IRB 98142, and both CD4 and CD8 for IRB 01160.
Assay for anti-lymphoma cytolytic activity
Cytolytic activity of CAR+ CTL clones against 51Cr-labeled human lymphoma Daudi cells was performed as previously described using a 4-hr chromium release assay (8). The pass criteria were defined as ≥ 50% specific lysis at an effector to target ratio of 25:1.
Viability
Viability was determined by standard trypan blue dye exclusion, with pass criteria defined as > 90 % viability.
Sensitivity to ganciclovir ablation (IRB 01160 only)
To test for acquired cytocidal sensitivity to ganciclovir (GCV), aliquots of clones were harvested from 5-day REM expansion cultures, then maintained for 14 days in 37.5 U/mL rhuIL-2 with or without 1μM GCV at which time cells were harvested and subjected to viability testing. Pass criteria were defined as ≤ 25% viability in the GCV treated cultures.
Assay for antigen/IL-2 independent growth
5×106 cells were washed and plated in antigen- and IL-2-free culture media at the end of a 14-day REM cycle. Parallel cultures of Jurkat T cells (ATCC, Manassas, VA) (IRB 98142) or T cells that were cultured in the presence of 37.5 U/mL rhuIL-2 (IRB 01160) served as controls for expansion and viability. For IRB 98142, following an 11-day incubation, cultures were harvested, counted using trypan blue, plated into 96-well plates at 6,000 viable cells per well, pulsed with 1 μCi of 3H-TdR and DNA was harvested following a 4-hr incubation at 37° C. For IRB 01160, viable cell numbers of 14-day cultures were determined by flow cytometry as previously described (11). Release criteria specified that cells must exhibit <10% of the Jurkat c.p.m. (IRB 98142) or <10% of the IL-2+ control cell number (IRB 01160).
Sterility
Sterility tests were performed based on an FDA Center for Biologics and Evaluation of Research mandated schedule. Aliquots of media from the T cell cultures were plated onto bacterial and fungal growth media; mycoplasma detection was conducted on media aliquots using the Gen-Probe Mycoplasma Tissue Culture-NI Rapid Detection System (San Diego, CA.); and endotoxin levels were determined by ELISA. Pass criteria were defined as negative bacterial, fungal and mycoplasma results, and an endotoxin level < 5 EU/kg recipient weight.
Adoptive Transfer of T Cells
Processed and cryopreserved cell banks were thawed and expanded in culture to the desired cell numbers prior to being resuspended in 0.9% NaCl with 2% human serum albumin in a clinical re-infusion bag. T cells were reinfused intravenously over 30 min. either through a central line or an age-appropriate sized intravenous catheter inserted into a peripheral vein. The infusion bag was gently mixed every 5 min. during the infusion. The intra-patient dose escalation plan is schematically presented in Figure S2. Fludarabine administration was performed after the first T cell infusion in IRB 01160 as a potential non-myeloablative immunosuppressive regimen for attenuating possible rejection responses against the transferred T cells. The guidelines provided in the NCI Common Toxicity Criteria version 2.0 (https://ctep.ifo.nih.gov/l) were followed for the monitoring of toxicity and adverse event reporting. Rules for dose escalation, de-escalation, and cancellation were strictly enforced and resulted in three of the four treated patients deviating from planned infusion cell dose escalation at least once.
In Vivo Persistence of Transferred T Cells
For IRB 98142, mononuclear cells from heparinized peripheral blood samples (PBMCs) were isolated and analyzed for percentage of transfected cells by Q-PCR as previously described (12) using primers and probes to quantify CD20R copy number (available upon request).
For IRB 01160, samples were received, processed, stored and analyzed in accordance with current good laboratory practice (GLP) guidelines. A validated Q-PCR-based assay to quantify CD19R plasmid vector DNA in samples was developed and performed using an MJ Research DNA engine with a Chromo 4™ Continuous Fluorescence Detector Q-PCR module (Bio-Rad Laboratories, Hercules, CA). Real-time PCR was performed in a 20-μL reaction mixture volume containing 50 ng DNA, 10 μL of IQ SYBR Green Supermix: (Bio-Rad Laboratories, cat. no. 170-8880), and 0.5 pmole of each primer. Quantification of the CD19R transgene sequence in DNA isolated from patient PBMC was evaluated using Q-PCR to amplify a 182 nucleotide fragment that spanned the CD4 transmembrane-zeta junction within the transgene coding sequence, and a standard curve derived by dilution of DNA isolated from a clone with a single integration of the CD19R transgene (primers, probes and amplification conditions available upon request). The qualification studies for this amplification reaction demonstrated no amplification from healthy donor-derived PBMC (n=5), whereas the transgene sequence could be quantified in a PBMC sample if the transgene containing DNA comprised as little as 0.1% of the total DNA sample.
Analysis of Anti-Transgene Rejection Responses
Pre- and Post-treatment PBMC (Days 75, 77, and 50 from UPN006, 009, and 035, respectively) were first stimulated with irradiated therapeutic T cells (3,000 rads) or LCL +/− pcDNA3.1(−) plasmid (8,000 rads) plus irradiated pre-treatment PBMC as feeder cells at a 10:1 responder to stimulator ratio in RPMI 1640 (Irvine Scientific, Santa Ana, CA) supplemented with 2 mM L-glutamine (Irvine Scientific), 25 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES, Irvine Scientific), 100 U/mL penicillin, 0.1 mg/mL streptomycin (Irvine Scientific), and 10% heat-inactivated human serum. One week later, the same irradiated stimulator as well as irradiated pre-treatment PBMC feeders (3,500 rads) were added at a 1:1:1 responder to stimulator to feeder ratio, and this stimulation schema continued up to two more times (once weekly) until sufficient numbers were obtained for chromium release assays. Cytolytic activity of these stimulated PBMC against 51Cr-labeled targets was performed as previously described using a 4-hr chromium release assay (8).
