

Abstract
Objective
Hyperlipidemia exacerbates ischemic stroke outcome and increased CD36 expression in the post-ischemic brain as well as in peripheral monocytes/macrophages. By exchanging bone marrow-derived cells between CD36-expressing and –deficient mice, this study investigates the contribution of peripheral CD36 vs brain CD36 to stroke pathology in hyperlipidemia.
Methods
Following bone marrow transplantation, mice were fed a high fat diet for 11 weeks, and then subjected to ischemic stroke. Stroke outcome, expression of brain CD36, MCP-1, CCR2, and plasma MCP-1 levels were determined at 3 days post-ischemia. CD36 and CCR2 expression were also determined in splenocytes incubated with serum obtained from CD36-expressing or CD36-deficient mice.
Results
Infiltrating immune cells from the periphery are the major source of CD36 in the post-ischemic brain and contribute to stroke-induced brain injury. This CD36 effect was dependent on the modulation of MCP-1 and CCR2 expression in peripheral immune cells as well as CD36-expressing cells in the host brain.
Interpretation
This study demonstrates that CD36 expressed in the periphery and brain synergize in ischemic brain injury through regulation of the MCP-1/CCR2 chemokine axis in hyperlipidemic conditions.
Introduction
The pathology of stroke involves multiple events including excitotoxicity, oxidative stress, and inflammation.1-3 Post-ischemic inflammation attracts mononuclear cells, including monocytes/macrophages, to the area of injury.4, 5 The persistent presence of mononuclear cells in the infarct zone suggests that peripheral immune cells play a role in ischemia-induced inflammation and acute brain injury as an inadvertent consequence of their function in controlling pathogens, clearing debris and wound healing.6-8
CD36, a class B scavenger receptor, functions in regulating inflammation, innate immunity, and lipid metabolism.9-13 Besides its expression on microglia and microvascular endothelial cells in the brain, CD36 is expressed on multiple cell types in the periphery including monocytes/macrophages, platelets, cardiomyocytes, hepatocytes, adipocytes and proximal tubule epithelial cells of the kidney.11, 14-18 CD36 recognizes a variety of structurally distinct ligands; among these are lipid-based ligands such as oxidized or modified low density lipoprotein (oxLDL, mLDL). These ligands are increased in hyperlipidemic conditions and in injured tissues where oxidative products from cells and tissues are released.19-21 In addition to its role in the pathogenesis of atherosclerosis and neurodegenerative diseases,9, 22, 23 we previously reported that this receptor is implicated in ischemic brain injury.24-26 CD36 was shown to be localized in CD11b+ microglia/macrophages in the infarct territory.25 Furthermore, our previous study suggested a role for peripheral CD36 in hyperlipidemia-exacerbated brain injury. CD36 expression is increased in peripheral monocytes/macrophages in hyperlipidemic mice, and the increase was associated with larger infarct size and up-regulation of CD36 and monocyte chemoattractant protein-1 (MCP-1) expression in the ischemic brain27.
MCP-1 is a member of the CC chemokine family and increases in expression upon tissue injury.28, 29 Major cell types that secrete chemokines include microvascular endothelial cells, microglia, astrocytes, neurons and monocytes/macrophages.30-37 In cells that express the G-protein coupled receptor CCR2, MCP-1 functions in the trafficking of inflammatory cells to the injury site.38 The importance of the MCP-1/CCR2 axis in monocyte/macrophage trafficking to the post-ischemic brain was shown by studies in which absence of CCR2 or MCP-1 reduced infarct size and monocyte/macrophage recruitment.39-41 In the hyperlipidemic condition, an involvement of CCR2 on monocyte recruitment in the injured tissue also has been reported.42 Several studies have indicated a link between CD36 and chemokine expression: CD36 ligands induced CC and CXC chemokine production, and the increases were attenuated in CD36-deficient mice.13, 27, 43
Macrophage CD36 has been implicated in promoting atherosclerosis through the uptake of lipid-based ligands and foam cell formation12, 13, 44-49, albeit these findings were initially controversial.43, 50 The contribution to ischemic injury of CD36 in infiltrating peripheral immune cells is confounded by the presence of CD36 in the brain as well as peripheral immune cells. The current study addresses the impact of peripheral CD36 on brain injury and defines a role for peripheral versus brain CD36 in hyperlipidemia-induced exacerbation of ischemic stroke outcomes. By exchanging bone marrow-derived cells between CD36-expressing hyperlipidemic ApoE knock-out (AKO) and CD36-deficient ApoE/CD36 double KO (DKO) mice, we report a necessary role for peripheral CD36 in hyperlipidemia-induced exacerbation of ischemic stroke outcomes and an additional requirement for brain CD36 to synergize the peripheral CD36 effect.
Subjects and Methods
Animals
The use of animals and procedures were approved by the Institutional Animal Care and Use Committees of Weill Medical College of Cornell University and Cleveland Clinic. Experiments were performed in ApoE KO (AKO), and ApoE/CD36 double KO (DKO) mice. These mice were backcrossed 7x into the C57BL/6 strain (99.22% C57BL/6 and the remainder 129SvJ.). Male mice were weaned at 4 weeks of age and fed normal chow (4.5% fat, 0% cholesterol; W.F. Fisher & Son) for 4 weeks. C57BL/6 (Jackson Lab, Bar Harbor, ME) and wild type (WT, 7x backcrossed with C57BL/6) mice were used for respective temporal gene expression following stroke and in vitro experiments.
Bone marrow (BM) transplantation and diet
Eight week old recipient male mice were subjected to whole body irradiation (11 grey from a cesium source) and 1×107 BM-derived cells from male donor mice were injected via the retro orbital sinus 4 hours later as we previously reported (Febbraio et al., 2004). The transplant groups were as follows: 1) AKO or DKO BM transplanted into AKO mice (AKOBM→AKO vs DKOBM→AKO), and 2) DKO or AKO BM transplanted into DKO mice (DKOBM→DKO vs AKOBM→DKO). Following BM transplantation, recipient mice were fed normal chow for 4 weeks and then switched to a high fat diet for 11 weeks (Harlan Teklad, 88137, 21% fat (wt/wt) adjusted calories and 0.15% (wt/wt) cholesterol). The high fat diet induces a hyperlipidemic condition in AKO and DKO mice.
Assessment of CD36 chimerism
To confirm engraftment of donor stem cells, genomic DNA from blood cells was obtained 3 days after focal ischemia. PCR was performed at 94°C for 1 min, 65°C for 1 min, and 72°C for 2 min for 30 cycles using WT primers (~600 bp), 5’ CAGCTCATACATTGCTGTTTATGCATG; 3’ GGTACAATCACAGTGTTTTCTACGTGG and CD36 KO primers (~ 800 bp), 5 ’ CAGCTCATACATTGCTGTTTATGCATG ; 3’ CCGCTTCCTCGTGCTTTACGGTATC.
