

Vascular plants invariably contain a class II diterpene cyclase (EC 5.5.1.x), as an ent-copalyl diphosphate synthase is required for gibberellin phytohormone biosynthesis. This has provided the basis for evolution of a functionally diverse enzymatic family.[1] These biocatalysts fold their substrate, the general diterpenoid precursor (E,E,E)-geranylgeranyl diphosphate (GGPP), to bring the terminal three carbon-carbon double bonds into proximity with each other, and then carry out bicyclization via a protonation-initiated carbocation cascade reaction. The resulting labda-15-en-8-yl+ diphosphate intermediate is most commonly quenched by deprotonation at an exocyclic methyl, as in the production of labdadienyl/copalyl diphosphate (Scheme 1). Alternatively, the bicyclized labda-15-en-8-yl+ diphosphate intermediate can be captured by water prior to deprotonation, to form hydroxylated compounds such as labda-15-en-8-ol diphosphate.[2] In addition, this intermediate can undergo subsequent rearrangement via 1,2-hydride and/or methyl shifts, starting with the hydrogen substituent on the neighboring endocyclic methine (C9).[3] However, terminating deprotonation at the neighboring endocyclic methylene (C7) has not previously been observed. Here we report that the lycophyte Selaginella moellendorffii contains a bifunctional diterpene synthase, SmCPSKSL1, which catalyzes just such a class II cyclization reaction. In particular, SmCPSKSL1 produces an endocyclic double bond isomer of copalyl diphosphate (CPP), as well as carries out subsequent replacement of the diphosphate by a hydroxyl group to form labda-7,13E-dien-15-ol. Although this is a known plant metabolite,[4] and a small family of bioactive derived natural products is known from a phylogenetically diverse group of plants,[4-5] its biosynthesis has not been previously investigated. Our results demonstrate that this diterpenoid can be generated by a single bifunctional diterpene synthase that directly generates the endocyclic double bond, as well as hydroxyl group.
Scheme 1.
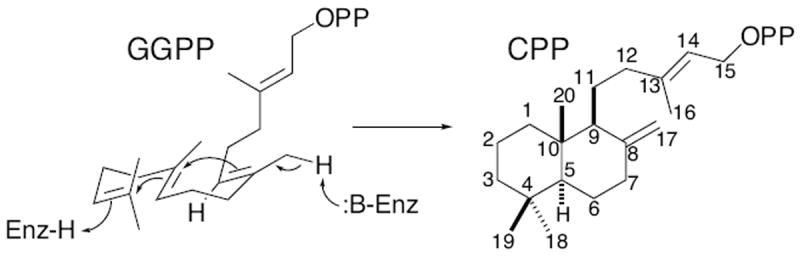
Cyclization of GGPP to CPP.
S. moellendorffii is a basal vascular land plant whose genome has been sequenced, and which contains a number of putative diterpene synthases.[6] At least one of these appears to be a bifunctional diterpene synthase (corresponding to the Joint Genome Institute gene accession Selmol 112927), as it contains both the DXDD motif associated with class II activity and DDXXD motif associated with allylic diphosphate ionization initiated class I activity.[7] In an attempt to determine the biochemical function of the encoded enzyme, a corresponding synthetic gene was obtained (Genscript), with codon-optimization for expression in Escherichia coli. Unfortunately, no diterpene synthase activity was observed with the resulting recombinant protein. The corresponding gene was then cloned from the lycophyte itself, revealing a few key differences from the predicted gene sequence, most notably a nine-nucleotide insertion. Expression of this native gene (SmCPSKSL1) in E. coli provided diterpene synthase activity. Specifically, upon co-expression with a GGPP synthase, using a previously described metabolic engineering system,[8] this bifunctional diterpene synthase predominantly led to production of an unrecognized diterpenoid by the recombinant bacteria, albeit with a few other minor products also observed (Figure 1).
Figure 1.