For IRB 01160, cellular anti-transgene immune responses were evaluated directly ex vivo using a combination of TCR Vβ spectratyping and CD107 degranulation assays. For the TCR Vβ spectratyping analysis, RNA was isolated from PBMC collected pre- and post- infusion using the RNAqueous-4 PCR Kit for Isolation of DNA-free RNA (Applied Biosystems/Ambion, Austin, TX), and cDNA was then synthesized using the iScript cDNA Synthesis Kit (Bio-Rad Laboratories). TCR Vβ spectratyping analysis was performed on cDNA samples essentially as described in (13) using pools of Vβ specific primers. A parallel series of amplifications using cDNA generated from pooled healthy donor PBMC was performed as a quality control for amplification of each Vβ family. Aliquots of the amplification mixes were run on sequencing gels, followed by analysis using Genemapper v3.7 software (Applied Biosystems, Foster City, CA). The CD107 degranulation/mobilization assay was performed essentially as previously described (14), using patient PBMC collected pre- and post- infusion as effectors, and infused T cell product or OKT3 expanded pre-infusion PBMC (i.e., autologous T cells) as targets. To detect spontaneous degranulation, a control sample without target cells was included in every experiment. FITC-conjugated anti-CD107a and anti-CD107b (BD Biosciences) were added directly to the tubes prior to incubation. After 5 hours co-incubation, cells were washed twice and stained with PE-Cy5-conjugated anti-CD8β and PE-conjugated anti-TCR Vβ23 (Beckman Coulter, Inc., Fullerton, CA) for 30 min at room temperature in the dark, washed again an analyzed on a FC500 (Beckman Coulter, Inc.) using FCS Express v3.0 software, gating on CD8β+, Vβ23+ lymphocytes.
Serologic Anti-CAR Immune Response Analysis
Serum was isolated from patient blood samples collected in red top (no additive) tubes using an established laboratory SOP and a qualified assay to detect CAR-specific serological responses in samples. Samples were allowed to clot for 2½ hours at room temperature, centrifuged at 1000 × g at 4°C for 15 minutes and serum was collected, aliquoted and frozen immediately at −80°C. Flow cytometric detection of potential serum antibody responses against the anti-CD19R transgene was performed using parental vs. CD19R-expressing Jurkat cell lines as indicator cell lines. The presence of antibodies in patient serum that specifically bound to CD19R+ Jurkat cells was evaluated by a subsequent incubation with FITC-conjugated AffiniPure F(ab′)2 fragment goat anti-human IgG, Fcγ, (Jackson ImmunoResearch Laboratories, Inc.). The cut off for a negative response was established by defining the 95% one-sided prediction interval using a pool of non-CAR-reactive serum samples from healthy volunteers.
Results
Patient Characteristics
Patients in these studies either had diffuse large cell lymphoma (BB-IND 8513/IRB 98142) or follicular non-Hodgkin lymphoma (BB-IND 11411/IRB 01160; Table S2). One of the four research participants had bulky disease, including sites at the neck, chest, lymph nodes and pelvis, at the time of enrollment. Three of the four participants had Rituximab (chimeric monoclonal antibody specific for CD20) therapy prior to the first infusion of therapeutic T cells. The average duration of time from leukapheresis to first infusion was 106 days, and was affected by either the time required to manufacture the T cell product and/or the timing of patient recovery from salvage therapy.
Generation of genetically modified T cells
Cell products meeting all quality control release tests (Table S1) were successfully generated for two of the five patients enrolled on IRB 98142, and each of the two patients enrolled on IRB 01160. Failure to release products for three of the patients enrolled on IRB 98142 was due to the failure to isolate T cell clones that expressed CD8, expressed endogenous TCR, or expanded adequately in vitro. The Southern blots indicating the desired single site insertions of the CD20R transgene within the released clones of IRB 98142 are depicted in Figure 1. Western blot and cell-surface expression profiles of T-cell products for both trials are also depicted, confirming expression of the CAR protein. These cells were further subjected to flow cytometric analysis for confirmation of the T-cell subset markers CD4, CD8 and either TCR-αβ or CD3. All of the cell products used in therapy also exhibited re-directed killing of CD19+ CD20+ human Daudi lymphoma targets in 4-hr chromium release assays. Furthermore, all of the cell lines retained their dependence on exogenous rhuIL-2 for survival and proliferation, and the HyTK expressing lines of IRB 01160 tested positive for sensitivity to ganciclovir-mediated ablation.
Figure 1. T cell products meet release requirements.
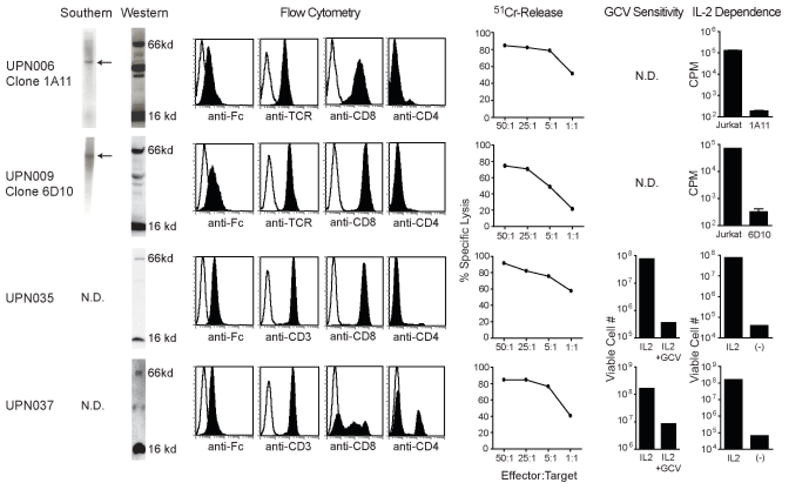
Depicted from left to right: Southern blots of T cell genomic DNA using an HyTK specific probe showing existence of single bands as indicated by arrows; Western blots revealing both the 16-kDA endogenous CD3ζ and the 66-kDA CE7R chimeric ζ bands detected with anti-human CD3ζ cytoplasmic tail specific antibody; flow cytometry analysis for surface expression of the chimeric receptor using anti-Fc antibody, or for the T cell markers TCR and, CD8, CD4 and/or CD3, where isotype control staining is indicated with the open histogram; ability of CTL clones to lyse CD19+ CD20+ Daudi targets was determined in a 4hr 51Cr release assay; ganciclovir (GCV) sensitivity using a flow cytometry based assay for viable cell numbers after 14 days of culture with either rhuIL-2 or rhuIL-2 + GCV; Assays for IL-2 dependence were performed using 3H-thymidine incorporation measurements (c.p.m) of Jurkat T cells vs. the indicated T cell clones after 11 days of culture in the absence of rhuIL-2 (UPN006, UPN009), or using a flow cytometry based assay for viable cell numbers after the T cell products were cultured in the presence vs. the absence of rhuIL-2 for 14 days (UPN035, UPN037).