Transient middle cerebral artery occlusion (MCAO) and infarct volume assessment
At the end of 11 weeks of the high fat diet, mice were subjected to transient focal ischemia by MCAO, as previously described.25-27 The detailed procedure is described in the supporting methods.
Tissue collection for infarct volume, gene, and protein assessment
Three days following MCAO, brains were excised, frozen and sectioned using an unbiased stereological sampling strategy. The infarct typically spans about 6 mm rostrocaudal, roughly from +2.8 mm to -3.8 mm based on the coordinates of the bregma. To collect tissue reflecting the infarct area, the entire infarct region was cryosectioned for infarct volume measurement (20 μm thickness) and collected serially at 600 micron intervals. Infarct volume and hemispheric swelling were measured using Axiovision software (Zeiss, Germany). Infarct volume was corrected for swelling by a method described previously.51 Tissues between sections used to measure infarct volume were serially cryosectioned for gene and protein assays.
Measurement of plasma cholesterol levels
Plasma cholesterol levels were determined by a colorimetric assay (Bio Vision, CA) from overnight fasted mice using tail blood collected in citrate buffer (25 mM citric acid, 75 mM sodium citrate, 136 mM glucose, citrate buffer:blood (1:8)). After the blood was centrifuged at 6,000 rpm for 5 min at room temperature, plasma was collected and stored at −80°C until the measurement.
Splenocyte isolation and serum treatment
Spleens from normolipidemic WT and hyperlipidemic AKO or DKO mice were collected in sterilized ice-cold Hank’s buffered Salt Solution (HBSS). Tissues were triturated in HBSS using an 18 gauge-needle and passed through a 70 μm filter (BD Bioscience, Bedford, MA). Spleen leukocytes were isolated by removing red blood cells (RBC) using RBC lysis buffer (Sigma, MO) and centrifugation at 300g for 10 min. Prior to tissue collection, trunk blood was collected from hyperlipidemic AKO or DKO mice and allowed to clot at room temperature for 30 min. Serum was obtained by centrifuging at 3,000 rpm for 10 min. The indicated serum was added to the splenocytes plated in 96-well plates (1.5×106 cells/well). Total RNA was isolated from splenocytes for the measurement of CD36 and CCR2 gene expression.
Real-time quantitative RT-PCR (qPCR) for gene expression
Gene expression levels were quantified by real-time quantitative RT-PCR (qPCR) using fluorescent TaqMan technology as described previously.27 The detailed procedure is described in the supporting methods.
MCP-1 and CD36 protein measurement
Brain and plasma MCP-1 protein levels were determined using a commercially available MCP-1 ELISA kit (R&D systems, MN), and brain CD36 protein levels were measured by western blot. The detailed procedures are described in the supporting methods.
Data Analysis
Infarct volume and percent hemispheric swelling were reported as mean ± 95% confidence interval (CI). Gene and protein levels were reported as mean ± SEM. Gene expression levels from in vivo studies were presented as the β-actin normalized value according to the formula,
Gene expression levels in in vitro studies were reported relative to control cultures and averaged from three independent experiments. Comparison between two groups was statistically evaluated using Student’s t-test. Multiple comparisons were made using ANOVA followed by a post hoc Newman-Keuls test. Differences were considered significant at p<0.05.
RESULTS
Assessment of CD36 chimerism
Engraftment analysis by assessing CD36 chimerism revealed that more than 99% of blood cells in the recipient (host) mice were repopulated with donor BM cells (Supporting Figure S1). We excluded mice that developed severe dermatitis during high fat diet intervention, since peripheral inflammation associated with skin lesions may potentially impact ischemic stroke outcome. The breakdown of the number of mice in each group (exclusion of animals due to dermatitis, success rate of MCAO, mortality following MCAO, and final number entered to study) is summarized (Supporting Table S1).
BM transplantation does not affect body weight gain, plasma cholesterol levels, and severity of ischemia
There were no significant differences in body weight among the BM transplanted groups at the time of MCAO (Table 1). Eleven weeks high fat diet intervention resulted in severe hyperlipidemia; plasma total cholesterol levels reached ~900 mg/dl (normal chow fed WT controls: 118.6±6.1 mg/dl, n=8, p<0.001), which is consistent with previous reports.27, 46, 52 Within transplantation groups, the cholesterol levels were moderately increased in DKO mice receiving AKOBM. There were no noticeable differences in blood flow reduction during ischemia or 10 min after reperfusion, suggesting that ischemic severity across and within each set of transplantation groups were comparable (Table 1).
Table 1.
Body weight, plasma cholesterol, and cerebral blood flow (CBF) during MCAO in BM transplanted mice.
| Host mice | Donor BM | Weight (g) | Cholesterol (mg/dl) | CBF reduction (%)* | CBF reperfusion (%)* |
|---|---|---|---|---|---|
| AKO | AKOBM | 29.4±0.7 | 935.6±29.7 | 85.9±2.6 | 120.8±8.9 |
| DKOBM | 28.7±0.5 | 902.2±27.0 | 84.4±1.1 | 115.4±3.6 | |
|
| |||||
| DKO | DKOBM | 29.1±0.5 | 808.4±52.6 | 81.0±1.7 | 110.3±4.1 |
| AKOBM | 30.1±0.6 | 965.1±19.7a | 86.1±1.5b | 101.9±4.4 | |
Two sets of BM TRANSPLANTATION.
Values indicate percentage of CBF reduction during ischemia and CBF reperfusion at 10 min compared to baseline CBF. Data are expressed as mean ± SEM; n=11-15/group,
p<0.001,
p<0.05 vs DKOBM; AKO, ApoE KO; DKO, ApoE/CD36 double KO.
Peripheral CD36 contributes to stroke-induced brain injury in hyperlipidemic AKO mice
CD36 was shown to be involved in exacerbating ischemic injury in hyperlipidmic conditions.27 Since CD36 is expressed both in the brain and peripheral immune cells, we investigated the contribution of peripheral CD36 in stroke-induced injury using hyperlipidemic AKO mice. A temporal gene expression in non-transplanted WT mice in the post-ischemic brain showed a significant increase in CD36 mRNA levels at 72h post-ischemia (Fig. 1A). We therefore selected this time point for the analysis of stroke outcome in BM studies. Stroke-induced increases in CD36 gene and protein expression in autologous transplanted mice (AKO mice that received AKOBM) were absent in mice receiving DKOBM (Fig. 1B and C). The data indicate that infiltrating cells from the periphery are the major source of CD36 in the post-ischemic brain. Analyses of stroke outcome revealed that AKO mice receiving DKOBM exhibited reduced infarct size and hemispheric swelling (Fig. 1D and E). Collectively, the data show a contributing role of peripheral CD36 to stroke-induced brain injury in hyperlipidemic AKO mice.
Figure 1. Effect of peripheral CD36 on stroke-induced CD36 expression and ischemic stroke outcomes in hyperlipidemic AKO mice.

