

Abstract
Activation of the transcription factor NF-E2 related factor 2 (Nrf2) by oxidative stress induces the expression of a variety of antioxidant and anti-inflammatory genes. Yet, genetic ablation of Nrf2 was shown to protect mice from high-fat diet (HFD)-induced obesity and insulin resistance. The mechanisms that underlay this seemingly paradoxical finding remain largely unexplored.
Here we examined whether Nrf2 deficiency in myeloid cells contributes to protection against HFD-induced metabolic changes by decreasing adipose tissue inflammation. In vitro, induction of IL-1β by inflammatory stimuli was significantly reduced in Nrf2-deficient macrophages. While, inflammatory gene expression in the stromal vascular fraction was reduced in both global and chimeric Nrf2 KO mice, only global Nrf2-deficient but not bone marrow-transplanted Nrf2 chimeric mice were protected against HFD-induced adipose tissue inflammation. While global Nrf2 deficiency resulted in significantly decreased expression of inflammatory genes and PPARγ2, there was no difference when Nrf2 was absent only in myeloid cells. In vitro co-culture with adipocytes demonstrated that macrophage Nrf2 regulated inflammatory gene expression in macrophages, however, was not required to induce inflammatory gene expression in adipocytes. Finally, in contrast to global Nrf2 knock-out, Nrf2 deficiency in myeloid cells did not protect against HFD-induced insulin resistance.
Together, our data demonstrate a dominant role of nonmyeloid Nrf2 in controlling HFD-induced adipose tissue inflammation and the development of insulin resistance.
Keywords: Nrf2, inflammation, insulin resistance, bone marrow transplantation, stromal vascular fraction and adipocyte/macrophage co-culture
INTRODUCTION
Oxidative stress is involved in the development of insulin resistance and diabetes [1]. Feeding a high-fat diet (HFD) to mice causes oxidative stress in adipose tissue, even before the onset of obesity and insulin resistance [2,3]. Oxidative tissue damage is counteracted by a battery of antioxidant and detoxifying enzymes such as heme oxygenese-1, glutamate-cysteine ligase modifier subunit, glutathione S-transferase and thioredoxin reductase 1 [4]. The production of these stress-responsive enzymes is largely regulated by binding of the redox-sensitive transcription factor NF-E2 related factor 2 (Nrf2) to the antioxidant response elements (ARE) that are present in the promoter regions of these genes [5]. Nrf2 is ubiquitously expressed and failure of expression of Nrf2 disturbs the redox homeostasis and aggravates many diseases related to oxidative stress and inflammation in mice [6-10]. Accordingly, Nrf2 deficiency is detrimental in mouse models of cancer, sepsis and toxic injury [11,12]. Paradoxically, Nrf2 deficiency has recently been shown to be beneficial in murine atherosclerosis [13,14] as well as in high-fat diet (HFD)-induced obesity [15] and insulin resistance [16]. The mechanisms and cell types that confer protection of Nrf2 deficient mice against effects of a HFD have not been elucidated.
Diet-induced obesity and insulin resistance are associated with low grade inflammation in adipose tissue and infiltration of bone marrow-derived macrophages [17,18]. In diet-induced obese mice, adipocytes are shown to produce pro-inflammatory cytokines such as IL-6, which contributes to the development of insulin resistance [19]. Adipose tissue macrophages (ATMs) form crown-like structures around dying hypertrophic adipocytes, which leads to exacerbation of adipose tissue inflammation with marked secretion of inflammatory factors such as TNFα, IL-6, MCP-1 and IL-1β [20,21]. ATMs switch their phenotype from anti-inflammatory “M2” to proinflammatory “M1”, which ultimately contribute to the development of insulin resistance [22,23]. Recently, we identified a novel macrophage phenotype “Mox”, formation of which is primarily dependent on Nrf2 [24].
Here, we test the hypothesis that Nrf2 deficiency in myeloid cells reduces HFD-induced inflammation in adipose tissue leading to protection from insulin resistance. We demonstrate that Nrf2 deficiency affects inflammatory gene expression in stromal vascular fraction of the adipose tissue; however, this was not sufficient to protect mice from HFD-induced insulin resistance. Collectively, our results show that Nrf2 in nonmyeloid cells plays a dominant role in regulating diet-induced inflammation and insulin resistance in mice.
MATERIALS AND METHODS
Animals
Nrf2-deficient mice (Nrf2 KO) originally constructed in 129/SvJ background [25] had been back-crossed for more than nine generations with C57BL/6J mice [26]. C57BL/6J mice (wild type) were obtained from the Jackson laboratories, USA. All mice were housed in a temperature-controlled pathogen-free facility with 12 h day/12 h night cycles. To induce obesity and insulin resistance, 8 to 12 weeks old male mice were fed with a HFD (HFD, Bioserv, Frenchtown, NJ, USA, product# F3282) containing 35.5% fats (60% of total calories is derived from fat) for 10 weeks. Control groups were fed a normal chow diet (Harlan Teklad 8728C, South Easton, MA, USA). For bone-marrow transplantation, 8 week old wild-type mice (recipients) were lethally irradiated with two doses of 200 Rad. Then, the mice were transplanted with bone marrow from wild-type mice or Nrf2 KO mice (donors) by tail vein injection. Bone marrow isolated from one donor mouse was transplanted into 10 recipient mice so that each recipient mouse received about 5×106 bone marrow cells. The mice were given Sulfa water 2 weeks before and 3 weeks after the irradiation. To investigate diet-induced insulin resistance, four weeks after transplantation, mice were fed with HFD or chow diet for 10 more weeks. All procedures were approved by the Animal Care and Use Committee at the University of Virginia.