Treatment experience
As depicted in Figure 2, the intra-patient dose escalations were carried out as planned (compare to Fig. S2) with the exception that the 1010/m2 cell dose was never given in IRB 98142 due to protocol toxicity dose modification rules (UPN006, UPN009). Indeed, because of grade 2 hepatic toxicities that were noticed with the first infusion dose of 108/m2 in UPN006, the second infusion was repeated at 108/m2, followed by an escalation to 109/m2 for the third infusion. Patient UPN009 exhibited a fall in hemoglobin following the second infusion that, while clinically insignificant (10.6 to 8.6) represented a CTC toxicity grading change from grade 1 to grade 2 anemia status, requiring that the 109/m2 dose be repeated for the third infusion based on the protocol’s defined rules for dose escalation. In UPN006 the second infusion was cancelled and rescheduled due to puncture of the bag which compromised the integrity of the T cell product. In UPN037 the last infusion was cancelled due to the detection of contaminated T cell product. Neither myeloablation followed by HSCT nor fludarabine resulted in absolute lymphocyte counts dropping below the normal range (Table S3).
Figure 2. Treatment regimens for each patient.
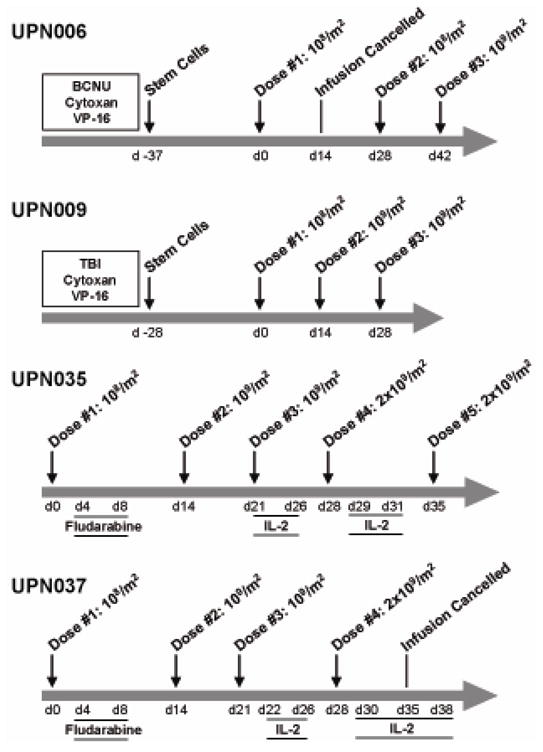
First i.v. infusions of T cells were administered on day 0 for each patient. For UPN006 and UPN009, fractionated total body irradiation (TBI) and/or myeloablative chemotherapies administered to UPN006 and UPN009 are indicated just prior to administration of CD34+ autologous stem cells. BCNU, bis-chloronitrosourea; Cytoxan, cyclophosphamide; VP-16, etoposide. For UPN035 and UPN037, administration of fludarabine (i.v. at 25mg/m2) occurred between days 4 and 8 after the first T cell infusion, and rhuIL-2 administration (5×105 IU/m2 BID) was initiated after the third T cell infusion.
There were no grade 3 or higher adverse events with possible correlation to administration of 108 T cells per m2. However, upon examination of the adverse events at 109 T cells per m2, there was one Grade 3 self-limited lymphopenia in both IRB 98142 and IRB 01160 with possible attribution to the cell administration (Table 1). At 2×109 T cells per m2, Grade 3 lymphopenia and Grade 3 eosinophelia each occurred once in IRB 01160. These all resolved spontaneously without adverse sequelae to the patients. Overall, the safety profile of this adoptive transfer therapy was acceptable.
Table 1.
Adverse Event Summary
| IRB Trial | T-cell dose (cells/m2) | Event* | Occurrence (# of times) |
|---|---|---|---|
| 98142 | 109 | Lymphopenia | 1 |
| 01160 | 109 | Lymphopenia | 1 |
| 2×109 | Lymphopenia | 1 | |
| Eosinophilia | 1 |
Only events of Grade 3 or higher, according to the NCI Common Toxicity Criteria, with possible attribution to the T cell administration are reported.
Follow-up clinical status of patients
Although this was a phase I clinical trial, with the primary purpose of determining safety, we also monitored the disease and survival status of each patient. For IRB 98142 (CD20R; diffuse large cell lymphoma), UPN006 (last infusion 02/24/00) relapsed in September of 2001 while UPN009 (last infusion 11/22/00) continues to be in remission post autologous stem cell transplantation. To date both UPN006 (after additional treatment) and UPN009 are alive and in remission. For IRB 01160 (CD19R; follicular lymphoma), UPN035 (last infusion 05/04/06) presented with a new diagnosis of CD19+/CD20+ diffuse large cell lymphoma in June of 2006, and was deceased in June of 2007. UPN037 (last infusion 02/15/07) displayed progression by CT scan in September 2007, and is currently alive and undergoing additional treatment.