A Temporal gene expression of CD36. CD36 mRNA levels were measured in the post-ischemic brain up to 72h in non-transplanted WT mice (n=3-4/group). Sham, sham-operated control (0h). B&C, Brain CD36 mRNA (B) and protein (C) levels were measured at 3d post-ischemia in BM transplanted AKO mice. D&E, Infarct volume (D) and % hemispheric swelling (E, % swelling) were measured at 3d post-ischemia in AKO mice that received AKOBM (open circle, n=12) or DKOBM (n=14, closed circle). AKOBM, BM-derived cells from AKO mice; DKOBM, BM-derived cells from DKO mice. *p<0.05, **p<0.01 vs contralateral hemisphere or AKOBM
Peripheral CD36 affects stroke-induced MCP-1 and CCR2 expression in AKO mice
An MCP-1 gradient generated in the injured tissue causes the homing of CCR2+ peripheral immune cells to the affected area.53-55 We first determined temporal expression of MCP-1 and CCR2 in non-transplanted WT mice in the post-ischemic brain. MCP-1 mRNA levels were increased in the early post-ischemic period as evidenced by a sustained increase from 6 to 72h post-ischemia (Fig. 2A). In contrast, peak CCR2 expression occurred at 72h, a time point at which a substantial infiltration of peripheral immune cells into the injury site occurs (Fig. 2B).56 Based on this temporal expression, we performed expression analyses at 3 days post-ischemia in BM transplanted AKO and DKO mice. Stroke increased brain MCP-1 gene expression and protein respectively (Fig. 2C and D) as well as CCR2 gene expression (Fig. 2E) in autologous transplanted AKO mice. The increases in MCP-1 and CCR2 expression were significantly reduced in AKO mice receiving DKOBM, suggesting that peripheral CD36 affects stroke-induced MCP-1 and CCR2 expression in the AKO brain (Fig. 2C-E).
Figure 2. Effect of peripheral CD36 on stroke-induced MCP-1 and CCR2 expression in AKO mice.

A&B, Temporal gene expression of MCP-1 (A) and CCR2 (B). MCP-1 and CCR2 mRNA levels were measured in the post-ischemic brain up to 72h in non-transplanted WT mice (n=3-4/group). Sham, sham-operated control (0h). C-E, Brain MCP-1 mRNA (C), MCP-1 protein measured by ELISA (D), and CCR2 mRNA (E) levels were determined at 72h post-ischemia in AKO mice transplanted with AKOBM (n=12) or DKOBM (n=14). AKOBM, BM-derived cells from AKO mice; DKOBM, BM-derived cells from DKO mice. *p<0.05, ***p<0.001 vs contralateral hemisphere or AKOBM
Peripheral CD36 does not affect CX3CL1/CX3CR1 expression in AKO mice
In response to tissue injury, early recruitment of the pro-inflammatory CCR2+ monocyte subset is followed by the late anti-inflammatory subset that expresses high levels of CX3CR1 for tissue repair/healing.57 We established expression profiles of the anti-inflammatory chemokine CX3CL1 (fractalkine) and the receptor CX3CR1 in non-transplanted WT mice following stroke. The expression of CX3CL1 was progressively reduced in the ipsilateral side of the brain (Fig. 3A), reflecting neuronal loss in the post-ischemic brain, as neurons are the major source of CX3CL1. The expression of CX3CR1 was relatively unchanged following stroke (Fig. 3B). In BM transplanted mice, CX3CL1 gene expression at 3 days post-ischemia was reduced in the ipsilateral hemisphere, but the extent of reduction was similar between mice that received either AKOBM or DKOBM (Fig. 3C). In addition, there were no differences in CX3CR1 expression between hemispheres or between transplantation groups (Fig. 3D). No changes of CX3CL1/CX3CR1 expression at this time suggest the selective involvement of pro-inflammatory axis in the setting of acute ischemia.
Figure 3. No effect of peripheral CD36 on stroke-induced CX3CL1/CX3CR1 expression in AKO mice.

A&B,Temporal gene expression of stroke-induced CX3CL1 (A) and CX3CR1 (B) in non-transplanted WT mice (n=3-4/group). CX3CL1 and CX3CR1 mRNA levels were measured in the post-ischemic brain up to 72h. Sham, sham-operated controls. C&D, CX3CL1 (C) and CX3CR1 (D) mRNA levels were determined at 72h post-ischemia in AKO mice transplanted with AKOBM (n=12) or DKO BM (n=14). AKOBM, BM-derived cells from AKO mice; DKOBM, BM-derived cells from DKO mice. **p<0.01, ***p<0.001 vs contralateral hemisphere
Host CD36 is required for the peripheral CD36 effect on ischemic stroke outcome and MCP-1/CCR2 expression
In order to address the sufficiency of peripheral CD36 in mediating ischemic brain injury, transplantation was performed using DKO mice as recipients. Brain CD36 mRNA levels 72 hours post-ischemia were undetectable in the DKO mice that underwent autologous transplantation (DKO mice receiving DKOBM). In contrast, transplantation of AKOBM significantly increased CD36 levels (Fig. 4A), confirming that CD36 expressed in the post-ischemic brain is from the periphery. Unexpectedly, AKOBM transfer to DKO mice did not increase infarct volume and percent hemispheric swelling compared to the control transplanted group (Fig. 4B and C). This lack of exacerbation of the ischemic stroke outcome was accompanied by no further increase in MCP-1 mRNA and protein levels or CCR2 mRNA levels (Fig. 4D-F). Gene expression of CX3CL1 and CX3CR1 was not different among the groups (Supporting Table S2). These data clearly indicate that peripheral CD36 alone is not sufficient for mediating ischemic brain injury, and that CD36 expressed in the host brain is additionally required for the peripheral CD36 effect to take place.
Figure 4. Effect of peripheral CD36 on ischemic stroke outcome and inflammatory gene expression in DKO mice.