Immunohistochemistry on adipose tissue
The adipose tissues were fixed in formalin and embedded in paraffin, sections were cut, deparaffinized and stained with Hematoxylin and Eosin. For staining the crown-like structures, deparaffinized adipose tissue sections were treated with 0.5% hydrogen peroxide in methanol, then with normal blocking serum (Vector Laboratories, Burlingame, CA, USA), then with purified anti-Mouse MAC-2 monoclonal antibody (Cedarlane, Ontario, Canada) overnight at 4°C. Thereafter, the sections were treated with biotinylated secondary antibody (Vector Laboratories) and an avidin-biotin peroxidase complex (Vector Laboratories). Finally, the sections were developed with 3, 3′-diaminobenzidine substrate kit (Fisher Scientific, Pittsburgh, PA, USA) and counterstained using hematoxylin.
Separation of the stromal vascular fraction from adipocytes
Harvested adipose tissue was treated with Krebs-Ringer bicarbonate buffer, pH 7.4, containing 30 mM HEPES, 1% (w/v) BSA, 3 mM glucose, 200 mM adenosine and 2 mg of collagenase per gram of adipose tissue, for 1 hour at 37 °C with shaking in a water bath. Thereafter, the suspension was filtered through a 200 μm nylon mesh and centrifuged at 100g for 5 min to separate the adipocytes from the stromal vascular fraction. The stromal vascular fractions were washed twice with the same buffer without collagenase.
Real-time RT-PCR
RNA from adipose tissue and isolated adipocytes was extracted by using TRIzol and purelink RNA mini kit from Invitrogen (Carlsbad, CA, USA), while RNA from the stromal vascular fraction was isolated by using RNeasy Mini Kit (Qiagen, Valencia, CA). The liver, pancreas, lung and heart tissue samples were homogenized in RLT buffer and RNA was isolated using RNeasy Mini Kit. From these RNA samples, cDNA was synthesized by using iScript cDNA Synthesis Kit (Biorad, Hercules, CA) by following the manufacturer’s protocol. The synthesized cDNA was diluted and used for real-time PCR. Primers for real-time PCR were designed by using Primer3 (available online “http://frodo.wi.mit.edu/”), synthesized (Integrated DNA Technologies, Inc, Skokie, IL, USA) and real-time PCR was performed using SYBR GreenER qPCR SuperMix (Invitrogen) and iCycler (Biorad, Hercules, CA). The sequences of the primers are given in supplemental table 1. Product formation was monitored by MyiQ Single Color Real-Time PCR Detection System (Biorad, Hercules, CA). PCR data were analyzed using the method described by Pfaffl [27], the expression of target molecules was normalized to the expression of β2-microglobulin.
In vitro generation of 3T3-L1 adipocytes
3T3-L1 fibroblasts (ATCC# CL-173, obtained from American Type Culture collection, Manassas, VA, USA) were grown to 100% confluency in DMEM high glucose (Gibco-BRL, Grand Island, NY) with 10% new born calf serum and antibiotic-antimycotic in a 24 well plate at 37 °C in a humidified chamber containing 5% CO2 and differentiated into adipocytes as described previously [28]. Differentiation into adipocytes was confirmed by oil Red O staining [29].
Isolation and culture of bone marrow-derived macrophages
Femurs and tibias from wild-type or Nrf2 KO mice were harvested, cut at the ends and bone marrow cells were extracted and bone marrow-derived macrophages were prepared as described previously [24]. To examine the effect of inflammatory mediators, serum-starved wild-type and Nrf2 KO macrophages were treated with LPS (1 μg/ml), TNFα (10 ng/ml), OxPAPC (oxidized 1-palmitoyl-2-arachidonoyl-sn-3-glycero-phosphorylcholine, 50 μg/ml [24]) or phorbol-12-myristate-13-acetate (PMA, 100 nM) for 4 hours and IL-1β gene expression was analyzed using real-time RT-PCR.
Adipocyte/macrophage co-culture
Wild-type or Nrf2 KO macrophages were co-cultured with differentiated 3T3-L1 adipocytes in a 24 well plate containing DMEM high glucose with 10% fetal bovine serum and 1% antibiotic-antimycotic, at a density of 105 cells in 12 mm inserts containing polyester membranes with pore size of 0.4 μm (Costar, Corning, NY, USA). For comparison, wild-type and Nrf2 KO macrophages, and differentiated adipocytes were cultured separately. The co-cultures and the individual cell cultures were kept for 12 or 24 h in a humidified chamber containing 5% CO2 at 37 °C. Thereafter, RNA was isolated from adipocytes and macrophages, using TRIzol and purelink RNA mini kit (Invitrogen), respectively. CDNA was synthesized and mRNA expression quantified by real-time RT-PCR, as described above.
Glucose and insulin tolerance tests
For glucose tolerance tests, mice were fasted for 12 h. Initial blood glucose levels were recorded by tail clipping and testing a drop of blood using One-touch Ultra blood glucose monitoring system (Milpitas, CA, USA). Mice were intraperitoneally injected with 1g/kg body weight of D-glucose and blood glucose levels were recorded at 10, 20, 30, 60, 90 and 120 min. For insulin tolerance tests, mice were fasted for 4 h, initial blood glucose levels were recorded, and 0.5 U/kg body weight of insulin (Humulin R, Eli Lilly and Company, Indianapolis, IN, USA) was injected intraperitoneally and blood glucose was monitored at 10 and 20 min as above.
Statistical analyses
Statistical analyses were performed using OriginPro 7.5 or Microsoft Excel and data were expressed as mean values with S.E.M. or S.D. To calculate statistical significance, one way analysis of variances (ANOVA) or Student’s t test was used. A p-value <0.05 was considered significant.