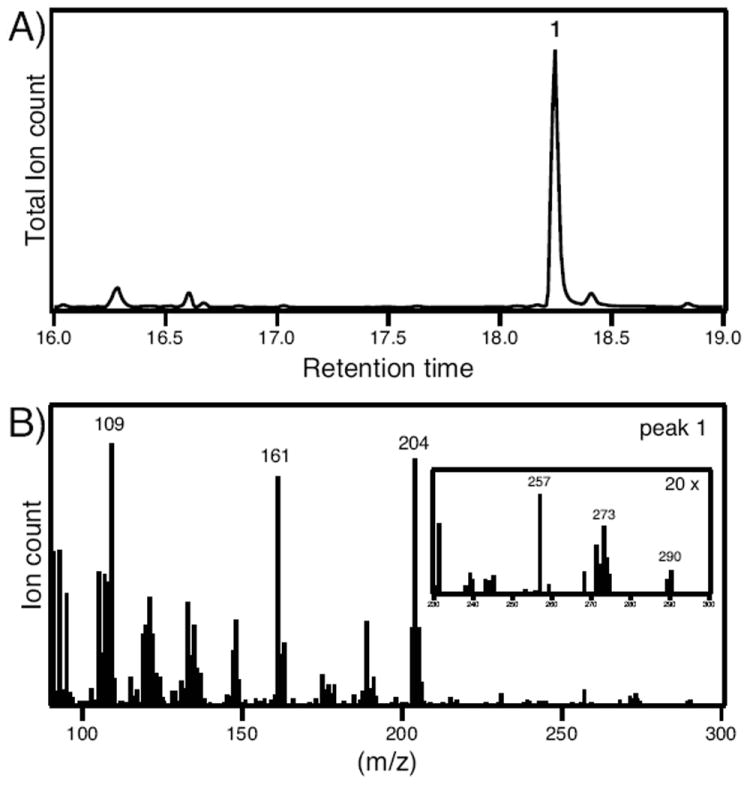
SmCPSKSL1 produces labda-7,13E-dien-15-ol (peak 1). A) GC-MS chromatogram of extract from E. coli engineered to co-express SmCPSKSL1 with a GGPP synthase. B) MS of peak 1 (labda-7,13E-dien-15-ol), with inset to clearly depict the molecular ion peak at m/z = 290.
The structure of this predominant diterpenoid, representing ~80% of the total product output, was determined by NMR. Sufficient amounts of material for this purpose was produced by increasing metabolic flux into isoprenoid/terpenoid metabolism in the metabolically engineered bacteria, as previously described,[9] and extraction from 2 L cultures of the resulting recombinant bacteria. Following purification via silica gel and reverse-phase high-performance liquid chromatography, 18 mg of this compound was obtained, enabling straightforward characterization. In particular, the structure was determined using HMBC, HSQC, and COSY data, leading to its assignment as labda-7,13E-dien-15-ol, which was confirmed by comparison to data in the literature.[4]
The presence of the primary C15 hydroxyl group essentially represents hydrolysis of the allylic diphosphate ester bond. While similar hydroxyl replacement of the diphosphate has been observed with other class I terpene synthases,[7] here it was possible that this was a result of direct hydrolysis catalyzed by endogenous phosphatases in the E. coli based metabolic engineering system. To verify that this was catalyzed by SmCPSKSL1, the recombinant enzyme was expressed with a 6×His tag, purified, and shown to catalyze direct production of labda-7,13E-dien-15-ol in vitro, even in the presence of phosphatase inhibitor. Thus, SmCPSKSL1 is bifunctional, catalyzing both class II bicyclization of GGPP and class I ionization of the resulting intermediate with the addition of water to C15 prior to deprotonation (Scheme 2). SmCPSKSL1 then provides the first example of a class I diterpene synthase that catalyzes such direct replacement of the diphosphate with a hydroxyl, although others that incorporate water following cyclization are known.[10] In addition, the mass spectra of the minor products suggests that these may correspond to production of labdatriene(s) and labda-7,14-dien-13-ol(s) – e.g., the apparent molecular ion peaks at m/z = 272 or 290, respectively – albeit of unknown configuration. While these will be more precisely characterized in future studies, production of such diterpenes is further consistent with class I mediated ionization of the postulated labda-7,13E-dienyl diphosphate intermediate produced by the class II active site, as analogous compounds are produced by other class I diterpene cyclases.[3a, 11]
Scheme 2.