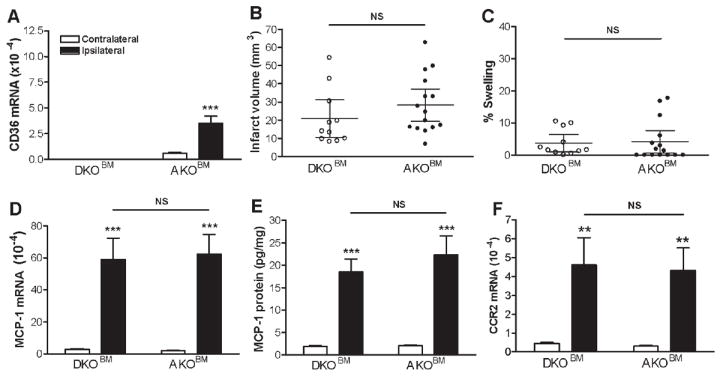
CD36 mRNA levels, ischemic stroke outcomes, MCP-1 and CCR2 expression were measured in the brain 3d post-ischemia in DKO mice that received either AKOBM or DKOBM. A, CD36 mRNA levels in both hemisphere, B&C, Measurement of infarct volume and % hemispheric swelling (% swelling) in DKO mice that received DKOBM (open circle, n=11) or AKOBM (n=15, closed circle). D-F, MCP-1 mRNA (D), MCP-1 protein (E), and CCR2 mRNA (F) were quantified in contralateral and ipsilateral side of the brain 3d after MCAO. AKOBM, BM-derived cells from AKO mice; DKOBM, BM-derived cells from DKO mice. **p<0.01, ***p<0.001 vs contralateral hemisphere
CD36 deficiency in the host brain attenuates stroke-induced MCP-1/CCR2 responses
To understand the contributing role of host brain CD36 in ischemic injury, correlation analyses among brain MCP-1 and CCR2 mRNA, and infarct volume were assessed. Significant correlations were observed among these three variables when all transplanted groups were included (data not shown). Sub-group analyses revealed that a correlation between MCP-1 and CCR2 expression in AKO mice was similar to that of DKO mice (Fig. 5A). In contrast, the DKO host showed reduced expression in MCP-1 (Fig. 5B, slope elevation between hosts p<0.05) and CCR2 (Fig. 5C, slope elevation between hosts p<0.01) expression at a given infarct volume. The dampened MCP-1 and CCR2 responses in the DKO host suggest that brain CD36 is involved in regulating MCP-1 expression in the post-ischemic brain, which subsequently influences recruitment of CCR2+ cells to the infarct.
Figure 5. Effect of host CD36 on stroke-induced MCP-1/CCR2 expression.

Correlation analyses among MCP-1 mRNA levels, CCR2 mRNA levels, and infarct volume in the post-ischemic brains were performed and presented separately by hosts. Positive correlations between MCP-1 and CCR2 mRNA levels (A), between MCP-1 mRNA and infarct volume (B), and between CCR2 and infarct volume (C). In all cases, the correlation was highly significant. Note the significant difference of slope elevations in MCP-1 mRNA (B) and CCR2 mRNA levels (C) in DKO host compared to AKO host. *p<0.05, **p<0.01 between hosts
CD36 regulates MCP-1 and CCR2 expression in the periphery
We next addressed whether host CD36 additionally regulates peripheral MCP-1/CCR2 expression. Assessment of plasma MCP-1 levels in AKO and DKO mice revealed host-specific responses. Regardless of the source of BM, DKO mice displayed significantly higher plasma MCP-1 levels (Fig. 6A). The host-specific but BM source-independent plasma MCP-1 levels suggest an intrinsic property of host CD36 in regulating plasma MCP-1 levels.
Figure 6. CD36 regulates MCP-1/CCR2 expression in periphery.

A. Plasma MCP-1 levels were measured in BM transplanted mice 3d post-ischemia. A dotted line indicates mean MCP-1 levels from plasma in WT mice. B.WT splenocytes were treated with WT, AKO, or DKO serum for 24h and CD36 and CCR2 mRNA were determined. C. AKO and DKO splenocytes were treated by AKO or DKO serum for 24h and CCR2 gene expression levels were measured. Three independent experiments were performed in quadruplicate. *p<0.05, **p<0.01 ***p<0.001 vs respective control conditions as indicated
The effect of host CD36 on CD36 and CCR2 expression in splenocytes was further investigated. The spleen contains half of the body’s monocytes and serves as an immediate monocyte reservoir that deploys in response to injury.58 WT splenocytes were incubated with control or hyperlipidemic serum obtained from AKO or DKO mice. Compared to WT serum, incubation with AKO serum increased CD36 and CCR2 expression in splenocytes; this effect was absent when cells were exposed to DKO serum (Fig. 6B). The changes were not due to lipid levels, since AKO and DKO plasma cholesterol levels were similar (~900 mg/dl). The results suggest that host CD36 induces specific changes in blood/serum that affect CD36 and CCR2 expression in circulating immune cells. To address whether the presence of CD36 in immune cells influences their intrinsic property to express CCR2, AKO or DKO splenocytes were incubated with host-specific serum. CCR2 expression in DKO splenocytes was significantly reduced compared to AKO splenocytes in the presence of AKO serum. Consistently, CCR2 expression in AKO splenocytes was higher than in DKO splenocytes when incubated with DKO serum (Fig. 6C), suggesting a necessity for splenocyte CD36 for increased splenocyte CCR2 expression. These findings imply that CCR2 expression in immune cells is regulated by CD36 directly in cells and indirectly by CD36 actions reflected in serum. Taken together, this study suggests that CD36 is involved in multiple layers of regulation of MCP-1/CCR2 expression.
Discussion
Although stroke-induced brain injury has been primarily viewed as a central nervous system (CNS) event, increasing evidence suggests that stroke outcomes are modulated by peripheral status of inflammation.59, 60 Previously, we reported that elevated plasma cholesterol levels in mice increases CD36 expression in monocytes/macrophages, exacerbates ischemic stroke outcome, and up-regulates CD36 expression in the ischemic brain.27 The current study directly addresses the extent to which peripheral CD36 influences ischemic brain injury in hyperlipidemia. The study utilizes a BM transplantation technique to exchange the genotypes of peripheral cells in the context of CD36-expressing (AKO) or - deficient (DKO) hyperlipidemic hosts prior to feeding mice a high fat diet for 11 weeks and then subjecting them to ischemic stroke. We report several key findings: stroke-induced CD36 expression in the brain originates predominantly from the periphery; peripheral CD36-expressing cells influence the extent of ischemic injury; the underlying mechanism(s) by which CD36 expression influences ischemic brain injury involves MCP-1/CCR2 expression; the presence of CD36 in the host brain is required for the peripheral CD36-mediated effect on stroke-induced injury.
CD36 is expressed in several cell types and tissues in the brain such as microvascular endothelial cells, neurons and microglia. In addition, astrocytes express CD36 in a temporally and spatially restricted pattern in the post-ischemic brain.25 In the peripheral circulation, CD36 is expressed predominantly by CD11b+ immune cells including monocytes/macrophages, dendritic cells, B cells and platelets. Through exchanging BM cells between AKO and DKO mice and subjecting these mice to stroke, we observed that stroke-induced CD36 expression was absent in AKO mice receiving DMOBM (Fig. 1B & C) but substantially increased in DKO mice receiving AKOBM (Fig. 4A). Since stroke causes trafficking of monocytes/macrophages, immune cells that express CD36 to the infarct, an important finding derived from this study is that the major source of CD36 expressed in the ischemic hemisphere is from the periphery. This finding is consistent with our previous report that showed elevated CD36 expression in peritoneal macrophages in hyperlipidemic mice and increased number of foam cells (CD36+ lipid laden macrophages) in the post-ischemic brain.27 Although it remains to be investigated whether platelet CD36 contributes to ischemic injury, the similarity in the platelet-induced prothrombotic phenotype as indicated by comparable ischemic severity and degree of reperfusion among BM transplanted groups (Table 1), suggests minimal involvement of platelet CD36 in the BM transplantation effect we observed.