RESULTS
Nrf2 is required for IL-1β expression in murine bone-marrow-derived macrophages
To investigate the role of Nrf2 in the regulation of inflammatory gene expression, we treated wild-type or Nrf2 KO macrophages with inflammatory mediators LPS, TNFα, OxPAPC or PMA. While PMA induced IL-1β expression in both wild-type and Nrf2 KO macrophages, LPS, TNFα and OxPAPC-induced IL-1β mRNA expression was significantly reduced in Nrf2 KO macrophages (Fig. 1). These data indicate that Nrf2 regulates transcription of IL-1β in macrophages.
Figure 1.

IL-1β gene expression is impaired in Nrf2 KO macrophages. Wild-type and Nrf2 KO macrophages were stimulated with control medium (co), 1 μg/ml of LPS, 10 ng/ml of TNFα, 50 μg/ml of OxPAPC or 100 nM of PMA for 4 hours and mRNA expression of IL-1β was examined by real-time RT-PCR. Values are expressed as means + SD and “*” indicates p<0.05, n=4.
Global but not myeloid Nrf2 deficiency attenuates HFD-induced adipose tissue inflammation
To examine the role of Nrf2 in myeloid cells in mice, we transplanted either Nrf2 KO or wild-type bone marrow into lethally irradiated wild-type mice resulting in Nrf2 KO chimeric mice (KO-WT) or controls (WT-WT), respectively. Successful transplantation was confirmed by significantly lower copy numbers of Nrf2 mRNA in the blood and stromal vascular fraction of adipose tissue of KO-WT chimeric mice compared to the WT-WT control mice (Suppl. Fig. 1A and B). No significant difference in the white blood cell count of the KO-WT and WT-WT mice, feeding either chow or HFD was observed (Suppl. Fig. 1C).
At the onset of feeding the HFD, age-matched Nrf2 KO mice already showed slightly lower body weight compared to the wild-type mice (25.5±0.7 g vs. 23.3±0.6 g, p=0.02). As reported earlier [15], Nrf2 KO mice were protected from high-fat diet-induced obesity (Suppl. Fig. 2A), as indicated by lower body weight gain (36.9±3.8 %) compared to wild-type mice (50.5±3.7 %, p=0.02). Moreover, we found significantly lower fat masses in Nrf2 KO mice feeding either chow or HFD (Suppl. Fig. 2B). Although HFD increased body weight of the bone marrow-transplanted mice at a similar rate to non-transplanted mice, we did not find significant differences in the body weight or epididymal fat weight of KO-WT and WT-WT mice (Suppl. Fig. 2C and D).
To study the effect of high-fat diet on expression of nonmyeloid Nrf2, we quantified Nrf2 mRNA in adipose tissue, isolated adipocytes from adipose tissue, liver, pancreas, lung and heart of KO-WT mice fed with chow or high-fat diet. The results revealed lower Nrf2 expression in adipose tissue, adipocytes and liver, whereas, no difference in pancreas, heart and lung of high-fat diet-fed mice compared to the chow diet-fed mice (Suppl. Fig. 3). These results suggest that high-fat diet regulates Nrf2 expression in adipocytes and liver.
Increased macrophage infiltration and appearance of crown-like structures (CLS) are hallmarks of high-fat diet-induced inflammation in adipose tissue [30]. Immunohistochemistry in wild-type mice on HFD using mac-2 antibodies on adipose tissue sections revealed the formation of CLS, which were barely detectable in Nrf2 KO mice (Fig. 2A). We did not find any CLS in the adipose tissue of mice on chow diet (not shown). Consistent with the number of CLS, mRNA expression of the macrophage marker genes CD68 and F4/80 in whole adipose tissue was significantly lower in Nrf2 KO mice (Fig. 2B). Moreover, mRNA levels of TNFα and monocyte chemotactic protein-1 (MCP-1) were significantly reduced in Nrf2 KO mice on HFD. However, in contrast to the global Nrf2 KO mice, no differences in the expression level of F4/80, CD68, TNFα and MCP-1 (Fig. 2C) were observed in the total adipose tissue of HFD-fed WT-WT and KO-WT mice.
Figure 2.
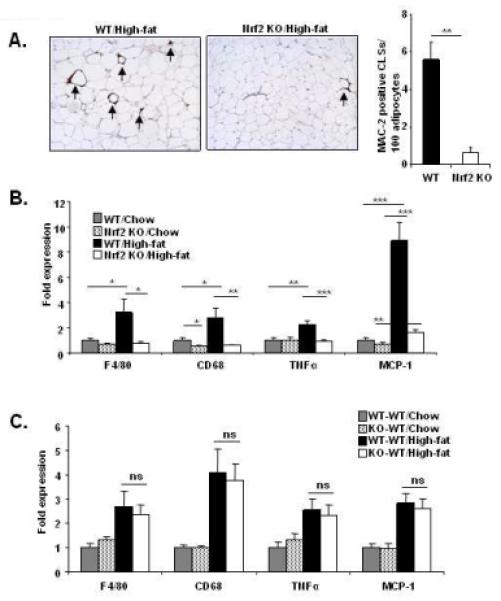
Global, but not the myeloid cell-specific Nrf2 deficiency protects mice from diet-induced adipose tissue inflammation. (A) Left and central panel: MAC-2 staining of adipose tissue cross-sections of wild-type and Nrf2 KO mice on HFD. Crown-like structures are indicated with arrows. Right panel: number of crown-like structures per 100 adipocytes (adipose tissues examined from 3 mice per group). (B) Messenger RNA expression of macrophage marker genes (CD68 and F4/80) and inflammatory genes TNFα and MCP-1 (n=9-11 per group) of wild-type (WT) and Nrf2 KO mice on chow or HFD. (C) Messenger RNA expression of CD68, F4/80, TNFα and MCP-1 (n=9-10 per group) of the control (WT-WT) and Nrf2 KO chimeric (KO-WT) mice on chow or HFD after bone-marrow transplantation. “n” represents number of mice. Values are expressed as means + SEM, and “*”, “**” and “***” indicate p<0.05, 0.01 and 0.001, respectively. “ns” indicates “not significantly different” with p-values ≥ 0.05.