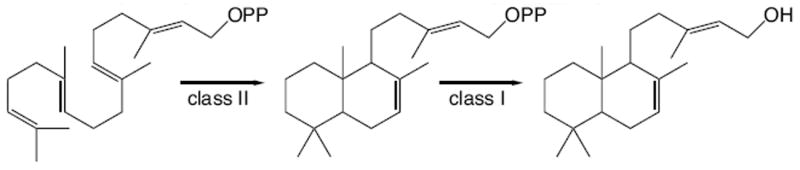
SmCPSKSL catalyzed production of labda-7,13E-dien-15-ol from GGPP.
Labdanes can be produced as several different stereoisomers, which presumably result from folding of GGPP into alternative prochiral conformations prior to class II diterpene cyclase catalyzed bicyclization.[1] While we can not assign the absolute stereochemistry of the SmCPSKSL1 labda-7,13E-dien-15-ol product, the observed NOE between the hydrogen substituents on C20 and C11 demonstrates that these C10 and C9 substituents exhibit a cis configuration. This then implies a pro-chair-chair conformation of GGPP in the class II active site. Based on analogous chemical shifts relative to those observed with other labdane-related diterpenes, as well as that of the previously characterized corresponding natural product,[4] we tentatively assign the SmCPSKLS1 product to the normal absolute configuration.
Regardless of absolute configuration, the production of an endocyclic double bond bicycle strongly indicates that SmCPSKSL1 catalyzes a unique class II reaction. Specifically, that the terminal labda-15-en-8-yl+ diphosphate intermediate is quenched by deprotonation at the neighboring C7 methylene. It is relatively easy to envision a concerted bicyclization reaction in the commonly catalyzed class II diterpene cyclase production of CPP, due to the evident ability to align the departing proton from the rotatable methyl group with the π orbitals of the relevant carbon-carbon double bonds (Scheme 1). Given the production of a C7 endocyclic double bond isomer observed here, the relative positioning of the C7 hydrogen substituents was examined. Notably, molecular modeling demonstrates that one of these hydrogens also can be closely aligned with the neighboring π orbital (e.g. C7α in the assumed “normal” configuration; Figure 2), enabling a similarly concerted bicyclization reaction, and providing insight into the conformation of the GGPP substrate in this class II active site.
Figure 2.
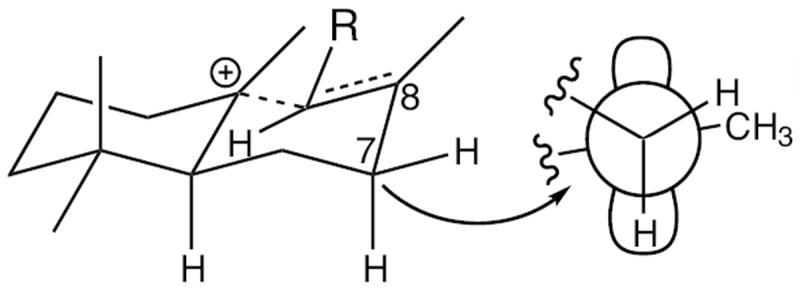
Relative configuration of C7 hydrogen substituents with π orbital of the sp3 hybridized C8 in SmCPSKSL1 catalyzed class II bicyclization. Depicted is putative moncyclic intermediate (R = allylic isoprenyl diphosphate “tail”), with Newman projection along the C7-C8 bond.