In vivo persistence of transferred T cells
Quantitative PCR was used to detect CD20R and CD19R plasmid copy numbers in the PBMC as a surrogate marker of the presence of adoptively transferred T cells and showed that the persistence of the T cells varied between patients (Fig. 3). Only one of the four patients (UPN006) had detectible levels of transferred T cells one week after the first infusion of 108 cells/m2. Detectable levels of transferred T cells one week after infusion of 109 cells/m2 were observed in only two of the seven higher doses (UPN006 infusion #3, UPN009 infusion #2), and at no time were transferred T cells detected at one week after infusion of 2×109 cells/m2. Thus, adoptively transferred T cell persistence did not appear to correlate with cell dose. Compared to the persistence after the initial infusion, patients 009, 035 and 037 also displayed significantly reduced levels of transferred T cells 24 hrs after each additional infusion, suggesting the possibility that an anti-transgene immune response had been mounted against the administered T cells (Fig. 3).
Figure 3. Transferred T cells do not persist long term in vivo.
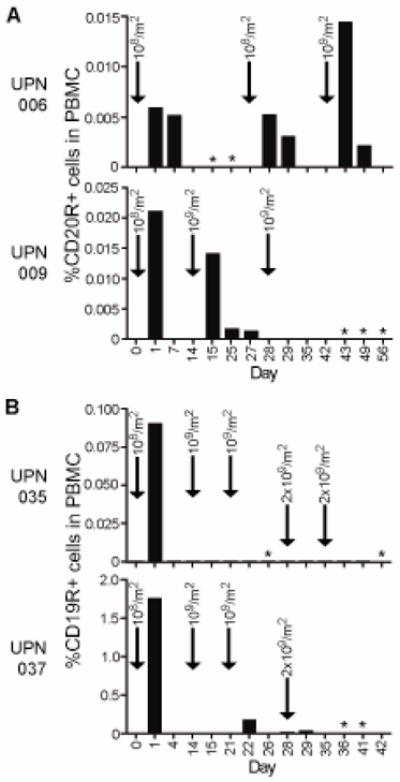
Using real time quantitative PCR, copy numbers of the CD20R (A) or CD19R (B) genes per μg of genomic DNA were determined as an indicator of the relative amount of chimeric receptor expressing T cells in the PBMC samples collected at the indicated days during the treatment schedule. Escalating infusion doses are indicated by arrows. *, cells not harvested.
Detection of transgene-specific immune responses
For IRB 98142 (CD20R; diffuse large cell lymphoma), the development of cellular immune responses against the infused T cell products was evaluated. For these analyses, PBMC were collected from UPN006 and UPN009 before and after T cell administration, and, following in vitro stimulation, were compared for their cytotoxic activity using chromium release assays (Fig. 4). Use of irradiated lymphoblastoid cells (LCLx) to stimulate the PBMC resulted in successful lysis of 51Cr-labeled LCL, indicating that functional effector cells could be derived from each patient’s PBMC samples. Interestingly, when the irradiated autologous T cell clones were used to stimulate the PBMC, cytotoxic responses were seen against the 51Cr-labeled T cell clone only with the post-treatment sample collected from UPN009 (Fig. 4A). This immunoreactivity against the T cell clone 6D10 used in therapy appeared to be specific for neomycin phosphotransferase, not the CAR, as cytotoxic responses could be observed against 51Cr-labeled LCL that had been transduced with the pcDNA3.1(−) vector which directs the expression of neomycin phosphotransferase, but lacks the CD20R transgene (Fig. 4B). To better analyze the specificity of the rejection response, UPN009 post-treatment PBMC that had been stimulated with irradiated 6D10 cells were cloned in limiting dilution and all clones were similarly specific for NeoR (Fig. 4C).
Figure 4. Transgene rejection response detected when T cells administered following hematopoietic stem cell transfer.
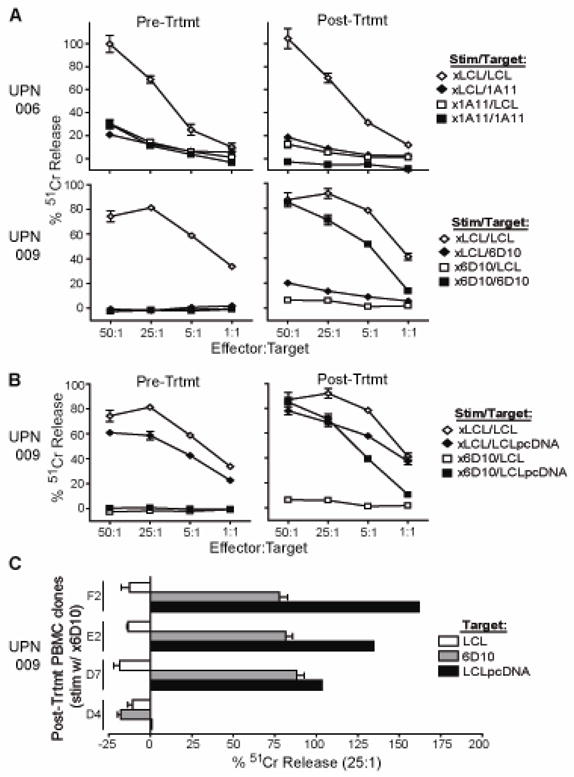
In the trial targeting CD20+ diffuse large cell lymphoma, PBMC collected prior to treatment and at day 75 (UPN006) or day 77 (UPN009) after initiation of treatment were stimulated in vitro with irradiated LCL as a control (xLCL) or the corresponding irradiated CTL clone that had been administered (i.e., x1A11 or x6D10). Effectors were then used in a 4-hour 51Cr-release assay using either LCL or the corresponding CTL as targets (A) or, in the case of UPN009, using LCL that had been transfected with the pcDNA3.1(−) vector lacking the CD20R transgene as targets (B). (C), Clones derived from UPN009 day 98 PBMC were also stimulated in vitro with irradiated 6D10 CTL and then analyzed for cytolytic activity against 51Cr-labeled LCL, 6D10 or pcDNA3.1(−) vector transfected LCL to determine specificity of transgene-specific response. Percent 51Cr-release at an E:T of 25:1 in each case is depicted for four representative clones.