Effects of global and myeloid Nrf2 deficiency on inflammatory gene expression in the stromal vascular fraction and adipocytes
The stromal vascular fraction (SVF) isolated from the adipose tissue is rich in macrophages and significantly contributes to adipose tissue inflammation [22]. In the SVF of global Nrf2 KO mice, expression of M1 (pro-inflammatory) genes IL-1β, IL-6, CXCL1, CXCL2 and MCP-1 was significantly lower, and TNFα showed a trend towards lower expression, when compared to the wild-type mice on HFD (Fig. 3A). Expression of Nrf2-dependent Mox macrophage markers Srxn1 and HO-1 [24] was also significantly lower in the SVF from Nrf2 KO mice. In contrast, no significant difference was observed in the expression of M2 (anti-inflammatory) genes Arg1, Mgl1 and Mgl2.
Figure 3.
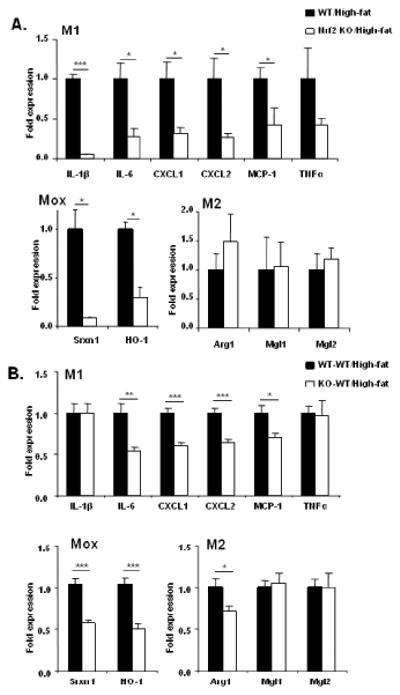
Both global and myeloid cell-specific Nrf2 deficiency decreases inflammation in the stromal vascular fraction (SVF) of adipose tissue. (A) Messenger RNA expression of M1, Mox and M2 macrophage marker genes in the SVF from wild-type and Nrf2 KO mice on HFD (n=5 per group). (B) Messenger RNA expression of M1, Mox and M2 macrophage marker genes in the SVF from control (WT-WT) and Nrf2 KO chimeric mice (KO-WT) on HFD (n=9-10 per group). “n” represents number of mice. Values are expressed as means + SEM and “*”, “**” and “***” indicate p<0.05, 0.01 and 0.001, respectively.
Analysis of SVF in the chimeric myeloid Nrf2 deficient (KO-WT) mice showed that expression of IL-6, MCP-1, CXCL1 and CXCL2 were significantly lower, while the levels of TNFα and IL-1β were not different (Fig. 3B). Moreover, expression of Nrf2-dependent Mox markers HO-1 and Srxn1 were significantly lower. Expression of M2 genes Mgl1 and Mgl2 was not different between the wild-type and Nrf2 KO chimeric mice, although the level of Arg1 was lower in the SVF from Nrf2 KO chimeric mice.
Examination of isolated adipocytes revealed that expression of TNFα, IL-6, IL-1β, and MCP-1 was significantly lower in adipocytes from global Nrf2 KO compared to wild-type mice on HFD. Expression of the adipogenic factor PPARγ was significantly reduced in global Nrf2 KO mice, as previously reported [15], expression of SREBP1c was lower, whereas expression of adiponectin and CEBPα was not different (Fig. 4A). In contrast, expression of none of these genes was different between myeloid Nrf2 KO (KO-WT) and control (WT-WT) bone marrow-transplanted mice (Fig. 4B). These results indicate that Nrf2 in macrophages is not required for cellular cross talk to adipocytes.
Figure 4.

Global, but not myeloid Nrf2 deficiency in mice attenuates diet-induced inflammation in adipocytes. (A) Messenger RNA expression of inflammatory and adipogenic genes in adipocytes isolated from wild-type and Nrf2 KO mice on HFD (n=5 per group). (B) Messenger RNA expression of inflammatory genes and adipogenic genes in adipocytes isolated from WT-WT and KO-WT mice on HFD (n=9-10 per group). Values are expressed as means + SEM, and “*”, “**” and “***” indicate p<0.05, 0.01 and 0.001, respectively.
To mimic cellular cross talk in inflamed adipose tissue, we co-cultured bone marrow-derived macrophages with 3T3-L1 adipocytes in vitro, so that we could analyze inflammatory gene expression in either of cell types separately (Fig. 5A). Co-culture with adipocytes induced mRNA expression of IL-1β and MCP-1 at 12 h and 24 h in wild-type macrophages, which was significantly reduced in Nrf2 deficient macrophages (Fig. 5B). Conversely, co-culture with wild-type or Nrf2 KO macrophages induced mRNA expression of IL-6 and MCP-1 in adipocytes by 12 h, residing to baseline by 24 h (Fig. 5C). These data corroborate our in vivo findings, demonstrating that cross talk between macrophages and adipocytes is not dependent on macrophage Nrf2.
Figure 5.