In conclusion, characterization of a bifunctional diterpene synthase from S. moellendorffii has revealed novel enzymatic activity. Most notably, this SmCPSKSL1 defines a new class II reaction, which further enabled some insight into GGPP substrate conformation, as well as providing the first example of a class I diterpene synthase carrying out replacement of the diphosphate with a hydroxyl. In addition, given the uncertain origins of the endocyclic double bond, along with that of the hydroxyl group, the biosynthesis of labda-7,13E-dien-15-ol was previously unclear, and potentially required several enzymatic steps. The results reported here demonstrate that labda-7,13E-dien-15-ol can instead be produced by a single (albeit bifunctional) enzyme. This highlights such simple biosynthesis, only requiring changes in a single enzymatic gene, as a potential mechanism enabling the repeated convergent evolution that presumably then underlies the observed phylogenetically scattered production of the derived family of natural products.
Experimental Section
SmCPSKSL1 was cloned from S. moellendorffii maintained in a terrarium under a 14/10 hr light/dark cycle at 22 °C. Detached plant material was induced using 5 mM methyl jasmonate for 36 hours, RNA extracted using a Plant RNA Purification kit (Invitrogen, Carlsbad CA), and cDNA produced with the Superscript system (Invitrogen). Primers were designed from putative bifunctional diterpene synthases (Selmol 120716 and 112927) found in the S. mollendorffii genome (http://genome.jgi-psf.org/Selmo1/Selmo1.home.html). SmCPSKSL1 was cloned using ATGATAGAGGAAATGAGAAAATTGCTTGC and TCATTCAGCTGCTTTATACAACACATT as the forward and reverse primers, respectively. The PCR product was cloned into pENTR/SD/D-Topo (Invitrogen) and completely sequenced, with deposition into Genbank (accession JN001323). SmCPSKSL1 was then transferred by directional recombination into pDEST15 and pDEST17 expression vectors.
SmCPSKSL1 activity was initially assessed by co-transforming E. coli strain C41 (Lucigen, Middleton, WI) with the pDEST15 construct and a previously described pGG vector.[8] This recombinant bacteria was grown in a 50 mL culture of TB media to a mid-log phase (OD600 ~0.6) at 37 °C, transferred to 16 °C for 1 hour prior to induction with 0.5 mM isopropylthiogalactoside (IPTG). Thereafter, they were fermented for an additional 72 hrs, and the culture then extracted with an equal volume of hexanes. This extract was dried under a stream of N2 and the residue redissolved in 200 μL of hexane for analysis by gas chromatography with mass spectrometry detection (GC-MS), using a Varian 3900 GC with Saturn 2100T ion-trap MS (Varian Inc., Palo Alto, CA), as previously described.[11a, 12]
A larger amount of the predominant unrecognized diterpenoid was obtained by additional transformation with a pIRS vector that increases the endogenous isoprenoid precursor supply.[9] The resulting bacterial was grown in 2 × 1 L cultures and extracted, as described above. The extract was dried by rotary evaporation, resuspended in 10 mL hexanes, and passed over a 10 mL column of silica gel, which was then eluted with 10 mL 20% (v/v) ethyl acetate in hexanes. This eluate was dried under a stream of N2 and resuspended in methanol, and the diterpenoid purified via reverse-phase chromatography using a C18 column on an Agilent 1100 series high-performance system (Santa Clara, CA), essentially as previously described.[11a, 12]