Close examination in the contralateral hemisphere revealed a slight reduction in CD36 mRNA levels in AKO mice receiving DKOBM (Fig. 1B) and a small but significant increase in DKO mice receiving AKOBM (Fig. 4A). This is likely due to the presence of residual blood in the hemisphere, since our PCR analyses elaborate confirmed host and BM status. Alternatively, during the 12 weeks from the point of BM transfer to stroke, there may have been differentiation of BM-derived stem cells to microglia and endothelial cells in the brain.61-67
CD36 expressed in peripheral immune cells had an important impact on ischemic stroke outcome. We observed that AKO mice receiving DKOBM displayed reduced inflammation and injury compared to autologous transplanted groups (Fig. 1D, E and 2C D). The improved ischemic stroke outcome in the absence of peripheral CD36 is supported by our previous finding in non-transplanted hyperlipidemic mice; compared to AKO mice, DKO mice exhibited significantly reduced brain MCP-1 and other pro-inflammatory markers (CCR2, TNFα and IL-1β) and had smaller infarcts.27 The underlying mechanisms for the peripheral CD36 effect include activation of CD36 in the presence of specific ligands generated in hyperlipidemic conditions and subsequent increased production of pro-inflammatory chemokines. Secretion of chemokines, including MCP-1, in response to pro-atherogenic modified/oxidized LDL, has been shown to be CD36-dependent.46, 68, 69 Of note, the peripheral CD36 effect observed in the current study occurred in a severe hyperlipidemic setting. Interestingly, in a much more mild hyperlipidemic setting, in which WT mice were transplanted with WTBM or CD36 KOBM and fed a high fat diet (plasma cholesterol levels: WTBM, 257.3 ± 5.0 mg/dl, n=12; KOBM, 234.6 ± 5.8 mg/dl, n=19), we did not observe a peripheral CD36 effect on ischemic stroke outcome nor inflammatory gene expression (WT vs CD36 KO: infarct, 20.3 ± 3.8 mm3 vs 26.1 ± 3.9 mm3, ns, % swelling, 13.8 ± 4.0 % vs 11.2 ± 3.4 %, ns, MCP-1 mRNA, 1.8×10-2 ± 2.6 ×10-3 vs 1.3×10-2 ± 1.8×10-3, ns, CCR2 mRNA, 1.4×10-3 ± 2.5×10-4 vs 1.2×10-3 ± 2.0×10-4, ns). These data suggest that exacerbated ischemic stroke outcome resulting from hyperlipidemia occurs via peripheral mechanisms that alter CD36 and cytokine expression, and that the CNS is less sensitive to lipid levels in terms of these parameters. A recent in vitro study reported that peripheral CD36 in immune cells, not brain CD36, are a major contributor for OGD induced tissue damage.70 Our finding of the added effect of brain CD36 likely results from the effect of immune cell mobilization at the system level and from a severe hyperlipidemic setting. Reduction of infarct size in normolipidemic CD36 KO mice25 suggests CNS CD36’s contribution to ischemic injury is multifactorial.
Multiple studies have established that there is heterogeneity in mouse monocyte subsets, and in tissue injury both a pro-inflammatory CCR2+ (LY-6Chi) subset exhibiting chemotaxis to MCP-1 produced in the inflammatory region and an anti-inflammatory CX3CR1+ (CCR2-/LY-6Clow) subset71-73 are sequentially recruited to the injury site in a controlled manner for inflammation and repair/healing.57, 71 The present study indicates that the effect of peripheral CD36 on ischemic injury selectively involves the MCP-1/CCR2 axis, since we observed no differences in CX3CL1/CX3CR1 in the same mice (Fig. 2 vs Fig. 3).
This study also revealed a novel requirement for host CD36 for the peripheral CD36 effect in hyperlipidemia: transplantation of AKOBM into DKO mice did not exacerbate ischemia-reperfusion brain injury (Fig. 4B and C, Supporting Table S2). This result was unexpected and differed from what has been observed with macrophage CD36 in atherosclerosis. In that BM transplant study, atherosclerotic lesion burden followed the expression of macrophage CD36.44 The discrepancy may be explained by the fact that stroke pathology also involves the CNS, which is in some respects an isolated and privileged site. In the CNS, CD36 is expressed on astrocytes, microglia and microvascular endothelial cells, which produce MCP-1 in response to various CD36 ligands generated in the injured brain.11, 14, 43 The MCP-1 gradient established in the infarcted tissue is critical for the recruitment of peripheral CCR2+ monocytes/macrophages. After AKOBM transplantation, we observed significantly less expression of MCP-1 and CCR2 in the post-ischemic brain of DKO mice compared to AKO mice: MCP-1 mRNA (62.4±11.5×10-4 vs 111.9±14.4×10-4 vs, p<0.05), MCP-1 protein (22.3± 4.1 pg/mg vs 31.0±3.4 pg/mg, p<0.05), and CCR2 mRNA levels (4.3±1.1×10-4 vs 9.7±1.7×10-4, p<0.05) (Supplemental Table S2). The overall decreased expression of MCP-1 and CCR2 in DKO mice at any given infarct volume, regardless of the source of BM (Fig. 5), further support the idea that host CD36 is an additional factor regulating the MCP-1/CCR2 axis in response to injury.
Our study also offers mechanistic insights into how host and peripheral CD36 regulate MCP-1 and CCR2 expression (Fig. 7). DKO mice had decreased MCP-1 expression in the brain, and significantly higher plasma MCP-1 levels, independent of BM source (Fig. 6A). The increased plasma MCP-1 levels in DKO mice could counteract the chemokine gradient produced at the injury site and reduce trafficking of CCR2+ cells to the infarct. We found that host CD36 also regulates CCR2 expression in peripheral immune cells: AKO or DKO serum increased or decreased CCR2 expression, respectively, in WT splenocytes (Fig. 6B). Moreover, the presence or absence of CD36 in splenocytes affected CCR2 expression. Consistent with this, studies showed that MCP-1-stimulated migration was reduced in CD36-deficient macrophages.46 Since chemotaxis is regulated by CCR2 expression in the migrating cells and an MCP-1 gradient at the injured site, 74, 75 the important aspect of CD36-dependnet CCR2 and MCP-1 expression thus may lies in the involvement of CD36 in immune cell trafficking into the infarct (Fig.7).
Figure 7.