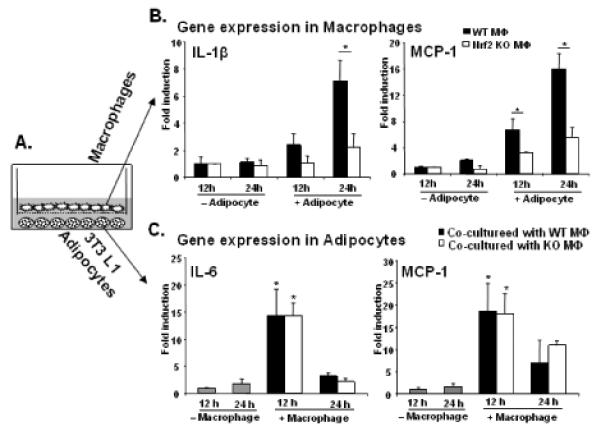
Nrf2 deficiency impairs inflammatory gene expression in macrophages in adipocytes/macrophage co-cultures. (A) Schematic presentation of 3T3-L1 adipocyte and macrophage co-culture. After 12 and 24 hrs of co-culture, gene expression was analyzed using real-time RT-PCR in macrophages (B) or in adipocytes (C). Values are expressed as means + SD and “*” indicates p<0.05, n=4.
Altogether, these results demonstrate that although expression of inflammatory genes is reduced in the SVF from Nrf2 KO mice or chimeric KO-WT mice, this was not sufficient to decrease overall adipose tissue inflammation. Moreover, these results strongly suggest that Nrf2 deficiency mainly in the nonmyeloid compartment protects mice from overall adipose tissue inflammation and thus may impact the development of insulin resistance.
Global but not myeloid-selective Nrf2 deficiency protects mice from diet-induced insulin resistance
To investigate whether global Nrf2 deficiency affects diet-induced insulin resistance, we examined fasting blood glucose and serum insulin, and performed glucose and insulin tolerance tests in chow or HFD-fed wild-type and global Nrf2 KO mice. The fasting blood glucose levels were significantly lower in Nrf2 KO mice (152±9 mg/dl) compared to wild-type mice on HFD (183±10 mg/dl, p=0.02), while there were no significant differences on chow diet. Glucose tolerance tests revealed that the Nrf2 KO mice cleared the excess glucose faster than the wild-type mice, irrespective of the diet (Fig. 6A). Despite the fact that fasting insulin levels were similar in Nrf2 KO and wild-type mice, insulin tolerance tests showed that the Nrf2 KO mice were more sensitive to insulin compared to the wild-type mice, irrespective of the diet (not shown).
Figure 6.

Global Nrf2 deficiency protects mice from diet-induced insulin resistance. (A) Glucose tolerance test (GTT) and area under the curve (AUC) in arbitrary units (AU) (n=17-18 per group) of wild-type (WT) and Nrf2 KO mice on chow or HFD. (B) Glucose tolerance test and area under the curve in arbitrary units of control (WT-WT) and Nrf2 KO chimeric mice (KO-WT) on chow or HFD (n=9-10 per group). “n” represents number of mice. Values are expressed as means ± SEM and “*” and “**” indicate p<0.05 and 0.01 respectively. “ns” indicates “not significantly different” with p-values ≥ 0.05.
We next examined whether Nrf2 deficiency in myeloid cells would prevent diet-induced insulin resistance, using bone marrow transplanted mice. In either of the diets, the fasting blood glucose and serum insulin levels of KO-WT and WT-WT mice were similar (not shown). Moreover, the KO-WT mice cleared glucose at a similar rate as the WT-WT mice (Fig. 6B). These results demonstrate that selective Nrf2 deficiency in myeloid cells was not sufficient to protect mice from diet-induced insulin resistance and support a dominant role of nonmyeloid Nrf2 in this process.
DISCUSSION
Oxidative stress and inflammation have both been linked to the development of chronic metabolic diseases such as type 2 diabetes [31,32]. Since global deficiency of the redox-sensitive transcription factor Nrf2 in mice was shown to be protective in a mouse model of diet-induced obesity and insulin resistance, we examined whether Nrf2 in macrophages contributes to this effect by controlling inflammation in obese adipose tissue.
Previous reports from our laboratory and others have demonstrated a role for Nrf2 in regulating inflammatory gene expression. We have shown that expression of IL-1β mRNA was significantly lower in Nrf2 KO macrophages compared to wild-type macrophages when treated with LPS and INFγ [24], and that Nrf2 regulates expression of the cytokine IL-8 in human dermal fibroblasts [33]. Treatment of macrophages with oxidized phospholipids not only induces IL-1β gene expression, it also enhances secretion of mature IL-1β [24]. A recent study showed Nrf2-mediated IL-1β protein synthesis is dependent on activation of NLRP3 inflammasome [34]. Here, we have shown attenuated IL-1β gene expression in Nrf2-deficient macrophages in response to oxidized phospholipids or upon stimulation with LPS or TNFα, but not with PMA, further supporting a regulatory role of Nrf2 in inflammation.
Several mechanisms may account for regulation of inflammatory gene expression by Nrf2: A recent report demonstrated ARE-dependent transcriptional regulation of IL-6 in liver carcinoma cell line HepG2 and in mice livers [35]. Another possible mechanism is trans-activation of IL-1β gene expression by Nrf2. We did not find a putative ARE in 5 kb upstream of IL-1β promoter. However, Nrf2 may regulate IL-1β promoter irrespective of presence of an ARE, as it has been shown for Nrf2-dependent gene epidermal fatty acid-binding protein [36] which does not contain an ARE. Alternatively, we have shown that Nrf2 deficiency led to decreased synthesis of glutathione in macrophages [24]. Murata et al. have shown that reduced content of intracellular glutathione in macrophages affects their inflammatory status [37]. These findings together suggest that Nrf2 may indirectly regulate inflammatory gene expression by modulating intra-cellular glutathione status.