Structural analysis by NMR also was carried out much as previously described.[11a, 12] Following identification of the product as labda-7,13E-dien-15-ol, the chemical shift and MS data was compared to that previously reported for this compound,[4] demonstrating good agreement. Thus, only the chemical shift data is reported here. Proton (700.13 MHz) chemical shifts and assignments: δ 0.919 ppm (1H, dt, 3.6 Hz, 13.0 Hz, H1a), 1.818 (1H, m, H1b), 1.414 (1H, m, H2a), 1.510 (1H, m, H2b), 1.132 (1H, d, 13.4 Hz, H3a), 1.384 (1H, t, H3b), 1.158 (1H, m, H5), 1.840 (1H, m, H6a), 1.945 (1H, m, H6b), 5.369 (1H, bs, H7), 1.591 (1H, s, H9), 1.256 (1H, m, H11a), 1.528 (1H, m, H11b), 1.935 (1H, m, H12a), 2.201 (1H, dt, 4.7 Hz, 12.4 Hz, H12b), 5.397 (1H, t, 6.8 Hz, H14), 4.134 (2H, d, 6.9 Hz, H15a-b), 1.669 (3H, s, H16), 1.674 (3H, s, H17), 0.830 (3H, s, H18 or H19), 0.852 (3H, s, H18 or H19), 0.731 (3H, s, H20). Carbon-13 (174.05 MHz) chemical shifts and assignments: δ (ppm) 39.35 (C1), 19.00 (C2), 42.51 (C3), 33.17 (C4), 50.35 (C5), 24.02 (C6), 122.53 (C7), 135.46 (C8), 54.66 (C9), 37.00 (C10), 25.79 (C11), 42.28 (C12), 140.61 (C13), 123.55 (C14), 59.63 (C15), 16.59 (C16), 22.40 (C17), 33.36 (C18 or C19), 22.05 (C18 or C19), 13.75 (C20). The cis configuration of the C9-10 substituents was assigned on the basis of the observed NOE correlations in a NOESY spectrum. NOE correlations for H11a and H11b (and their relative intensities observed), are H1b (s) and H20 (s). The assigned E configuration of C13 was based on a strong NOE observed between H15 and H16.
In vitro assays with SmCPSKSL1 were carried out using the pDEST17 construct in C41 E. coli. The recombinant bacteria were grown in NZY liquid media to a mid-log phase (OD600=0.6) at 37°C, transferred to 16°C for 1 hr prior to induction with IPTG to a final concentration of 0.5 mM, and fermented overnight (14–16 hrs). Cells were harvested by centrifugation and suspended in lysis buffer (10 mM Tris–Cl, pH 6.8, 10% glycerol, 1 mM dithiothreitol) for sonication. The resulting lysates were clarified via centrifugation (15 min × 15,000×g). The recombinant protein was then purified over Ni-NTA Superflow resin (Novagen) following the manufacturers protocols. The purified enzyme was characterized in assay buffer (50 mM HEPES, pH 7.8, 100 mM KCl, 5 mM MgCl2 and 10% glycerol) with 10 μM GGPP as substrate, 50 μL of the purified enzyme, and in some cases 100 mM PhosStop phosphatase inhibitor (Roche Diagnostics), in 1 mL assays. These were incubated 10 min at 30 °C, and the reaction stopped by addition of 110 μL of 0.2 M N-ethylmaleimide for 5 min at room temperature, which was then quenched by the addition of 10 μL DTT. The production of labda-7,13E-dien-15-ol was verified by extraction with an equal volume of hexanes, which was dried under a stream of N2, resuspended in 50 μL hexanes, and then analyzed by GC-MS.
Acknowledgments
We thank Prof. Robert M. Coates for assistance with preparation of Figure 2. This work was supported by grants from the NIH (GM076324) and NSF (MCB0919735) to R.J.P.