For IRB 01160 (CD19R; follicular lymphoma), the development of both antibody and cellular immune responses against the infused T cell products was evaluated. For the antibody analyses, serum collected from UPN035 and UPN037 at enrollment and after T-cell administration were both negative for antibody reactivity against surface expressed CD19R transgene using a flow cytometry based assay (Fig. 5A). However, evidence was again found for cell-mediated immuno-reactivity against the infused T cells. Examination of the T cell receptor (TCR) Vβ gene repertoire through spectratyping sequence analysis of PBMC that were collected from UPN035 and UPN037 both before and after T cell administration revealed alterations in the Vβ profiles of the post-treatment PBMC with appearance of unique clonotypes in the post-infusion samples that are indicative of a new immunoreactive response (Fig. 5B). Furthermore, flow cytometric analysis of the TCR Vβ23+ and Vβ14+ subpopulations in the UPN035 post-treatment PBMC collected 2 weeks after the first infusion showed significant surface CD107 expression, as an indicator of lysis-associated degranulation, upon co-culture with the infused T cell product (Fig. 5C, and data not shown). This specific degranulation was not observed with the pre-treatment PBMC, nor when the post-treatment PBMC were co-cultured with control T cells. Similar flow cytometric assays could not be carried out with the UPN037 PBMC due to the lack of commercially available antibodies specific for the TCR Vβ genes (Vβ15, Vβ21) that arose in this patient. The pre- and post-treatment PBMC from UPN035 were also stimulated in vitro with irradiated LCL or infused T-cell product and compared for their cytotoxic activity using chromium release assays (Fig. 5D). As seen with UPN009 in IRB 98142, functional effector cells were derived from both UPN035 PBMC samples, but cytolytic activity against the 51Cr-labeled T cells was observed only with the post-treatment sample. Together these data suggest that, at least in some cases, the lack of T cell persistence observed in these two trials was due to immune rejection responses that are mounted by the patients’ endogenous T cells.
Figure 5. Rejection response detected when T cells administered following fludarabine administration.
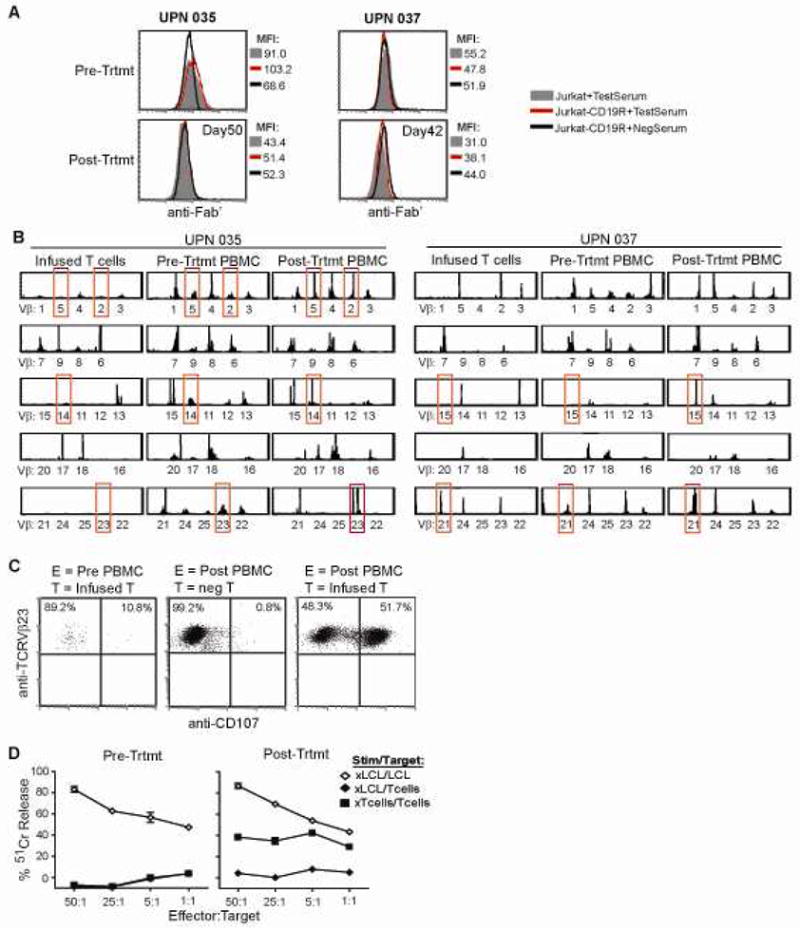
(A), In the trial targeting CD19+ follicular lymphoma, serum collected at the time of patient enrollment (Pre-Trtmt) and at day 50 (UPN035) or day 42 (UPN037) after initiation of treatment was examined for immunoreactivity against Jurkat cells expressing the CD19R (red line) in a flow cytometry based assay. Parental Jurkat cells (grey histogram), and a known non- reactive serum (black line) were used as negative controls. (B), TCR Vβ profiles of the infused T cell product, day 0 PBMC (collected just prior to first T cell infusion; Pre-Trtmt), and day 14 PBMC (collected prior to the second T cell infusion, Post-Trtmt) were determined by spectratyping analysis. Alterations in Vβ usage that were observed pre- vs. post-treatment are highlighted with red boxes. (C), The TCR Vβ23+ population of pre-treatment and day 14 PBMC from UPN035 was further analyzed by flow cytometry for surface CD107 expression as a marker of degranulation upon co-culture with the infused T cell product (infused T), or non-modified autologous T cells (neg T). (D), Pre-treatment and day 50 PBMC from UPN035 were stimulated in vitro with irradiated LCL as a control (xLCL) or with the irradiated T cell product (xTcells). Effectors were then used in a 4-hour 51Cr-release assay using either LCL or T cells as targets.