To examine whether Nrf2-deficiency in macrophages is sufficient to protect mice from diet-ginduced adipose tissue inflammation, we transplanted Nrf2 KO or wild-type bone marrow into lethally irradiated wild-type mice. Challenging chimeric mice with HFD revealed no differences in development of obesity, while global Nrf2 deficiency protected mice from diet-induced obesity.
Global Nrf2 deficiency resulted in drastically reduced inflammation in adipose tissue of mice on a HFD: adipose tissue from Nrf2 KO mice contained fewer macrophages, demonstrating a marked decrease in expression levels of macrophage marker genes F4/80 and CD68. Moreover, expression of MCP-1 was significantly lower compared to the wild-type mice. MCP-1 has been shown to attract CCR2-positive monocytes to invade adipose tissue [38] and aggravate insulin resistance [39]. Lower expression of MCP-1 could account for the decreased macrophage content in the adipose tissue of Nrf2 KO mice. In the HFD-fed bone-marrow transplanted mice, despite similar expression levels of F4/80 and CD68, inflammatory gene expression was reduced in the macrophage-rich SVF of KO-WT mice. Since, there were no weight differences in the WT-WT and KO-WT chimeric mice, decreased inflammation in SVF of KO-WT mice can not be attributed to reduced diet-induced obesity, and, thus would be an effect of Nrf2 deficiency in myeloid cells. However, decreased inflammation in SVF was not sufficient to protect mice from overall adipose tissue inflammation.
While adipocytes isolated from global Nrf2 KO mice on HFD displayed markedly decreased inflammatory gene expression compared to wild-type mice, adipocytes from the bone marrow-transplanted mice did not show any differences. These data indicate an important role for macrophage/adipocyte cross talk in adipose tissue inflammation. To further investigate the role of Nrf2 in macrophages in cellular cross talk, we mimicked the in vivo situation of inflamed adipose tissue using an in vitro adipocyte/macrophage co-culture model. While mere co-culture induced expression of inflammatory genes in both cell types, Nrf2-deficient macrophages synthesized significantly lower levels of pro-inflammatory chemokines compared to the wild-type macrophages. These data demonstrate that cross talk between macrophages and adipocyte is not dependent on the presence of Nrf2 in macrophages and supports a dominant role of nonmyeloid Nrf2 in adipose tissue inflammation.
Examining gene expression of master regulators of adipogenesis, we found that expression of PPARγ2 was almost abrogated in adipocytes isolated from Nrf2 KO mice on HFD. These data are in agreement with supports a previous findings by Pi et al. demonstrating that Nrf2 regulates PPARγ2 expression [15]. However, PPARγ2 expression was similar in adipocytes of bone marrow chimeras on HFD, suggesting a role of Nrf2 in nonmyeloid cells in the regulation of diet-induced adipogenesis.
Recently, Chartoumpekis et al. reported that Nrf2 deficiency protects mice from insulin resistance on long-term (25.7 weeks/180 days) HFD feeding [16]. When we fed HFD to wild-type and Nrf2 KO mice for 10 weeks, we observed significant differences in insulin resistance: wild-type mice developed insulin resistance in response to HFD, while global Nrf2 KO mice were protected. However, the WT-WT and KO-WT bone marrow transplanted mice developed insulin resistance and adipose tissue inflammation to a similar level in response to HFD. These data demonstrate that though Nrf2 deficiency decreased inflammatory gene expression in myeloid cells, it was not sufficient to protect mice from insulin resistance. Moreover, this also suggests a dominant role of nonmyeloid Nrf2 in development of insulin resistance.
Collectively, we show that global Nrf2 deficiency protects mice from insulin resistance and adipose tissue inflammation. We suggest both the reduced macrophage infiltration in the adipose tissue and impaired inflammatory gene expression in macrophages may account for protection from insulin resistance in the global Nrf2 KO mice. Our studies reveal a permissive role of Nrf2 in the myeloid cells and a dominant role of Nrf2 in nonmyeloid compartment in the development of diet-induced adipose tissue inflammation and insulin resistance.
Supplementary Material
Highlights.
Nrf2 deficiency impaired diet-induced inflammation and insulin resistance.
Myeloid Nrf2 deficiency did not affect pro-inflammatory gene expression in adipocytes.
Cross talk between macrophages and adipocytes was independent of macrophage Nrf2.
Myeloid Nrf2 deficiency was not sufficient to protect mice from diet-induced insulin resistance.
ACKNOWLEDGEMENTS
We thank Rama Satya Vani Nandula, Jenny Han and Dr. Suseela Srinivasan for technical support. We further thank Dr. Susanna R. Keller for help with glucose tolerance tests, and adipocyte and SVF isolation from adipose tissue; Dr. Thurl E. Harris for help with adipocyte differentiation of 3T3-L1 fibroblasts and Dr. Kyle L. Hoehn for consulting on insulin tolerance tests. This work was supported by the National Institute of Health (R01-HL-084422-01, to N.L.) and the American Heart Association (09POST2210019, to A.K.M.). No potential conflicts of interest relevant to this article were reported.