Hypothesized models depicting a synergy between host and peripheral CD36 in immune cell trafficking into the infarct through the MCP-1/CCR2 axis. A, The diagram shows the peripheral CD36 effect. CD36-expressing (CD36+/+) host mice (AKO) generate a higher MCP-1 gradient in the brain. Higher and lower expression of CCR2 in AKOBM or DKOBM led to increased or reduced infiltration of CCR2+ cells into ischemic injury. The net results are increased MCP-1 and CCR2 expression in the post-ischemic brain. B, The diagram shows the involvement of host CD36 in the recruitment of peripheral CCR2+ cells. CD36-deficient (CD36-/-) host mice (DKO) generate a lower MCP-1 gradient in the brain and higher plasma MCP-1 levels. This may counteract the chemokine gradient produced in the injury site and reduce the trafficking of CCR2+ bearing cells from the periphery to infarct. Lower and higher expression of CCR2 in DKOBM or AKOBM may not lead to reduced or increased infiltration of CCR2+ cells into ischemic injury due to a higher MCP-1 gradient in the plasma. Net results will be reduced MCP-1 (host) and CCR2 expression in the post-ischemic brain.
The current study demonstrates a necessary role for peripheral CD36 in hyperlipidemia-induced exacerbation in ischemic stroke outcomes and a requirement for CNS CD36 to attain the peripheral CD36 effect. The findings suggest that host brain and peripheral CD36 synergistically regulate immune cell trafficking through modulation of the MCP-1/CCR2 axis. Thus, modulation of the MCP-1/CCR2 axis by targeting host and peripheral CD36 may provide a novel strategy to achieve neuroprotection from stroke-induced acute brain injury. With much attention given to the mobilization of BM derived stem cells for stroke therapy,76, 77 future studies using adoptive transfers of CD36+/+ or CD36-/- BM-derived cells into the CD36-expressing or -deficient host are warranted to define the contribution of CD36 to stem cell engraftment and therapeutic utility for acute stroke.
Supplementary Material
Acknowledgments
funding sources: This work was supported by NIH grants HL82511, HL82511-04S1 (SC), and the Burke Foundation.
Footnotes
Conflict of interest
None
References
- 1.Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003 May;4(5):399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- 2.Huang J, Upadhyay UM, Tamargo RJ. Inflammation in stroke and focal cerebral ischemia. Surg Neurol. 2006 Sep;66(3):232–245. doi: 10.1016/j.surneu.2005.12.028. [DOI] [PubMed] [Google Scholar]
- 3.Iadecola C, Alexander M. Cerebral ischemia and inflammation. Curr Opin Neurol. 2001 Feb;14(1):89–94. doi: 10.1097/00019052-200102000-00014. [DOI] [PubMed] [Google Scholar]
- 4.Amantea D, Nappi G, Bernardi G, et al. Post-ischemic brain damage: pathophysiology and role of inflammatory mediators. Febs J. 2009 Jan;276(1):13–26. doi: 10.1111/j.1742-4658.2008.06766.x. [DOI] [PubMed] [Google Scholar]
- 5.del Zoppo G, Ginis I, Hallenbeck JM, et al. Inflammation and stroke: putative role for cytokines, adhesion molecules and iNOS in brain response to ischemia. Brain Pathol. 2000 Jan;10(1):95–112. doi: 10.1111/j.1750-3639.2000.tb00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin P, Leibovich SJ. Inflammatory cells during wound repair: the good, the bad and the ugly. Trends Cell Biol. 2005 Nov;15(11):599–607. doi: 10.1016/j.tcb.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 7.Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004 Mar 5;303(5663):1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 8.Leibovich SJ, Ross R. The role of the macrophage in wound repair. A study with hydrocortisone and antimacrophage serum. Am J Pathol. 1975 Jan;78(1):71–100. [PMC free article] [PubMed] [Google Scholar]
- 9.El Khoury JB, Moore KJ, Means TK, et al. CD36 mediates the innate host response to beta-amyloid. J Exp Med. 2003 Jun 16;197(12):1657–1666. doi: 10.1084/jem.20021546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Febbraio M, Abumrad NA, Hajjar DP, et al. A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J Biol Chem. 1999 Jul 2;274(27):19055–19062. doi: 10.1074/jbc.274.27.19055. [DOI] [PubMed] [Google Scholar]
- 11.Jimenez B, Volpert OV, Crawford SE, et al. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat Med. 2000 Jan;6(1):41–48. doi: 10.1038/71517. [DOI] [PubMed] [Google Scholar]
- 12.Rahaman SO, Lennon DJ, Febbraio M, et al. A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab. 2006 Sep;4(3):211–221. doi: 10.1016/j.cmet.2006.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stewart CR, Stuart LM, Wilkinson K, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010 Feb;11(2):155–161. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coraci IS, Husemann J, Berman JW, et al. CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer’s disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am J Pathol. 2002 Jan;160(1):101–112. doi: 10.1016/s0002-9440(10)64354-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Talle MA, Rao PE, Westberg E, et al. Patterns of antigenic expression on human monocytes as defined by monoclonal antibodies. Cell Immunol. 1983 May;78(1):83–99. doi: 10.1016/0008-8749(83)90262-9. [DOI] [PubMed] [Google Scholar]
- 16.Bordessoule D, Jones M, Gatter KC, et al. Immunohistological patterns of myeloid antigens: tissue distribution of CD13, CD14, CD16, CD31, CD36, CD65, CD66 and CD67. Br J Haematol. 1993 Mar;83(3):370–383. doi: 10.1111/j.1365-2141.1993.tb04659.x. [DOI] [PubMed] [Google Scholar]
- 17.Greenwalt DE, Scheck SH, Rhinehart-Jones T. Heart CD36 expression is increased in murine models of diabetes and in mice fed a high fat diet. J Clin Invest. 1995 Sep;96(3):1382–1388. doi: 10.1172/JCI118173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Nieuwenhoven FA, Verstijnen CP, Abumrad NA, et al. Putative membrane fatty acid translocase and cytoplasmic fatty acid-binding protein are co-expressed in rat heart and skeletal muscles. Biochem Biophys Res Commun. 1995 Feb 15;207(2):747–752. doi: 10.1006/bbrc.1995.1250. [DOI] [PubMed] [Google Scholar]