References
- 1.Peters RJ. Nat Prod Rep. 2010;27:1521–1530. doi: 10.1039/c0np00019a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Falara V, Pichersky E, Kanellis AK. Plant Physiol. 2010;154:301–310. doi: 10.1104/pp.110.159566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Hamano Y, Kuzuyama Y, Itoh N, Furihata K, Seto H, Dairi T. J Biol Chem. 2002;277:37098–37104. doi: 10.1074/jbc.M206382200. [DOI] [PubMed] [Google Scholar]; b) Nakano C, Okamura T, Sato T, Dairi T, Hoshino T. Chem Commun (Camb) 2005;2005:1016–1018. doi: 10.1039/b415346d. [DOI] [PubMed] [Google Scholar]
- 4.a) Suzuki H, Noma M, Kawashima N. Phytochemistry. 1983;22:1294–1295. [Google Scholar]; b) Demetzos C, Harvala C, Philianos SM, Skaltsounis AL. J Nat Prod. 1990;53:1365–1368. [Google Scholar]
- 5.a) Rodilla JML, de Mendonca DIM, Urones JG, Moro RF. Phytochemistry. 1998;49:817–822. [Google Scholar]; b) Feresin GE, Tapia A, Gimenez A, Ravelo AG, Zacchino S, Sortino M, Schmeda-Hirschmann G. J Ethnopharmacol. 2003;89:73–80. doi: 10.1016/s0378-8741(03)00259-9. [DOI] [PubMed] [Google Scholar]; c) Aoki W, Ohtsuki T, Sadhu SK, Sato M, Koyano T, Preeprame S, Kowithayakorn T, Ishibashi M. J Nat Med. 2007;61:77–79. [Google Scholar]; d) Bevan CWL, Ekong DEU, Okogun JI. J Chem Soc C. 1968:1067–1070. [Google Scholar]; e) Rodriguez B. Phytochemistry. 1978;17:281–286. [Google Scholar]
- 6.Chen F, Tholl D, Bohlmann J, Pichersky E. Plant J. 2011;66:212–229. doi: 10.1111/j.1365-313X.2011.04520.x. [DOI] [PubMed] [Google Scholar]
- 7.Christianson DW. Chem Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- 8.Cyr A, Wilderman PR, Determan M, Peters RJ. J Am Chem Soc. 2007;129:6684–6685. doi: 10.1021/ja071158n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morrone D, Lowry L, Determan MK, Hershey DM, Xu M, Peters RJ. Appl Microbiol Biotechnol. 2010;85:1893–1906. doi: 10.1007/s00253-009-2219-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Hayashi K, Kawaide H, Notomi M, Sakigi Y, Matsuo A, Nozaki H. FEBS Lett. 2006;580:6175–6181. doi: 10.1016/j.febslet.2006.10.018. [DOI] [PubMed] [Google Scholar]; b) Toyomasu T, Niida R, Kenmoku H, Kanno Y, Miura S, Nakano C, Shiono Y, Mitsuhashi W, Toshima H, Oikawa H, Hoshino T, Dairi T, Kato N, Sassa T. Biosci Biotechnol Biochem. 2008;72:1038–1047. doi: 10.1271/bbb.70790. [DOI] [PubMed] [Google Scholar]; c) Oikawa H, Toyomasu T, Toshima H, Ohashi S, Kawaide H, Kamiya Y, Ohtsuka M, Shinoda S, Mitsuhashi W, Sassa T. J Am Chem Soc. 2001;123:5154–5155. doi: 10.1021/ja015747j. [DOI] [PubMed] [Google Scholar]; d) Herde M, Gartner K, Kollner TG, Fode B, Boland W, Gershenzon J, Gatz C, Tholl D. Plant Cell. 2008;20:1152–1168. doi: 10.1105/tpc.106.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Morrone D, Hillwig ML, Mead ME, Lowry L, Fulton DB, Peters RJ. Biochem J. 2011;435:589–595. doi: 10.1042/BJ20101429. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Maugel N, Mann FM, Hillwig ML, Peters RJ, Snider BB. Org Lett. 2010;12:2626–2629. doi: 10.1021/ol100832h. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Nakano C, Ootsuka T, Takayama K, Mitsui T, Sato T, Hoshino T. Bioscience, biotechnology, and biochemistry. 2011;75:75–81. doi: 10.1271/bbb.100570. [DOI] [PubMed] [Google Scholar]
- 12.a) Morrone D, Xu M, Fulton DB, Determan MK, Peters RJ. J Am Chem Soc. 2008;130:5400–5401. doi: 10.1021/ja710524w. [DOI] [PubMed] [Google Scholar]; b) Swaminathan S, Morrone D, Wang Q, Fulton DB, Peters RJ. Plant Cell. 2009;21:3315–3325. doi: 10.1105/tpc.108.063677. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gao W, Hillwig ML, Huang L, Cui G, Wang X, Kong J, Yang B, Peters RJ. Org Lett. 2009;11:5170–5173. doi: 10.1021/ol902051v. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Wang Q, Hillwig ML, Peters RJ. Plant J. 2011;65:87–95. doi: 10.1111/j.1365-313X.2010.04408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]