ABBREVIATIONS
- Nrf2
NF-E2 related factor 2
- WT
Wild-type
- KO
Knock out
- PAPC
1-palmitoyl-2-arachidonoyl-sn-3-glycero-phosphorylcholine
- OxPAPC
Oxidized PAPC
- HO-1
Heme oxygenase −1
- Srxn1
Sulfiredoxin-1
- IL-1β
Interleukin-1β
- IL-6
Interleukin-6
- CXCL
Chemokine (C-X-C motif) ligand
- MCP-1
Monocyte chemotactic protein-1
- TNFα
Tumor necrosis factor α
- Arg1
Arginase 1
- Mgl
Macrophage galactose N-acetylgalactosamine-specific lectin
- LPS
Lipopolysaccharide
- PMA
Phorbol 12-myristate 13-acetate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Rains JL, Jain SK. Oxidative stress, insulin signaling, and diabetes. Free Radic. Biol. Med. 2011;50:567–575. doi: 10.1016/j.freeradbiomed.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hoehn KL, Salmon AB, Hohnen-Behrens C, Turner N, Hoy AJ, Maghzal GJ, Stocker R, Van RH, Kraegen EW, Cooney GJ, Richardson AR, James DE. Insulin resistance is a cellular antioxidant defense mechanism. Proc. Natl. Acad. Sci. U. S. A. 2009;106:17787–17792. doi: 10.1073/pnas.0902380106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Matsuzawa-Nagata N, Takamura T, Ando H, Nakamura S, Kurita S, Misu H, Ota T, Yokoyama M, Honda M, Miyamoto K, Kaneko S. Increased oxidative stress precedes the onset of high-fat diet-induced insulin resistance and obesity. Metabolism. 2008;57:1071–1077. doi: 10.1016/j.metabol.2008.03.010. [DOI] [PubMed] [Google Scholar]
- [4].Itoh K, Ishii T, Wakabayashi N, Yamamoto M. Regulatory mechanisms of cellular response to oxidative stress. Free Radic. Res. 1999;31:319–324. doi: 10.1080/10715769900300881. [DOI] [PubMed] [Google Scholar]
- [5].Zhang DD. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab Rev. 2006;38:769–789. doi: 10.1080/03602530600971974. [DOI] [PubMed] [Google Scholar]
- [6].Aleksunes LM, Manautou JE. Emerging role of Nrf2 in protecting against hepatic and gastrointestinal disease. Toxicol. Pathol. 2007;35:459–473. doi: 10.1080/01926230701311344. [DOI] [PubMed] [Google Scholar]
- [7].Innamorato NG, Rojo AI, Garcia-Yague AJ, Yamamoto M, de Ceballos ML, Cuadrado A. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J. Immunol. 2008;181:680–689. doi: 10.4049/jimmunol.181.1.680. [DOI] [PubMed] [Google Scholar]
- [8].Li YJ, Takizawa H, Azuma A, Kohyama T, Yamauchi Y, Takahashi S, Yamamoto M, Kawada T, Kudoh S, Sugawara I. Disruption of Nrf2 enhances susceptibility to airway inflammatory responses induced by low-dose diesel exhaust particles in mice. Clin. Immunol. 2008;128:366–373. doi: 10.1016/j.clim.2008.05.005. [DOI] [PubMed] [Google Scholar]
- [9].Mochizuki M, Ishii Y, Itoh K, Iizuka T, Morishima Y, Kimura T, Kiwamoto T, Matsuno Y, Hegab AE, Nomura A, Sakamoto T, Uchida K, Yamamoto M, Sekizawa K. Role of 15-deoxy delta(12,14) prostaglandin J2 and Nrf2 pathways in protection against acute lung injury. Am. J. Respir. Crit Care Med. 2005;171:1260–1266. doi: 10.1164/rccm.200406-755OC. [DOI] [PubMed] [Google Scholar]
- [10].Kim J, Cha YN, Surh YJ. A protective role of nuclear factor-erythroid 2-related factor-2 (Nrf2) in inflammatory disorders. Mutat. Res. 2010;690:12–23. doi: 10.1016/j.mrfmmm.2009.09.007. [DOI] [PubMed] [Google Scholar]
- [11].Reddy NM, Suryanarayana V, Kalvakolanu DV, Yamamoto M, Kensler TW, Hassoun PM, Kleeberger SR, Reddy SP. Innate immunity against bacterial infection following hyperoxia exposure is impaired in NRF2-deficient mice. J. Immunol. 2009;183:4601–4608. doi: 10.4049/jimmunol.0901754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, Biswal S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Invest. 2006;116:984–995. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sussan TE, Jun J, Thimmulappa R, Bedja D, Antero M, Gabrielson KL, Polotsky VY, Biswal S. Disruption of Nrf2, a key inducer of antioxidant defenses, attenuates ApoE-mediated atherosclerosis in mice. PLoS. One. 2008;3:e3791. doi: 10.1371/journal.pone.0003791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Barajas B, Che N, Yin F, Rowshanrad A, Orozco LD, Gong KW, Wang X, Castellani LW, Reue K, Lusis AJ, Araujo JA. NF-E2-Related Factor 2 Promotes Atherosclerosis by Effects on Plasma Lipoproteins and Cholesterol Transport That Overshadow Antioxidant Protection. Arterioscler. Thromb. Vasc. Biol. 2010 doi: 10.1161/ATVBAHA.110.210906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pi J, Leung L, Xue P, Wang W, Hou Y, Liu D, Yehuda-Shnaidman E, Lee C, Lau J, Kurtz TW, Chan JY. Deficiency in the nuclear factor E2-related factor-2 transcription factor results in impaired adipogenesis and protects against diet-induced obesity. J. Biol. Chem. 2010;285:9292–9300. doi: 10.1074/jbc.M109.093955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chartoumpekis DV, Ziros PG, Psyrogiannis AI, Papavassiliou AG, Kyriazopoulou VE, Sykiotis GP, Habeos IG. Nrf2 Represses FGF21 During Long-Term High-Fat Diet-Induced Obesity in Mice. Diabetes. 2011;60:2465–2473. doi: 10.2337/db11-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J. Clin. Invest. 2006;116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sabio G, Das M, Mora A, Zhang Z, Jun JY, Ko HJ, Barrett T, Kim JK, Davis RJ. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science. 2008;322:1539–1543. doi: 10.1126/science.1160794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lagathu C, Yvan-Charvet L, Bastard JP, Maachi M, Quignard-Boulange A, Capeau J, Caron M. Long-term treatment with interleukin-1beta induces insulin resistance in murine and human adipocytes. Diabetologia. 2006;49:2162–2173. doi: 10.1007/s00125-006-0335-z. [DOI] [PubMed] [Google Scholar]