DISCUSSION
Over 55,000 new cases of NHL are diagnosed each year in the United States and the incidence of this disease is increasing (15, 16). Intermediate grade B-cell lymphomas (diffuse large cell, mantle cell, marginal zone) and low grade follicular lymphomas are the most common sub-types of NHL and account for approximately 80% of cases. The majority of patients have widespread disease at the time of diagnosis and are treated with combinations of chemotherapy, radiation therapy, and rituximab. Unfortunately, over two thirds will relapse with their disease and only 10% of these patients can be salvaged (17). Efforts to improve survival for NHL recurrent disease focus primarily on the use of myeloablative conditioning and autologous stem cell transplantation (18–21) and this strategy is curative in approximately 46% of selected patients. The selected group of salvageable patients (age <60 years, complete remission after primary treatment, and no known marrow or central nervous system disease), however, constitutes less than one-third of those with relapsed intermediate grade lymphomas. Patients with chemotherapy-resistant recurrent disease have a <15% 5-year event-free survival following stem cell transplantation and those with refractory disease at the time of transplant are rarely cured. Similarly, patients with mantle cell lymphoma and low-grade follicular lymphoma whose disease becomes refractory to chemotherapy and radiation have a poor prognosis despite high-intensity salvage therapy (22–25). These findings have prompted the evaluation of additional strategies to eradicate lymphoma MRD after cytoreductive chemotherapy/radiation/rituximab, including the immunotherapeutic targeting of malignant B-cells with adoptively transferred antigen-specific T cells.
Here we describe our initial experience testing the feasibility and safety of lymphoma adoptive therapy with CTLs genetically modified to express re-directing CD20 and CD19 specific CARs. Our T cell production platform relied on plasmid vector electrotransfer into patient peripheral blood mononuclear cell preparations. While this approach facilitated regulatory approval and diminished expenses relative to the use of a viral vector platform, the low efficiency of chromosomal integration and sustained transgene expression encumbered the production platform to multiple rounds of activation/propagation in selection drugs (G418 and hygromycin B). Nevertheless, for each of the seven enrolled patients drug-resistant CAR+ T cells were isolated. The reason three of the enrolled subjects on protocol 98142 did not have clones released was that only CD4+CD8- clones were isolated, when the release criteria specified CD4-CD8+. This skewed result in production runs was specific to lymphoma patients on this trial and likely reflects the repertoire changes in these patients due to disease and/or prior therapy at the time of apheresis for T-cell production. Another observed limitation in the production platform during generation of polyclonal lines in protocol 01160 was the discordance between CAR expression and hygromycin resistance. The plasmid vector employed in the trial drives the CAR and HyTK from two separate promoters allowing chromosomal integration events that result in deletion of the CAR-encoding portion of the vector or transcriptional repression of the CAR’s promoter. As a result we found that only a subset of polyclonal cell preps had demonstrable CAR expression. This problem can potentially be resolved in plasmid vectors using single promoter systems in which the two transgene open reading frames are separated by an IRES element or directly integrated into a single polypeptide with a cleavable linker element. Our group has now redesigned our platform to use SIN lentiviral vectors and shortened ex vivo culture duration (~28 days), improving the percentage of CAR expressing T cells in polyclonal cell preps, and eliminating the need for bacterial drug-resistance gene co-expression.
The primary focus of these studies was to establish the safety of this approach. In this regard the T cell infusions were well tolerated up to 2×109 cells/m2. The most common event that could be attributed to T cell infusion was transient self-limited lymphopenia lasting less than seven days. We suspect, however, that this phenomenon is related to redistribution of the endogenous circulating repertoire as a consequence of infused cell product; whether it is based on cytokine/chemokine elaboration upon activation or other mechanisms remains to be delineated. In the two patients where immunologic rejection of the infused cell product was demonstrated, the third and subsequent cell doses in these two patients elicited a self limited (<24hr) febrile response with rigors. Despite the dramatic systemic febrile response patients did not exhibit cardiovascular instability or other over toxicities associated with a “cytokine storm” syndrome. A toxic death proximal to cell infusion has been recently reported (26) in a patient with bulky CLL who received cyclophosphamide prior to administration of re-directed T cells expressing a CD19 specific CAR having both CD28 co-stimulatory and CD3-ζ activation signaling domains. At present, the lack of serious toxicities observed in our patients may be due to the limited numbers of circulating B cells at the time of T cell infusion; 98142 patients were 28-days from a myeloablative autologous HSCT, while 01160 patients had B cell reduction as a consequence of Rituxan. However, analysis of the peripheral blood after the last T cell infusion, as well as the follow-up clinical status of these patients, indicates that this strategy did not result in sustained B cell lymphopenia, as would be expected based on the transient engraftment of infused effector cells. Another possible explanation for the observed lack of toxicity is an attenuated cytokine response to activation based on the CAR having only a CD3-ζ activation domain. Carefully designed future trials can test the effects of these parameters as they relate to the tolerability of cell infusions.
The NCI surgical branch has demonstrated that the frequency and magnitude of melanoma-reactive tumor-infiltrating lymphocyte (TIL) engraftment can be enhanced by rendering patients lymphopenic prior to adoptive transfer and administering high-dose rhuIL-2 following transfer. TIL products that previously were difficult to detect following AT engraft at very high numbers in about 50% of patients treated with the most intensive lymphodepleting regimens consisting of myeloablative chemotherapy/TBI with CD34-selected stem cell rescue. Despite having received myeloablative HSCT, patients on 98142 were not lymphopenic on transplant day +28 when infusions of their CD20-specific CD8+ clones commenced. Similarly, patients on 01160 were given a 5-day course of fludarabine at 25mg/m2/dose without achieving lymphopenia prior to T cell transfer. Given the compelling data from the NCI melanoma trials, we plan to administer T cell products to lymphoma patients in conjunction with their autologous HSCT procedure on Day +2 when lymphopenia is profound.