- 19.Febbraio M, Hajjar DP, Silverstein RL. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest. 2001 Sep;108(6):785–791. doi: 10.1172/JCI14006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenberg ME, Sun M, Zhang R, et al. Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J Exp Med. 2006 Nov 27;203(12):2613–2625. doi: 10.1084/jem.20060370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Podrez EA, Byzova TV, Febbraio M, et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat Med. 2007 Sep;13(9):1086–1095. doi: 10.1038/nm1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Febbraio M, Podrez EA, Smith JD, et al. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest. 2000 Apr;105(8):1049–1056. doi: 10.1172/JCI9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Su X, Maguire-Zeiss KA, Giuliano R, et al. Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol Aging. 2008 Nov;29(11):1690–1701. doi: 10.1016/j.neurobiolaging.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cho S, Kim E. CD36: a multi-modal target for acute stroke therapy. J Neurochem. 2009 May;109(Suppl 1):126–132. doi: 10.1111/j.1471-4159.2009.05801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cho S, Park EM, Febbraio M, et al. The class B scavenger receptor CD36 mediates free radical production and tissue injury in cerebral ischemia. J Neurosci. 2005 Mar 9;25(10):2504–2512. doi: 10.1523/JNEUROSCI.0035-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cho S, Szeto HH, Kim E, et al. A novel cell-permeable antioxidant peptide, SS31, attenuates ischemic brain injury by down-regulating CD36. J Biol Chem. 2007 Feb 16;282(7):4634–4642. doi: 10.1074/jbc.M609388200. [DOI] [PubMed] [Google Scholar]
- 27.Kim E, Tolhurst AT, Qin LY, et al. CD36/fatty acid translocase, an inflammatory mediator, is involved in hyperlipidemia-induced exacerbation in ischemic brain injury. J Neurosci. 2008 Apr 30;28(18):4661–4670. doi: 10.1523/JNEUROSCI.0982-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim JS, Gautam SC, Chopp M, et al. Expression of monocyte chemoattractant protein-1 and macrophage inflammatory protein-1 after focal cerebral ischemia in the rat. J Neuroimmunol. 1995 Feb;56(2):127–134. doi: 10.1016/0165-5728(94)00138-e. [DOI] [PubMed] [Google Scholar]
- 29.Minami M, Satoh M. Chemokines and their receptors in the brain: pathophysiological roles in ischemic brain injury. Life Sci. 2003 Dec 5;74(2-3):321–327. doi: 10.1016/j.lfs.2003.09.019. [DOI] [PubMed] [Google Scholar]
- 30.Rollins BJ. Chemokines. Blood. 1997 Aug 1;90(3):909–928. [PubMed] [Google Scholar]
- 31.Rock RB, Hu S, Gekker G, et al. Mycobacterium tuberculosis-induced cytokine and chemokine expression by human microglia and astrocytes: effects of dexamethasone. J Infect Dis. 2005 Dec 15;192(12):2054–2058. doi: 10.1086/498165. [DOI] [PubMed] [Google Scholar]
- 32.Meng SZ, Oka A, Takashima S. Developmental expression of monocyte chemoattractant protein-1 in the human cerebellum and brainstem. Brain Dev. 1999 Jan;21(1):30–35. doi: 10.1016/s0387-7604(98)00065-5. [DOI] [PubMed] [Google Scholar]
- 33.Coughlan CM, McManus CM, Sharron M, et al. Expression of multiple functional chemokine receptors and monocyte chemoattractant protein-1 in human neurons. Neuroscience. 2000;97(3):591–600. doi: 10.1016/s0306-4522(00)00024-5. [DOI] [PubMed] [Google Scholar]
- 34.Flugel A, Hager G, Horvat A, et al. Neuronal MCP-1 expression in response to remote nerve injury. J Cereb Blood Flow Metab. 2001 Jan;21(1):69–76. doi: 10.1097/00004647-200101000-00009. [DOI] [PubMed] [Google Scholar]
- 35.White FA, Sun J, Waters SM, et al. Excitatory monocyte chemoattractant protein-1 signaling is up-regulated in sensory neurons after chronic compression of the dorsal root ganglion. Proc Natl Acad Sci U S A. 2005 Sep 27;102(39):14092–14097. doi: 10.1073/pnas.0503496102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Che X, Ye W, Panga L, et al. Monocyte chemoattractant protein-1 expressed in neurons and astrocytes during focal ischemia in mice. Brain Res. 2001 Jun 1;902(2):171–177. doi: 10.1016/s0006-8993(01)02328-9. [DOI] [PubMed] [Google Scholar]
- 37.Banisadr G, Gosselin RD, Mechighel P, et al. Highly regionalized neuronal expression of monocyte chemoattractant protein-1 (MCP-1/CCL2) in rat brain: evidence for its colocalization with neurotransmitters and neuropeptides. J Comp Neurol. 2005 Aug 29;489(3):275–292. doi: 10.1002/cne.20598. [DOI] [PubMed] [Google Scholar]
- 38.Chen Y, Hallenbeck JM, Ruetzler C, et al. Overexpression of monocyte chemoattractant protein 1 in the brain exacerbates ischemic brain injury and is associated with recruitment of inflammatory cells. J Cereb Blood Flow Metab. 2003 Jun;23(6):748–755. doi: 10.1097/01.WCB.0000071885.63724.20. [DOI] [PubMed] [Google Scholar]
- 39.Babcock AA, Kuziel WA, Rivest S, et al. Chemokine expression by glial cells directs leukocytes to sites of axonal injury in the CNS. J Neurosci. 2003 Aug 27;23(21):7922–7930. doi: 10.1523/JNEUROSCI.23-21-07922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dimitrijevic OB, Stamatovic SM, Keep RF, et al. Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke. 2007 Apr;38(4):1345–1353. doi: 10.1161/01.STR.0000259709.16654.8f. [DOI] [PubMed] [Google Scholar]
- 41.Hughes PM, Allegrini PR, Rudin M, et al. Monocyte chemoattractant protein-1 deficiency is protective in a murine stroke model. J Cereb Blood Flow Metab. 2002 Mar;22(3):308–317. doi: 10.1097/00004647-200203000-00008. [DOI] [PubMed] [Google Scholar]
- 42.Schober A, Zernecke A, Liehn EA, et al. Crucial role of the CCL2/CCR2 axis in neointimal hyperplasia after arterial injury in hyperlipidemic mice involves early monocyte recruitment and CCL2 presentation on platelets. Circ Res. 2004 Nov 26;95(11):1125–1133. doi: 10.1161/01.RES.0000149518.86865.3e. [DOI] [PubMed] [Google Scholar]
- 43.Moore KJ, El Khoury J, Medeiros LA, et al. A CD36-initiated signaling cascade mediates inflammatory effects of beta-amyloid. J Biol Chem. 2002 Dec 6;277(49):47373–47379. doi: 10.1074/jbc.M208788200. [DOI] [PubMed] [Google Scholar]
- 44.Febbraio M, Guy E, Silverstein RL. Stem cell transplantation reveals that absence of macrophage CD36 is protective against atherosclerosis. Arterioscler Thromb Vasc Biol. 2004 Dec;24(12):2333–2338. doi: 10.1161/01.ATV.0000148007.06370.68. [DOI] [PubMed] [Google Scholar]
- 45.Harb D, Bujold K, Febbraio M, et al. The role of the scavenger receptor CD36 in regulating mononuclear phagocyte trafficking to atherosclerotic lesions and vascular inflammation. Cardiovasc Res. 2009 Jul 1;83(1):42–51. doi: 10.1093/cvr/cvp081. [DOI] [PubMed] [Google Scholar]
- 46.Kuchibhotla S, Vanegas D, Kennedy DJ, et al. Absence of CD36 protects against atherosclerosis in ApoE knock-out mice with no additional protection provided by absence of scavenger receptor A I/II. Cardiovasc Res. 2008 Apr 1;78(1):185–196. doi: 10.1093/cvr/cvm093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park YM, Febbraio M, Silverstein RL. CD36 modulates migration of mouse and human macrophages in response to oxidized LDL and may contribute to macrophage trapping in the arterial intima. J Clin Invest. 2009 Jan;119(1):136–145. doi: 10.1172/JCI35535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stewart CR, Tseng AA, Mok YF, et al. Oxidation of low-density lipoproteins induces amyloid-like structures that are recognized by macrophages. Biochemistry. 2005 Jun 28;44(25):9108–9116. doi: 10.1021/bi050497v. [DOI] [PubMed] [Google Scholar]