- [21].Strissel KJ, Stancheva Z, Miyoshi H, Perfield JW, DeFuria J, Jick Z, Greenberg AS, Obin MS. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes. 2007;56:2910–2918. doi: 10.2337/db07-0767. [DOI] [PubMed] [Google Scholar]
- [22].Lumeng CN, Deyoung SM, Bodzin JL, Saltiel AR. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes. 2007;56:16–23. doi: 10.2337/db06-1076. [DOI] [PubMed] [Google Scholar]
- [23].Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest. 2007;117:175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kadl A, Meher AK, Sharma PR, Lee MY, Doran AC, Johnstone SR, Elliott MR, Gruber F, Han J, Chen W, Kensler T, Ravichandran KS, Isakson BE, Wamhoff BR, Leitinger N. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ. Res. 2010;107:737–746. doi: 10.1161/CIRCRESAHA.109.215715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- [26].Shin S, Wakabayashi J, Yates MS, Wakabayashi N, Dolan PM, Aja S, Liby KT, Sporn MB, Yamamoto M, Kensler TW. Role of Nrf2 in prevention of high-fat diet-induced obesity by synthetic triterpenoid CDDO-imidazolide. Eur. J. Pharmacol. 2009;620:138–144. doi: 10.1016/j.ejphar.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Frost SC, Lane MD. Evidence for the involvement of vicinal sulfhydryl groups in insulin-activated hexose transport by 3T3-L1 adipocytes. J. Biol. Chem. 1985;260:2646–2652. [PubMed] [Google Scholar]
- [29].Tobe K, Kasuga M, Kitasato H, Takaku F, Takano T, Segawa K. Differential effects of DNA tumor virus nuclear oncogene products on adipocyte differentiation. FEBS Lett. 1987;215:345–349. doi: 10.1016/0014-5793(87)80175-8. [DOI] [PubMed] [Google Scholar]
- [30].West M. Dead adipocytes and metabolic dysfunction: recent progress. Curr. Opin. Endocrinol. Diabetes Obes. 2009;16:178–182. doi: 10.1097/med.0b013e3283292327. [DOI] [PubMed] [Google Scholar]
- [31].Rains JL, Jain SK. Oxidative stress, insulin signaling, and diabetes. Free Radic. Biol. Med. 2011;50:567–575. doi: 10.1016/j.freeradbiomed.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 2011;11:98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- [33].Gruber F, Mayer H, Lengauer B, Mlitz V, Sanders JM, Kadl A, Bilban M, de MR, Wagner O, Kensler TW, Yamamoto M, Leitinger N, Tschachler E. NF-E2-related factor 2 regulates the stress response to UVA-1-oxidized phospholipids in skin cells. FASEB J. 2010;24:39–48. doi: 10.1096/fj.09-133520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Freigang S, Ampenberger F, Spohn G, Heer S, Shamshiev AT, Kisielow J, Hersberger M, Yamamoto M, Bachmann MF, Kopf M. Nrf2 is essential for cholesterol crystal-induced inflammasome activation and exacerbation of atherosclerosis. Eur. J. Immunol. 2011;41:2040–2051. doi: 10.1002/eji.201041316. [DOI] [PubMed] [Google Scholar]
- [35].Wruck CJ, Streetz K, Pavic G, Goetz ME, Tohidnezhad M, Brandenburg LO, Varoga D, Eickelberg O, Herdegen T, Trautwein C, Chan K, Kan YW, Pufe T. Nrf2 induces interleukin-6 (IL-6) expression via an antioxidant response element within the IL-6 promoter. J. Biol. Chem. 2010;286:4493–9. doi: 10.1074/jbc.M110.162008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kitteringham NR, Abdullah A, Walsh J, Randle L, Jenkins RE, Sison R, Goldring CE, Powell H, Sanderson C, Williams S, Higgins L, Yamamoto M, Hayes J, Park BK. Proteomic analysis of Nrf2 deficient transgenic mice reveals cellular defence and lipid metabolism as primary Nrf2-dependent pathways in the liver. J. Proteomics. 2010;73:1612–1631. doi: 10.1016/j.jprot.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Murata Y, Amao M, Yoneda J, Hamuro J. Intracellular thiol redox status of macrophages directs the Th1 skewing in thioredoxin transgenic mice during aging. Mol. Immunol. 2002;38:747–757. doi: 10.1016/s0161-5890(01)00111-0. [DOI] [PubMed] [Google Scholar]
- [38].Chen A, Mumick S, Zhang C, Lamb J, Dai H, Weingarth D, Mudgett J, Chen H, MacNeil DJ, Reitman ML, Qian S. Diet induction of monocyte chemoattractant protein-1 and its impact on obesity. Obes. Res. 2005;13:1311–1320. doi: 10.1038/oby.2005.159. [DOI] [PubMed] [Google Scholar]
- [39].Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashira K, Kasuga M. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Invest. 2006;116:1494–1505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.