Our experience clearly delineates the issue of transgene immunogenicity as one mechanism that limits persistence. The plasmid electrotransfer platform required that we drug select for stably integrated clones/lines. The rejection response observed was cellular and focused on NeoR in UPN-009, while the transgene-specificity of the rejection response in UPN035 and UPN037 was not further characterized. The ability of transgenes expressed in T cells to elicit immune responses following adoptive transfer was clearly established by the findings of S.R. Riddell and colleagues in which the limited persistence of adoptively transferred HyTK+ T cells correlates with anti-HyTK transgene-specific immune responses (27). Furthermore, while studies by this same group suggest that a non-myeloablative immunosuppressive regimen could prolong the in vivo persistence of the T cells by attenuating possible rejection responses (28), fludarabine, applied after the first dose of T cells in IRB 01160, apparently fails as an immunopreparative strategy to delete anti-transgene specific responses. Thus, more effective immunosuppressive/lymphodepleting regimens might be advantageous for future clinical application. Use of more efficient vector transduction systems (e.g. lentiviral vectors or the Sleeping Beauty system (29)) that would negate the requirement for ex vivo selection might also allow the omission of NeoR and HyTK. Even without the selection markers, the CARs themselves are expected to be immunogenic based on mouse scFvs and fusion sites in the chimera. We expect that providing a window for cells to evade immunologic rejection by patient lymphodepletion will be the most practical strategy to limit early rejection responses, while late rejection responses have the advantage of eliminating the gene-modified cells that, if timed appropriately, could be exploited as a safety feature.
Poor in vivo persistence is the major problem in the cancer AT field in general, and likely relates primarily to the intrinsic programming of T cells to survive after adoptive transfer. Effector T cells are inherently short-lived (30) and it has been suggested that acquisition of an effector phenotype during in vitro culture and expansion is a major reason for the poor survival of transferred T cells (31). However, the adoptive transfer of virus-specific effector T cells from memory cell precursors can result in long-term repopulation in humans. Thus, the use of memory T cells, which are known to self-renew (32), and/or virus specific T cells, which would receive optimal costimulation after engagement of their native receptors, as populations for genetic redirection has become of increasing interest. Indeed, it has recently been reported in neuroblastoma patients that Epstein Barr virus (EBV)-specific T cells engineered to co-express tumor-specific receptors survived longer than those lacking virus specificity, and were associated with tumor regression or necrosis in half of the subjects tested (33). It has also recently been found in a macaque model of adoptive transfer, that antigen-specific CD8+ T cell clones derived from central memory T (TCM) cells persist long-term in vivo, reacquiring phenotypic and functional properties of memory T cells and occupying memory T cell niches (34). Accordingly, we have begun to develop a clinically compatible immunomagnetic selection system to isolate central memory T cells from human PBMC for subsequent processing to generate CAR re-directed effector cells. We propose viral-specific, TCM-derived, anti-tumor effector cell infusion into lymphoma patients during profound lymphopenia shortly following auto-HSCT, as the next logical iteration of our translational research in lymphoma immunotherapy.
Supplementary Material
Acknowledgments
This work was supported by NIH P01 CA30206, P50 CA107399, General Clinical Research Center M01 RR0004, Lymphoma Research Foundation, Marcus Foundation, Tim Nesvig Family Foundation. The authors would like to thank Christine Wright, Araceli Hamlett, Cherrilyn Bautista, members of the COH Center for Biomedicine & Genetics, and Dr. Shu Mi, Dr. Ludmila Krymkaya and Vivi Tran of the COH Clinical Immunobiology Correlative Studies Laboratory for their technical assistance; and Merlita Alvarez, Lior Lewensztain and Jamie Wagner for their help in compiling data.
Footnotes
Financial Disclosures
MCJ: has major ownership interest ($10,000 or more) as a patent holder
LJC: has minor ownership interest (< $10,000) as founder and majority owner of InCellerate, Inc - a company that commercializes genetically modified T cells.
None of the other authors have anything to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kofler DM, Mayr C, Wendtner CM. Current status of immunotherapy in B cell malignancies. Curr Drug Targets. 2006;7:1371–1374. doi: 10.2174/138945006778559120. [DOI] [PubMed] [Google Scholar]
- 2.Molina A. A decade of rituximab: improving survival outcomes in non-Hodgkin’s lymphoma. Annu Rev Med. 2008;59:237–250. doi: 10.1146/annurev.med.59.060906.220345. [DOI] [PubMed] [Google Scholar]
- 3.Cesco-Gaspere M, Morris E, Stauss HJ. Immunomodulation in the treatment of haematological malignancies. Clin Exp Med. 2009;9:81–92. doi: 10.1007/s10238-009-0037-1. [DOI] [PubMed] [Google Scholar]
- 4.Timmerman JM. Immunotherapy for lymphomas. Int J Hematol. 2003;77:444–455. doi: 10.1007/BF02986612. [DOI] [PubMed] [Google Scholar]
- 5.Sadelain M, Brentjens R, Riviere I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. 2009;21:215–223. doi: 10.1016/j.coi.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riddell SR. Engineering antitumor immunity by T-cell adoptive immunotherapy. Hematology Am Soc Hematol Educ Program. 2007:250–256. doi: 10.1182/asheducation-2007.1.250. [DOI] [PubMed] [Google Scholar]