- 49.Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. Apr 29;464(7293):1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Witztum JL. You are right too! J Clin Invest. 2005 Aug;115(8):2072–2075. doi: 10.1172/JCI26130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin TN, He YY, Wu G, et al. Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke. 1993 Jan;24(1):117–121. doi: 10.1161/01.str.24.1.117. [DOI] [PubMed] [Google Scholar]
- 52.Guy E, Kuchibhotla S, Silverstein R, et al. Continued inhibition of atherosclerotic lesion development in long term Western diet fed CD36o /apoEo mice. Atherosclerosis. 2007 May;192(1):123–130. doi: 10.1016/j.atherosclerosis.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 53.Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006 Feb 9;354(6):610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- 54.Boring L, Gosling J, Chensue SW, et al. Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J Clin Invest. 1997 Nov 15;100(10):2552–2561. doi: 10.1172/JCI119798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fantuzzi L, Borghi P, Ciolli V, et al. Loss of CCR2 expression and functional response to monocyte chemotactic protein (MCP-1) during the differentiation of human monocytes: role of secreted MCP-1 in the regulation of the chemotactic response. Blood. 1999 Aug 1;94(3):875–883. [PubMed] [Google Scholar]
- 56.Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol. May;87(5):779–789. doi: 10.1189/jlb.1109766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nahrendorf M, Swirski FK, Aikawa E, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007 Nov 26;204(12):3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Swirski FK, Nahrendorf M, Etzrodt M, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009 Jul 31;325(5940):612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Denes A, Humphreys N, Lane TE, et al. Chronic systemic infection exacerbates ischemic brain damage via a CCL5 (regulated on activation, normal T-cell expressed and secreted)-mediated proinflammatory response in mice. J Neurosci. Jul 28;30(30):10086–10095. doi: 10.1523/JNEUROSCI.1227-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Langdon KD, Maclellan CL, Corbett D. Prolonged, 24-h delayed peripheral inflammation increases short- and long-term functional impairment and histopathological damage after focal ischemia in the rat. J Cereb Blood Flow Metab. Aug;30(8):1450–1459. doi: 10.1038/jcbfm.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wehner T, Bontert M, Eyupoglu I, et al. Bone marrow-derived cells expressing green fluorescent protein under the control of the glial fibrillary acidic protein promoter do not differentiate into astrocytes in vitro and in vivo. J Neurosci. 2003 Jun 15;23(12):5004–5011. doi: 10.1523/JNEUROSCI.23-12-05004.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lawson LJ, Perry VH, Gordon S. Turnover of resident microglia in the normal adult mouse brain. Neuroscience. 1992;48(2):405–415. doi: 10.1016/0306-4522(92)90500-2. [DOI] [PubMed] [Google Scholar]
- 63.Simard AR, Rivest S. Bone marrow stem cells have the ability to populate the entire central nervous system into fully differentiated parenchymal microglia. Faseb J. 2004 Jun;18(9):998–1000. doi: 10.1096/fj.04-1517fje. [DOI] [PubMed] [Google Scholar]
- 64.Eglitis MA, Mezey E. Hematopoietic cells differentiate into both microglia and macroglia in the brains of adult mice. Proc Natl Acad Sci U S A. 1997 Apr 15;94(8):4080–4085. doi: 10.1073/pnas.94.8.4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shi Q, Rafii S, Wu MH, et al. Evidence for circulating bone marrow-derived endothelial cells. Blood. 1998 Jul 15;92(2):362–367. [PubMed] [Google Scholar]
- 66.Takahashi T, Kalka C, Masuda H, et al. Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat Med. 1999 Apr;5(4):434–438. doi: 10.1038/7434. [DOI] [PubMed] [Google Scholar]
- 67.Asahara T, Masuda H, Takahashi T, et al. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res. 1999 Aug 6;85(3):221–228. doi: 10.1161/01.res.85.3.221. [DOI] [PubMed] [Google Scholar]
- 68.Manning-Tobin JJ, Moore KJ, Seimon TA, et al. Loss of SR-A and CD36 activity reduces atherosclerotic lesion complexity without abrogating foam cell formation in hyperlipidemic mice. Arterioscler Thromb Vasc Biol. 2009 Jan;29(1):19–26. doi: 10.1161/ATVBAHA.108.176644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kennedy DJ, Kuchibhotla SD, Guy E, et al. Dietary cholesterol plays a role in CD36-mediated atherogenesis in LDLR-knockout mice. Arterioscler Thromb Vasc Biol. 2009 Oct;29(10):1481–1487. doi: 10.1161/ATVBAHA.109.191940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhou P, Qian L, Gallo EF, et al. The scavenger receptor CD36 contributes to the neurotoxicity of bone marrow-derived monocytes through peroxynitrite production. Neurobiol Dis. 2011 Jun;42(3):292–299. doi: 10.1016/j.nbd.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Auffray C, Fogg DK, Narni-Mancinelli E, et al. CX3CR1+ CD115+ CD135+ common macrophage/DC precursors and the role of CX3CR1 in their response to inflammation. J Exp Med. 2009 Mar 16;206(3):595–606. doi: 10.1084/jem.20081385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003 Jul;19(1):71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 73.Gordon S. Macrophage heterogeneity and tissue lipids. J Clin Invest. 2007 Jan;117(1):89–93. doi: 10.1172/JCI30992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tsou CL, Peters W, Si Y, et al. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest. 2007 Apr;117(4):902–909. doi: 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Isoda K, Folco E, Marwali MR, et al. Glycated LDL increases monocyte CC chemokine receptor 2 expression and monocyte chemoattractant protein-1-mediated chemotaxis. Atherosclerosis. 2008 Jun;198(2):307–312. doi: 10.1016/j.atherosclerosis.2007.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Borlongan CV. Bone marrow stem cell mobilization in stroke: a ‘bonehead’ may be good after all! Leukemia. 2011 Nov;25(11):1674–1686. doi: 10.1038/leu.2011.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Borlongan CV, Glover LE, Tajiri N, et al. The great migration of bone marrow-derived stem cells toward the ischemic brain: therapeutic implications for stroke and other neurological disorders. Prog Neurobiol. 2011 Oct;95(2):213–228. doi: 10.1016/j.pneurobio.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.