- 7.Jensen M, Tan G, Forman S, Wu AM, Raubitschek A. CD20 is a molecular target for scFvFc: zeta receptor redirected T cells: implications for cellular immunotherapy of CD20+ malignancy. Biol Blood Marrow Transplant. 1998;4:75–83. doi: 10.1053/bbmt.1998.v4.pm9763110. [DOI] [PubMed] [Google Scholar]
- 8.Cooper LJ, Topp MS, Serrano LM, et al. T-cell clones can be rendered specific for CD19: toward the selective augmentation of the graft-versus-B-lineage leukemia effect. Blood. 2003;101:1637–1644. doi: 10.1182/blood-2002-07-1989. [DOI] [PubMed] [Google Scholar]
- 9.Jensen MC, Clarke P, Tan G, et al. Human T lymphocyte genetic modification with naked DNA. J Mol Ther. 2000;1:49–55. doi: 10.1006/mthe.1999.0012. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez S, Naranjo A, Serrano LM, Chang WC, Wright CL, Jensen MC. Genetic engineering of cytolytic T lymphocytes for adoptive T-cell therapy of neuroblastoma. Journal of Gene Medicine. 2004;6:704–711. doi: 10.1002/jgm.489. [DOI] [PubMed] [Google Scholar]
- 11.Cooper LJ, Ausubel L, Gutierrez M, et al. Manufacturing of gene-modified cytotoxic T lymphocytes for autologous cellular therapy for lymphoma.[see comment] Cytotherapy. 2006;8:105–117. doi: 10.1080/14653240600620176. [DOI] [PubMed] [Google Scholar]
- 12.Park JR, Digiusto DL, Slovak M, et al. Adoptive transfer of chimeric antigen receptor redirected cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. 2007;15:825–833. doi: 10.1038/sj.mt.6300104. [DOI] [PubMed] [Google Scholar]
- 13.Akatsuka Y, Martin EG, Madonik A, Barsoukov AA, Hansen JA. Rapid screening of T-cell receptor (TCR) variable gene usage by multiplex PCR: application for assessment of clonal composition. Tissue Antigens. 1999;53:122–134. doi: 10.1034/j.1399-0039.1999.530202.x. [DOI] [PubMed] [Google Scholar]
- 14.Betts MR, Brenchley JM, Price DA, et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 15.Alexander DD, Mink PJ, Adami HO, et al. The non-Hodgkin lymphomas: a review of the epidemiologic literature. Int J Cancer. 2007;120 (Suppl 12):1–39. doi: 10.1002/ijc.22719. [DOI] [PubMed] [Google Scholar]
- 16.Rogers BB. Overview of non-Hodgkin’s lymphoma. Semin Oncol Nurs. 2006;22:67–72. doi: 10.1016/j.soncn.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 17.Arora NK, Hamilton AS, Potosky AL, et al. Population-based survivorship research using cancer registries: a study of non-Hodgkin’s lymphoma survivors. J Cancer Surviv. 2007;1:49–63. doi: 10.1007/s11764-007-0004-3. [DOI] [PubMed] [Google Scholar]
- 18.Gisselbrecht C, Bethge W, Duarte RF, et al. Current status and future perspectives for yttrium-90 ((90)Y)-ibritumomab tiuxetan in stem cell transplantation for non-Hodgkin’s lymphoma. Bone Marrow Transplant. 2007;40:1007–1017. doi: 10.1038/sj.bmt.1705868. [DOI] [PubMed] [Google Scholar]
- 19.Paolo C, Lucia F, Anna D. Hematopoietic stem cell transplantation in peripheral T-cell lymphomas. Leuk Lymphoma. 2007;48:1496–1501. doi: 10.1080/10428190701435275. [DOI] [PubMed] [Google Scholar]
- 20.Santos ES, Kharfan-Dabaja MA, Ayala E, Raez LE. Current results and future applications of radioimmunotherapy management of non-Hodgkin’s lymphoma. Leuk Lymphoma. 2006;47:2453–2476. doi: 10.1080/10428190600923140. [DOI] [PubMed] [Google Scholar]
- 21.Zinzani PL. Autologous hematopoietic stem cell transplantation in Non-Hodgkin’s lymphomas. Acta Haematol. 2005;114:255–259. doi: 10.1159/000088416. [DOI] [PubMed] [Google Scholar]
- 22.Bertoni F, Zucca E, Cavalli F. Mantle cell lymphoma. Curr Opin Hematol. 2004;11:411–418. doi: 10.1097/01.moh.0000138682.13354.da. [DOI] [PubMed] [Google Scholar]
- 23.Brody J, Advani R. Treatment of mantle cell lymphoma: current approach and future directions. Crit Rev Oncol Hematol. 2006;58:257–265. doi: 10.1016/j.critrevonc.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 24.Goy A, Feldman T. Expanding therapeutic options in mantle cell lymphoma. Clin Lymphoma Myeloma. 2007;7 (Suppl 5):S184–191. doi: 10.3816/clm.2007.s.021. [DOI] [PubMed] [Google Scholar]
- 25.Smith MR. Mantle cell lymphoma: advances in biology and therapy. Curr Opin Hematol. 2008;15:415–421. doi: 10.1097/MOH.0b013e328302c9c5. [DOI] [PubMed] [Google Scholar]
- 26.Brentjens RJ, Riviere I, Hollyman D, et al. Unexpected toxicity of cyclophosphamide followed by adoptively transferred CD19-targeted T cells in a patient with bulky CLL. Mol Ther. 2009;17:S157. [Google Scholar]
- 27.Berger C, Flowers ME, Warren EH, Riddell SR. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood. 2006;107:2294–2302. doi: 10.1182/blood-2005-08-3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berger C, Huang ML, Gough M, Greenberg PD, Riddell SR, Kiem HP. Nonmyeloablative immunosuppressive regimen prolongs In vivo persistence of gene-modified autologous T cells in a nonhuman primate model. J Virol. 2001;75:799–808. doi: 10.1128/JVI.75.2.799-808.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh H, Manuri PR, Olivares S, et al. Redirecting specificity of T-cell populations for CD19 using the Sleeping Beauty system. Cancer Res. 2008;68:2961–2971. doi: 10.1158/0008-5472.CAN-07-5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wherry EJ, Teichgraber V, Becker TC, et al. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol. 2003;4:225–234. doi: 10.1038/ni889. [DOI] [PubMed] [Google Scholar]
- 31.Gattinoni L, Powell DJ, Jr, Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol. 2006;6:383–393. doi: 10.1038/nri1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fearon DT, Manders P, Wagner SD. Arrested differentiation, the self-renewing memory lymphocyte, and vaccination. Science. 2001;293:248–250. doi: 10.1126/science.1062589. [DOI] [PubMed] [Google Scholar]
- 33.Pule MA, Savoldo B, Myers GD, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8 T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118:294–305. doi: 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.